Abstract
Tetrasomy 9p is a chromosomal disorder characterized by the presence of a supernumerary chromosome. This rare abnormality exhibits a broad phenotypic variability and is not clearly distinguishable from other more frequent aneuploidies in the prenatal setting. We present two prenatal cases with dissimilar phenotypes, one with solely increased fetal nuchal translucency and one with multiple congenital anomalies, and discuss prior studies. These cases illustrate the difficulty of prenatally diagnosing this condition based on phenotypic information alone. We conclude that invasive prenatal diagnosis with (molecular) karyotyping is the best choice for the prenatal detection of tetrasomy 9p.
1. Introduction
Tetrasomy 9p is a chromosomal disorder characterized by the presence of a supernumerary chromosome, isochromosome 9p, originating from the short arm of chromosome 9 [1]. This rare imbalance was first described by Ghymers et al. in 1973 [2]. Since then, 107 cases have been reported, 22 of which were diagnosed prenatally [3,4].
It has been stated that tetrasomy 9p is a clinically recognizable syndrome with several recurrent characteristics, including craniofacial dysmorphism, intra-uterine growth restriction (IUGR), and Dandy–Walker malformation [5,6]. Yet, there is considerable phenotypic variability among patients, which is assumed to correlate with the degree of mosaicism and the tissue distribution of the partial tetrasomy; a correlation between disease severity and the size of the isochromosome has been suggested but never proven [3,5,7]. Especially in a prenatal context, it is challenging to distinguish this partial aneuploidy exclusively on the basis of ultrasound findings [7].
In this work, we present two prenatal cases of tetrasomy 9p, with different clinical manifestations, thereby contributing to the existing knowledge on this rare chromosomal aberration.
2. Case Presentation
2.1. Case 1
A 34-year-old, G2P1A0, of Middle Eastern descent, was referred at 14+2 weeks amenorrhoea because of increased fetal nuchal translucency (NT). A prenatal ultrasound revealed an increased nuchal fold of 7 mm (Figure 1A) and a single umbilical artery (Figure 1B). The biometric findings were unremarkable. Amniocentesis was performed. The single nucleotide polymorphism (SNP) microarray (cytoSNP12-v2.1, Illumina, San Diego, CA, USA) demonstrated a 39Mb triplication of the 9p24.3p13.1 region in this female fetus: arr[GRCh37] 9p24.3p13.1(206132_38771460) × 4. As the SNP array data were nearly identical for cases 1 and 2, we chose to show only those of Case 2 (see Figure 2). Additional karyotyping on cultured amniocytes demonstrated that the extra material was present as an extra derivative chromosome 9, consisting of the short arm of chromosome 9, the centromere, a part of the long arm (up to chromosome band 9q21), followed by most of the short arm (Figure 3). The apparent difference between the SNP array result and the karyotyping can be explained by the absence of SNPs in the heterochromatic region around the centromere of chromosome 9 (9p11.2–9q13).
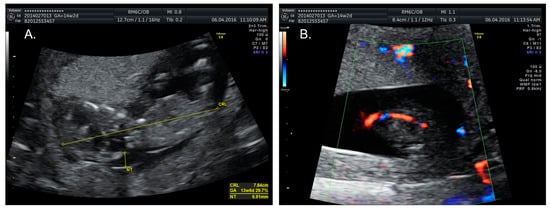
Figure 1.
Prenatal ultrasound images of Case 1, taken during the first trimester. (A) Nuchal fold measurement shows increased fetal nuchal translucency (NT) of almost 7 mm; (B) Single umbilical artery.
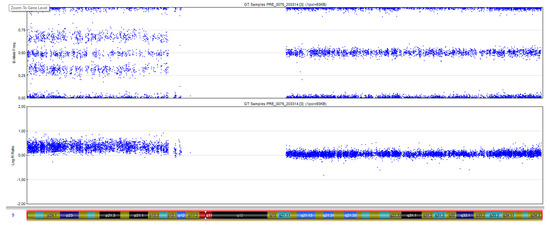
Figure 2.
SNP array result of chromosome 9 of Case 2. Upper panel: B allele frequency, showing a ‘split’ along the short arm of chromosome 9 because of the additional genotypes AAAB, AABB, and ABBB. Lower panel: The logR ratio of the SNPs on the short arm of chromosome 9 is increased as compared to those on the long arm, given that 4 copies are present instead of 2.
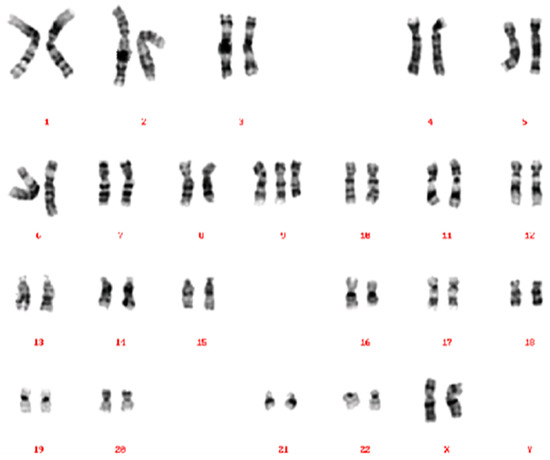
Figure 3.
Karyogram of Case 1, showing a 47,XX,+der(9)(pter→q21::p12→pter) karyotype. The derivative chromosome 9 consists of the short arm of chromosome 9, the centromere, a part of the long arm (up to chromosome band 9q21), followed by an additional copy of most of the short arm.
The couple received genetic counseling and, as a result of the variable phenotype with the possibility of delayed psychomotor development and intellectual disability, the parents chose, after approval by the ethics committee, to terminate the pregnancy at 18 weeks of gestation. An autopsy was refused for religious reasons. Karyotyping of both parents showed no structural abnormalities that could account for the tetrasomy 9p observed in the fetus. The recurrence risk was thus estimated to be low. Amniocentesis in a subsequent pregnancy showed no abnormalities on the SNP array.
2.2. Case 2
A healthy 36-year-old, G4P2A1, was referred at 13 weeks of gestation for prenatal diagnostics because of hydrops fetalis (Figure 4A). An abdominal ultrasound revealed cerebellar aplasia and an absent nasal bone, along with branchial cleft cysts and hypoplastic left heart syndrome with ventricular septum defect, in addition to the known hydrops fetalis (Figure 4B). An SNP microarray on DNA that was isolated from a chorion villus biopsy revealed a 39 Mb triplication of the short arm of chromosome 9 (9p24.3p13.1) in this male fetus, corresponding to a tetrasomy 9p: arr[GRCh37] 9p24.3p13.1(46587_38771460) × 4) (Figure 2). No additional karyotyping was performed, thus precluding definite proof of the presence of a supernumerary marker chromosome, nor of its size, like in Case 1. Due to the extent of the fetal malformations and the poor prognosis, the couple chose to terminate the pregnancy at 15 weeks of gestation (Figure 4C) after approval by the ethics committee. This couple also declined fetal autopsy for reasons of faith. There was no consanguinity, and the karyotyping in the parents showed no structural anomalies.
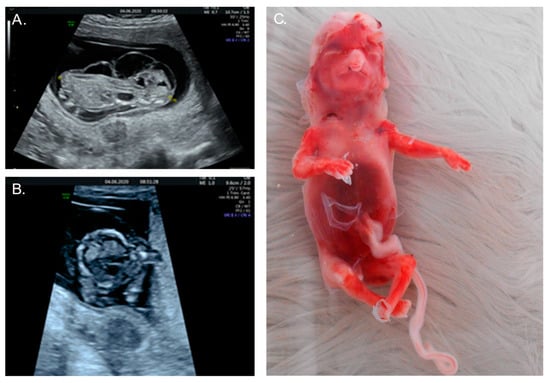
Figure 4.
Images of Case 2. (A) Prenatal ultrasound at 13 weeks shows hydrops fetalis; (B) Hypoplastic left heart syndrome at 13 weeks; (C) 15-week fetus postpartum showing loose skin as a result of extreme hydrops. Also visible are typical dysmorphic facial features, including hypertelorism, small chin, bulbous nose, and cleft lip.
3. Discussion
In this paper, we describe two prenatal cases with a molecularly proven tetrasomy 9p. The supernumerary chromosome in this rare abnormality can have one of three different constitutions: the entire short arm, the entire short arm and part of the heterochromatic region of the long arm, or the entire short arm and part of the long arm extending into the euchromatic region [5,6].
Historically, although several different mechanisms have been hypothesized to cause supernumerary chromosomes, all recent studies agree with the theory of maternal meiosis II nondisjunction, followed by a rearrangement or centromeric misdivision, resulting in the duplication of the short arm and loss of the long arm of the chromosome [3,6]. This phenomenon arises predominantly de novo; hence, the recurrence risk for future pregnancies is low [1]. In 2015 however, El Khattabi et al. described the first case of possible parental germline mosaicism, reporting two siblings with tetrasomy 9p [1].
It is generally accepted that the risk for aneuploidies caused by errors in meiosis I and II (e.g., trisomy 13, 16, 18, and 21) increases with maternal age [8,9,10]. This effect was confirmed by an in vitro chromosomal study on 1397 oocytes, which detected a significant increase in aneuploidies at maternal ages above 36 years [8]. The contribution of maternal age to the occurrence of tetrasomy 9p nevertheless remains a matter of debate; some authors assume that advanced maternal age contributes to meiotic nondisjunction, while others state that the low mean maternal age for all known tetrasomy 9p cases (32.3 years) demonstrates that maternal age does not seem to influence the occurrence of this syndrome [1,11].
According to prior studies, tetrasomy 9p is emerging as an easily recognizable syndrome with many recurring aberrations [1,6,12]. On ultrasound, IUGR (45%), central nervous system abnormalities (55%), such as ventriculomegaly and Dandy–Walker malformation, cleft lip and/or palate (45%), and skeletal defects, such as wide sutures and limb defects (55%) are frequently observed [3]. At birth, facial dysmorphias can be observed, including hypertelorism, ear and ocular abnormalities, micrognathia, a broad nasal root or bulbous/beaked nose, congenital heart defects, urogenital defects, and gastrointestinal malformations [1,13]. In Table 1, we summarized the prenatal abnormalities described in previous cases and related them to the two new cases presented here.

Table 1.
Prenatally detected ultrasound abnormalities in our cases, compared to those in the 22 published prenatal tetrasomy 9p cases.
For Case 1, the elevated fetal NT early in the second trimester was the reason for referral and amniocentesis, as fetuses with increased NT are at risk of aneuploidy or other chromosomal anomalies [3]. Sporadic case studies have reported the association between increased NT and tetrasomy 9p. This finding is not imperative to the diagnosis, however, as it is present in only 18% of all cases. On the other hand, this marker is found in many fetuses with chromosomal defects [5,7]. For Case 2, although multiple abnormalities were suspected on the first-trimester ultrasound, the main reason for further investigation was the presence of hydrops fetalis. Of the 22 prenatally diagnosed cases of tetrasomy 9p described by previous studies, only one other case mentions hydrops fetalis as the primary phenotype [6].
Another finding that has recently been associated with tetrasomy 9p is an absent nasal bone in the first trimester [13]. The absence of the nasal bone can be a marker of different chromosomal abnormalities, including trisomy 13, trisomy 18, and, most notably, trisomy 21 or Down syndrome. The association with tetrasomy 9p is a relatively new finding, however, having first been described by Podolsky et al. in 2011 [13]. To date, only one additional case of a fetus with an absent nasal bone has been reported [1]. Here, we present another case (Case 2) of a fetus with an absent nasal bone on ultrasound, underscoring that this finding makes inclusion of tetrasomy 9p in the differential diagnosis necessarywhen noted.
The cases presented in this report—one presenting solely with increased fetal NT, the other presenting with multiple anomalies—underscore the large phenotypic variability of the tetrasomy 9p syndrome.
Currently, very little is known about the contribution of individual genes in the triplicated region to the different symptoms. The minimal region that is triplicated in our patients contains 264 genes, among which are several genes associated with neurodevelopment (GLDC, KANK1, DOCK8, FOXD4, CBWD1, and PTPRD), but none of these have definitively been shown to be triplosensitive (see https://www.clinicalgenome.org/, accessed on 12 February 2022), meaning that the additional copy is not expected to cause a phenotype. It has been suggested that the severity of the tetrasomy 9p phenotype increases with the involvement of (genes on) the q arm since this region’s involvement is more often associated with cardiac malformations, intellectual disability, and death, however, this statement is controversial, as it is mostly based on karyotype results with a poor resolution [1,3]. Most authors state that no clear correlation can be found between the length or type of isochromosome and the phenotypic outcome [12]. So far, none of the genes located in the maximally triplicated region (up to chromosome band 9q21.11) have received a high triplosensitivity score. In this study, Case 1, to whom the triplication extends up to 9q21, shows a relatively mild phenotype, implying that factors other than the size of the aberration contribute to the severity of the phenotype. With the increased use of SNP array/array CGH instead of classic karyotyping, one would assume that the boundaries of the triplication could be defined more precisely. However, depending on the array platform used, very little to no SNPs are present in the heterochromatic region surrounding the centromere, hampering a direct comparison between cases analyzed by an array. In a large series of patients investigated by El Khattabi et al., the smallest region involved was 39 Mb, similar to the region triplicated in our patients, however, the duplicated region was as large as 73 Mb in some patients. Notably, the diagnosis in the 12 described cases was made on four different array platforms [1]. In general, dosage sensitivity and genes that are known to be triplosensitive, thus, duplication intolerant can be found through the genome and cross-disorder dosage sensitivity maps of the genome begin to emerge [26].
The effect of the degree of mosaicism on the phenotypic outcome seems more solid; prognosis in non-mosaic individuals is poor, with a high mortality rate in the neonatal period—two-thirds of the infants die within the first two months [12]. In cases with mosaicism for tetrasomy 9p on the other hand, no early deaths have been reported and 8.4% of the detected tetrasomy 9p cases are not associated with major adverse health outcomes [4,12]. Both cases described here appear to be non-mosaic, although these results are based on the analysis of one sample type (amniocentesis and chorion villus biopsy, respectively), yet presented with a different phenotype. At present, we have no explanation for this difference in outcome.
Prenatal genetic testing is crucial to reach a definite diagnosis so that parents can be informed in a timely manner on the aspects of the disorder and make a well-informed decision concerning whether or not to carry the pregnancy to term. Prenatal testing can be either non-invasive (NIPT) or invasive (amniocentesis or chorion villus biopsy). Despite the fact that, given its size and nature (~40 Mb triplication), tetrasomy 9p should be detectable with a genome-wide NIPT, and that with the wide implementation of NIPT, the first-trimester ultrasound is frequently limited to a dating scan, we advocate the preservation of the first-trimester fetal anomaly scan, prior to the venipuncture for the NIPT, so that it can be replaced with an invasive test in case of anomalies. First, since the material tested by NIPT is placental in origin, a confirmatory invasive test is nonetheless required after a positive NIPT to confirm the presence of the aberration in the fetus and to determine the grade of mosaicism and the exact size of the aberration [27]. Second, a (molecular) karyotype on an invasively obtained sample can detect all sub-chromosomal aberrations at a much higher resolution than NIPT. Thirdly, in case the karyotype is normal, the invasively obtained DNA can be used for whole exome analysis to look for genetic defects at the single base-pair level. In case of ultrasound abnormalities (as in Case 2), the diagnosis of non-mosaic tetrasomy 9p in chorionic villi can be considered conclusive. In the absence of ultrasound anomalies, a mosaic tetrasomy 9p in chorionic villus sampling (CVS) can be indicative of confined placental mosaicism, warranting confirmation with amniocentesis.
In both cases described here, the parents opted for termination of the pregnancy after extensive multidisciplinary counseling and approval by the ethical committee of the hospital.
Author Contributions
Conceptualization, B.B., K.J. and M.Z.; investigation, B.B. and K.J.; writing—original draft preparation, B.B., K.J. and M.Z.; writing—review and editing, B.B., K.J., M.Z., W.H. and P.L.; visualization, M.Z. and W.H.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Ethical review and approval were waived for this study by the local Ethics Committee as this is not required for case reports and review papers.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study. Written informed consent has been obtained from the patients to publish this paper.
Data Availability Statement
All data are stored in the medical files of the patients.
Acknowledgments
We thank the patients for their willingness to participate in this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- El Khattabi, L.; Jaillard, S.; Andrieux, J.; Pasquier, L.; Perrin, L.; Capri, Y.; Benmansour, A.; Toutain, A.; Marcorelles, P.; Vincent-Delorme, C.; et al. Clinical and molecular delineation of Tetrasomy 9p syndrome: Report of 12 new cases and literature review. Am. J. Med. Genet. Part A 2015, 167, 1252–1261. [Google Scholar] [CrossRef] [PubMed]
- Ghymers, D.; Hermann, B.; Distèche, C.; Frederic, J. Partial tetrasomy of number 9 chromosome, and mosaicism in a child with multiple malformations (author’s transl). Humangenetik 1973, 20, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Vinkšel, M.; Volk, M.; Peterlin, B.; Lovrecic, L. A systematic clinical review of prenatally diagnosed tetrasomy 9p. Balk. J. Med. Genet. 2019, 22, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Liehr, T.; Al-Rikabi, A. Mosaicism: Reason for normal phenotypes in carriers of small supernumerary marker chromosomes with known adverse outcome. A systematic review. Front. Genet. 2019, 10, 1131. [Google Scholar] [CrossRef]
- Nakamura-Pereira, M.; Cima, L.C.; Llerena, J.C., Jr.; Guerra, F.A.; Peixoto-Filho, F.M. Sonographic findings in a case of tetrasomy 9p associated with increased nuchal translucency and Dandy-Walker malformation. J. Clin. Ultrasound 2009, 37, 471–474. [Google Scholar] [CrossRef]
- Henriques-Coelho, T.; Oliva-Teles, N.; Fonseca-Silva, M.L.; Tibboel, D.; Guimarães, H.; Correia-Pinto, J. Congenital diaphragmatic hernia in a patient with tetrasomy 9p. J. Pediatr. Surg. 2005, 40, e29–e31. [Google Scholar] [CrossRef]
- Lazebnik, N.; Cohen, L. Prenatal diagnosis and findings of tetrasomy 9p. J. Obstet. Gynaecol. Res. 2015, 41, 997–1002. [Google Scholar] [CrossRef]
- Pellestor, F. Maternal age and chromosomal abnormalities in human oocytes. Med. Sci. M/S 2004, 20, 691–696. [Google Scholar] [CrossRef][Green Version]
- Eichenlaub-Ritter, U. Parental age-related aneuploidy in human germ cells and offspring: A story of past and present. Environ. Mol. Mutagen. 1996, 28, 211–236. [Google Scholar] [CrossRef]
- Nicolaides, K.H. Screening for fetal aneuploidies at 11 to 13 weeks. Prenat. Diagn. 2011, 31, 7–15. [Google Scholar] [CrossRef]
- De Azevedo Moreira, L.M.; Freitas, L.M.; Gusmão, F.A.; Riegel, M. New case of non-mosaic tetrasomy 9p in a severely polymalformed newborn girl. Birth Defects Res. Part A Clin. Mol. Teratol. 2003, 67, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Dhandha, S.; Hogge, W.A.; Surti, U.; McPherson, E. Three cases of tetrasomy 9p. Am. J. Med. Genet. 2002, 113, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Podolsky, R.; Saltzman, D.; Auerbach, M.; Roman, A.S. Absent nasal bone as a marker of tetrasomy 9p. Prenat. Diagn. 2011, 31, 1313. [Google Scholar] [CrossRef] [PubMed]
- McDowall, A.A.; Blunt, S.; Berry, A.C.; Fensom, A.H. Prenatal diagnosis of a case of tetrasomy 9p. Prenat. Diagn 1989, 9, 809–811. [Google Scholar] [CrossRef]
- Schaefer, G.B.; Domek, D.B.; Morgan, M.A.; Muneer, R.S.; Johnson, S.F. Tetrasomy of the short arm of chromosome 9: Prenatal diagnosis and further delineation of the phenotype. Am. J. Med. Genet. 1991, 38, 612–615. [Google Scholar] [CrossRef]
- Van Hove, J.; Kleczkowska, A.; De Bruyn, M.; Bekaert, J.; Fryns, J.P. Tetrasomy 9p: Prenatal diagnosis and fetopathological findings in a second trimester male fetus. Ann. Genet. 1994, 37, 139–142. [Google Scholar]
- Cazorla Calleja, M.R.; Verdú, A.; Félix, V. Dandy-Walker malformation in an infant with tetrasomy 9p. Brain Dev. 2003, 25, 220–223. [Google Scholar] [CrossRef]
- Deurloo, K.L.; Cobben, J.M.; Heins, Y.M.; de Ru, M.; Wijnaendts, L.C.; van Vugt, J.M. Prenatal diagnosis of tetrasomy 9p in a 19-week-old fetus with Dandy-Walker malformation: A case report. Prenat. Diagn. 2004, 24, 796–798. [Google Scholar] [CrossRef]
- Tang, W.; Boyd, B.K.; Hummel, M.; Wenger, S.L. Prenatal diagnosis of tetrasomy 9p. Am. J. Med. Genet. A 2004, 126A, 328. [Google Scholar] [CrossRef]
- Hengstschläger, M.; Bettelheim, D.; Drahonsky, R.; Repa, C.; Deutinger, J.; Bernaschek, G. Prenatal diagnosis of tetrasomy 9p with Dandy-Walker malformation. Prenat. Diagn. 2004, 24, 623–626. [Google Scholar] [CrossRef]
- Di Vera, E.; Liberati, M.; Celentano, C.; Calabrese, G.; Guanciali-Franchi, P.E.; Morizio, E.; Rotmensch, S. Rhombencephalosynapsis in a severely polymalformed fetus with non-mosaic tetrasomy 9p, in intracytoplasmic-sperm-injection pregnancy. J. Assist. Reprod. Genet. 2008, 25, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Dutly, F.; Balmer, D.; Baumer, A.; Binkert, F.; Schinzel, A. Isochromosomes 12p and 9p: Parental origin and possible mechanisms of formation. Eur. J. Hum. Genet. 1998, 6, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.Q.; Chen, X.M.; Hu, L.; Guan, X.Y.; Lu, G.X. Prenatal diagnosis of nonmosaic tetrasomy 9p by microdissection and FISH: Case report. Chin. Med. J. 2007, 120, 1281–1283. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.P.; Wang, L.K.; Chern, S.R.; Wu, P.S.; Chen, Y.T.; Kuo, Y.L.; Chen, W.L.; Lee, M.S.; Wang, W. Mosaic tetrasomy 9p at amniocentesis: Prenatal diagnosis, molecular cytogenetic characterization, and literature review. Taiwan. J. Obstet. Gynecol. 2014, 53, 79–85. [Google Scholar] [CrossRef]
- Wang, H.; Xie, L.S.; Wang, Y.; Mei, J. Prenatal diagnosis of mosaic tetrasomy 9p in a fetus with isolated persistent left superior vena cava. Taiwan. J. Obstet. Gynecol. 2015, 54, 204–205. [Google Scholar] [CrossRef]
- Collins, R.L.; Glessner, J.T.; Porcu, E.; Niestroj, L.-M.; Ulirsch, J.; Kellaris, G.; Howrigan, D.P.; Everett, S.; Mohajeri, K.; Nuttle, X.; et al. A cross-disorder dosage sensitivity map of the human genome. MedRxiv 2021. [Google Scholar] [CrossRef]
- Van Opstal, D.; Srebniak, M.I. Cytogenetic confirmation of a positive NIPT result: Evidence-based choice between chorionic villus sampling and amniocentesis depending on chromosome aberration. Expert Rev. Mol. Diagn. 2016, 16, 513–520. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).