
A General Overview of Transthyretin Cardiac Amyloidosis and Summary of Expert Opinions on Pre-Symptomatic Testing and Management of Asymptomatic Patients with a Focus on Transthyretin V122I


Abstract
1. Introduction
2. TTR-Cardiac Amyloidosis
2.1. Pathogenesis
2.2. Clinical Manifestations
2.2.1. Wild Type TTR-CA
2.2.2. Hereditary TTR-CA
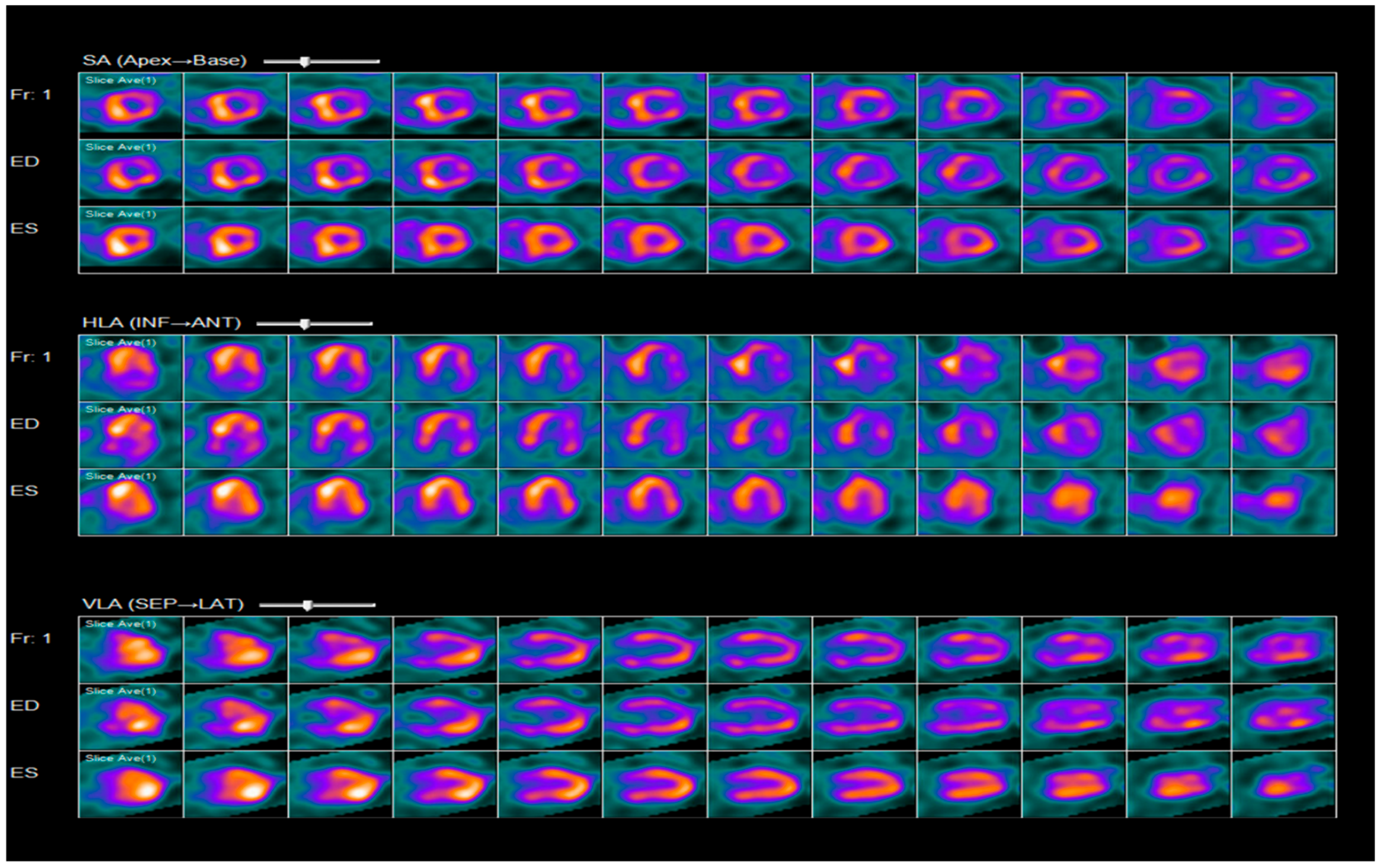
2.3. Diagnosis
2.4. Treatment
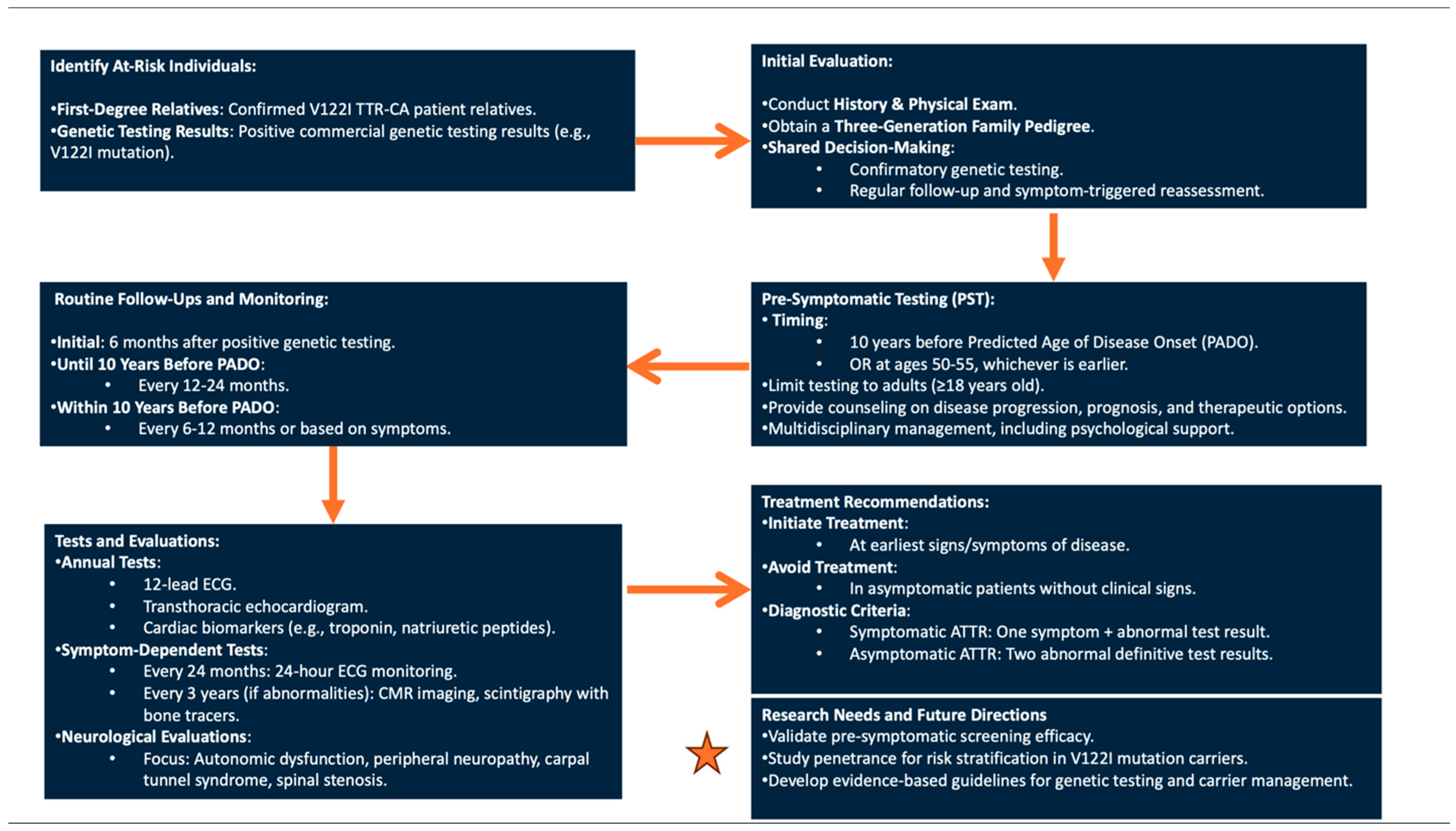
3. V122I TTR-CA: Pre-Symptomatic Genetic Testing Protocol and Management of Asymptomatic Carriers
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ash, S.; Shorer, E.; Ramgobin, D.; Vo, M.; Gibbons, J.; Golamari, R.; Jain, R.; Jain, R. Cardiac amyloidosis-A review of current literature for the practicing physician. Clin. Cardiol. 2021, 44, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.S.; Hanna, M.; Grogan, M.; Dispenzieri, A.; Witteles, R.; Drachman, B.; Judge, D.P.; Lenihan, D.J.; Gottlieb, S.S.; Shah, S.J.; et al. Genotype and Phenotype of Transthyretin Cardiac Amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J. Am. Coll. Cardiol. 2016, 68, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Shije, J.Z.; Bautista, M.A.B.; Smotherman, C. The Frequency of V122I Transthyretin Mutation in a Cohort of African American Individuals With Bilateral Carpal Tunnel Syndrome. Front. Neurol. 2022, 13, 949401. [Google Scholar] [CrossRef] [PubMed]
- Madhani, A.; Sabogal, N.; Massillon, D.; Paul, L.D.; Rodriguez, C.; Fine, D.; Helmke, S.; Winburn, M.; Kurian, D.; Raiszadeh, F.; et al. Clinical Penetrance of the Transthyretin V122I Variant in Older Black Patients With Heart Failure: The SCAN-MP (Screening for Cardiac Amyloidosis With Nuclear Imaging in Minority Populations) Study. J. Am. Heart Assoc. 2023, 12, e028973. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J.; Alexander, A.; Koziol, J.; Tagoe, C.; Fox, E.; Kitzman, D. Significance of the amyloidogenic transthyretin Val 122 Ile allele in African Americans in the Arteriosclerosis Risk in Communities (ARIC) and Cardiovascular Health (CHS) Studies. Am. Heart J. 2010, 159, 864–870. [Google Scholar] [CrossRef]
- Damrauer, S.M.; Chaudhary, K.; Cho, J.H.; Liang, L.W.; Argulian, E.; Chan, L.; Dobbyn, A.; Guerraty, M.A.; Judy, R.; Kay, J.; et al. Association of the V122I Hereditary Transthyretin Amyloidosis Genetic Variant With Heart Failure Among Individuals of African or Hispanic/Latino Ancestry. JAMA 2019, 322, 2191–2202. [Google Scholar] [CrossRef]
- Haring, B.; Hunt, R.P.; Shadyab, A.H.; Eaton, C.; Kaplan, R.; Martin, L.W.; Panjrath, G.; Kuller, L.H.; Assimes, T.; Kooperberg, C.; et al. Cardiovascular Disease and Mortality in Black Women Carrying the Amyloidogenic V122I Transthyretin Gene Variant. JACC Heart Fail. 2023, 11, 1189–1199. [Google Scholar] [CrossRef]
- Quarta, C.C.; Buxbaum, J.N.; Shah, A.M.; Falk, R.H.; Claggett, B.; Kitzman, D.W.; Mosley, T.H.; Butler, K.R.; Boerwinkle, E.; Solomon, S.D. The Amyloidogenic V122I Transthyretin Variant in Elderly Black Americans. N. Engl. J. Med. 2015, 372, 21–29. [Google Scholar] [CrossRef]
- Parcha, V.; Malla, G.; Irvin, M.R.; Armstrong, N.D.; Judd, S.E.; Lange, L.A.; Maurer, M.S.; Levitan, E.B.; Goyal, P.; Arora, G.; et al. Association of Transthyretin Val122Ile Variant with Incident Heart Failure Among Black Individuals. JAMA 2022, 327, 1368–1378. [Google Scholar] [CrossRef]
- Gilstrap, L.G.; Dominici, F.; Wang, Y.; El-Sady, M.S.; Singh, A.; Di Carli, M.F.; Falk, R.H.; Dorbala, S. Epidemiology of Cardiac Amyloidosis—Associated Heart Failure Hospitalizations Among Fee-for-Service Medicare Beneficiaries in the United States. Circ. Heart Fail. 2019, 12, e005407. [Google Scholar] [CrossRef]
- Garcia-Pavia, P.; Rapezzi, C.; Adler, Y.; Arad, M.; Basso, C.; Brucato, A.; Burazor, I.; Caforio, A.L.; Damy, T.; Eriksson, U.; et al. Diagnosis and treatment of cardiac amyloidosis. A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. J. Heart Fail. 2021, 23, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Ruberg, F.L.; Berk, J.L. Transthyretin (TTR) Cardiac Amyloidosis. Circulation 2012, 126, 1286–1300. [Google Scholar] [CrossRef]
- Scirpa, R.; Russo, D.; Tini, G.; Sclafani, M.; Tropea, A.; Cava, F.; Autore, C.; Musumeci, B. Clinical translation of genetic testing in TTR Amyloidosis: Genotype-phenotype correlations, management of asymptomatic carriers and familial screening. Vessel. Plus 2022, 6, 52. [Google Scholar] [CrossRef]
- Grandis, M.; Obici, L.; Luigetti, M.; Briani, C.; Benedicenti, F.; Bisogni, G.; Canepa, M.; Cappelli, F.; Danesino, C.; Fabrizi, G.M.; et al. Recommendations for pre-symptomatic genetic testing for hereditary transthyretin amyloidosis in the era of effective therapy: A multicenter Italian consensus. Orphanet J. Rare Dis. 2020, 15, 348. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Zahra, F. Transthyretin Amyloid Cardiomyopathy (ATTR-CM). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: http://www.ncbi.nlm.nih.gov/books/NBK574531/ (accessed on 3 November 2022).
- Buxbaum, J.N.; Ruberg, F.L. Transthyretin V122I (pV142I)* cardiac amyloidosis: An age-dependent autosomal dominant cardiomyopathy too common to be overlooked as a cause of significant heart disease in elderly African Americans. Anesth. Analg. 2017, 19, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Grogan, M.; Scott, C.G.; Kyle, R.A.; Zeldenrust, S.R.; Gertz, M.A.; Lin, G.; Klarich, K.W.; Miller, W.L.; Maleszewski, J.J.; Dispenzieri, A. Natural History of Wild-Type Transthyretin Cardiac Amyloidosis and Risk Stratification Using a Novel Staging System. J. Am. Coll. Cardiol. 2016, 68, 1014–1020. [Google Scholar] [CrossRef]
- Ruberg, F.L.; Grogan, M.; Hanna, M.; Kelly, J.W.; Maurer, M.S. Transthyretin Amyloid Cardiomyopathy. J. Am. Coll. Cardiol. 2019, 73, 2872–2891. [Google Scholar] [CrossRef]
- Kittleson, M.M.; Maurer, M.S.; Ambardekar, A.V.; Bullock-Palmer, R.P.; Chang, P.P.; Eisen, H.J.; Nair, A.P.; Nativi-Nicolau, J.; Ruberg, F.L.; On behalf of the American Heart Association Heart Failure; et al. Cardiac Amyloidosis: Evolving Diagnosis and Management: A Scientific Statement From the American Heart Association. Circulation 2020, 142, E7–E22. [Google Scholar] [CrossRef]
- Dispenzieri, A.; Coelho, T.; Conceição, I.; Waddington-Cruz, M.; Wixner, J.; Kristen, A.V.; Rapezzi, C.; Planté-Bordeneuve, V.; Gonzalez-Moreno, J.; Maurer, M.S.; et al. Clinical and genetic profile of patients enrolled in the Transthyretin Amyloidosis Outcomes Survey (THAOS): 14-year update. Orphanet J. Rare Dis. 2022, 17, 236. [Google Scholar] [CrossRef]
- Chandrashekar, P.; Alhuneafat, L.; Mannello, M.; Al-Rashdan, L.; Kim, M.M.; Dungu, J.; Alexander, K.; Masri, A. Prevalence and Outcomes of p.Val142Ile TTR Amyloidosis Cardiomyopathy: A Systematic Review. Circ. Genom. Precis. Med. 2021, 14, e003356. [Google Scholar] [CrossRef]
- Griffin, J.M.; Rosenthal, J.L.; Grodin, J.L.; Maurer, M.S.; Grogan, M.; Cheng, R.K. ATTR Amyloidosis: Current and Emerging Management Strategies. Cardio Oncol. 2021, 3, 488–505. [Google Scholar] [CrossRef] [PubMed]
- Pour-Ghaz, I.; Bath, A.; Kayali, S.; Alkhatib, D.; Yedlapati, N.; Rhea, I.; Khouzam, R.N.; Jefferies, J.L.; Nayyar, M. A Review of Cardiac Amyloidosis: Presentation, Diagnosis, and Treatment. Curr. Probl. Cardiol. 2022, 47, 101366. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.S.; Kale, P.; Fontana, M.; Berk, J.L.; Grogan, M.; Gustafsson, F.; Hung, R.R.; Gottlieb, R.L.; Damy, T.; González-Duarte, A.; et al. Patisiran Treatment in Patients with Transthyretin Cardiac Amyloidosis. N. Engl. J. Med. 2023, 389, 1553–1565. [Google Scholar] [CrossRef] [PubMed]
- Milandri, A.; Farioli, A.; Gagliardi, C.; Longhi, S.; Salvi, F.; Curti, S.; Foffi, S.; Caponetti, A.G.; Lorenzini, M.; Ferlini, A.; et al. Carpal tunnel syndrome in cardiac amyloidosis: Implications for early diagnosis and prognostic role across the spectrum of aetiologies. Eur. J. Heart Fail. 2020, 22, 507–515. [Google Scholar] [CrossRef]
- Fosbøl, E.L.; Rørth, R.; Leicht, B.P.; Schou, M.; Maurer, M.S.; Kristensen, S.L.; Kober, L.; Gustafsson, F. Association of Carpal Tunnel Syndrome With Amyloidosis, Heart Failure, and Adverse Cardiovascular Outcomes. J. Am. Coll. Cardiol. 2019, 74, 15–23. [Google Scholar] [CrossRef]
- Sperry, B.W.; Reyes, B.A.; Ikram, A.; Donnelly, J.P.; Phelan, D.; Jaber, W.A.; Shapiro, D.; Evans, P.J.; Maschke, S.; Kilpatrick, S.E.; et al. Tenosynovial and Cardiac Amyloidosis in Patients Undergoing Carpal Tunnel Release. J. Am. Coll. Cardiol. 2018, 72, 2040–2050. [Google Scholar] [CrossRef]
- Westin, O.; Fosbøl, E.L.; Maurer, M.S.; Leicht, B.P.; Hasbak, P.; Mylin, A.K.; Rørvig, S.; Lindkær, T.H.; Johannesen, H.H.; Gustafsson, F. Screening for Cardiac Amyloidosis 5 to 15 Years After Surgery for Bilateral Carpal Tunnel Syndrome. J. Am. Coll. Cardiol. 2022, 80, 967–977. [Google Scholar] [CrossRef]
- Arno, S.; Cowger, J. The genetics of cardiac amyloidosis. Heart Fail. Rev. 2021, 27, 1485–1492. [Google Scholar] [CrossRef]
- Alexander, K.M.; Orav, J.; Singh, A.; Jacob, S.A.; Menon, A.; Padera, R.F.; Kijewski, M.F.; Liao, R.; Di Carli, M.F.; Laubach, J.P.; et al. Geographic Disparities in Reported US Amyloidosis Mortality From 1979 to 2015: Potential Underdetection of Cardiac Amyloidosis. JAMA Cardiol. 2018, 3, 865. [Google Scholar] [CrossRef]
- Ueda, M.; Yamashita, T.; Misumi, Y.; Masuda, T.; Ando, Y. Origin of sporadic late-onset hereditary ATTR Val30Met amyloidosis in Japan. Amyloid 2018, 25, 143–147. [Google Scholar] [CrossRef]
- Pinto, M.V.; Pinto, L.F.; Dias, M.; Rosa, R.S.; Mundayat, R.; Pedrosa, R.C.; Waddington-Cruz, M. Late-onset hereditary ATTR V30M amyloidosis with polyneuropathy: Characterization of Brazilian subjects from the THAOS registry. J. Neurol. Sci. 2019, 403, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Luigetti, M.; Romano, A.; Di Paolantonio, A.; Bisogni, G.; Sabatelli, M. Diagnosis and Treatment of Hereditary Transthyretin Amyloidosis (hATTR) Polyneuropathy: Current Perspectives on Improving Patient Care. Ther. Clin. Risk Manag. 2020, ume 16, 109–123. [Google Scholar] [CrossRef]
- Waddington-Cruz, M.; Wixner, J.; Amass, L.; Kiszko, J.; Chapman, D.; Ando, Y. Characteristics of Patients with Late- vs. Early-Onset Val30Met Transthyretin Amyloidosis from the Transthyretin Amyloidosis Outcomes Survey (THAOS). Neurol. Ther. 2021, 10, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Sattianayagam, P.T.; Hahn, A.F.; Whelan, C.J.; Gibbs, S.D.; Pinney, J.H.; Stangou, A.J.; Rowczenio, D.; Pflugfelder, P.W.; Fox, Z.; Lachmann, H.J.; et al. Cardiac phenotype and clinical outcome of familial amyloid polyneuropathy associated with transthyretin alanine 60 variant. Eur. Heart J. 2011, 33, 1120–1127. [Google Scholar] [CrossRef] [PubMed]
- Quarta, C.C.; Gonzalez-Lopez, E.; Gilbertson, J.A.; Botcher, N.; Rowczenio, D.; Petrie, A.; Rezk, T.; Youngstein, T.; Mahmood, S.; Sachchithanantham, S.; et al. Diagnostic sensitivity of abdominal fat aspiration in cardiac amyloidosis. Eur. Hear. J. 2017, 38, 1905–1908. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Damy, T.; FontAna, M.; Hutchinson, M.; Lachmann, H.J.; Martinez-Naharro, A.; Quarta, C.C.; Rezk, T.; Whelan, C.J.; Gonzalez-Lopez, E.; et al. A new staging system for cardiac transthyretin amyloidosis. Eur. Heart J. 2018, 39, 2799–2806. [Google Scholar] [CrossRef]
- Dispenzieri, A.; A Gertz, M.; Kyle, R.A.; Lacy, M.Q.; Burritt, M.F.; Therneau, T.M.; Greipp, P.R.; Witzig, T.E.; Lust, J.A.; Rajkumar, S.V.; et al. Serum Cardiac Troponins and N-Terminal Pro-Brain Natriuretic Peptide: A Staging System for Primary Systemic Amyloidosis. J. Clin. Oncol. 2004, 22, 3751–3757. [Google Scholar] [CrossRef]
- Longhi, S.; Quarta, C.C.; Milandri, A.; Lorenzini, M.; Gagliardi, C.; Manuzzi, L.; Bacchi-Reggiani, M.L.; Leone, O.; Ferlini, A.; Russo, A.; et al. Atrial fibrillation in amyloidotic cardiomyopathy: Prevalence, incidence, risk factors and prognostic role. Amyloid 2015, 22, 147–155. [Google Scholar] [CrossRef]
- Cyrille, N.B.; Goldsmith, J.; Alvarez, J.; Maurer, M.S. Prevalence and Prognostic Significance of Low QRS Voltage Among the Three Main Types of Cardiac Amyloidosis. Am. J. Cardiol. 2014, 114, 1089–1093. [Google Scholar] [CrossRef]
- Mussinelli, R.; Salinaro, F.; Alogna, A.; Boldrini, M.; Raimondi, A.; Musca, F.; Palladini, G.; Merlini, G.; Perlini, S. Diagnostic and Prognostic Value of Low QRS Voltages in Cardiac AL Amyloidosis. Ann. Noninvasive Electrocardiol. 2013, 18, 271–280. [Google Scholar] [CrossRef]
- Quarta, C.C.; Zheng, J.; Hutt, D.; Grigore, S.F.; Manwani, R.; Sachchithanantham, S.; Mahmood, S.A.; Whelan, C.J.; Fontana, M.; Martinez-Naharro, A.; et al. 99mTc-DPD scintigraphy in immunoglobulin light chain (AL) cardiac amyloidosis. Eur. Heart J. Cardiovasc. Imaging 2021, 22, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.S.; Elliott, P.; Comenzo, R.; Semigran, M.; Rapezzi, C. Addressing Common Questions Encountered in the Diagnosis and Management of Cardiac Amyloidosis. Circulation 2017, 135, 1357–1377. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Naharro, A.; Treibel, T.A.; Abdel-Gadir, A.; Bulluck, H.; Zumbo, G.; Knight, D.S.; Kotecha, T.; Francis, R.; Hutt, D.F.; Rezk, T.; et al. Magnetic Resonance in Transthyretin Cardiac Amyloidosis. J. Am. Coll. Cardiol. 2017, 70, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Boldrini, M.; Cappelli, F.; Chacko, L.; Restrepo-Cordoba, M.A.; Lopez-Sainz, A.; Giannoni, A.; Aimo, A.; Baggiano, A.; Martinez-Naharro, A.; Whelan, C.; et al. Multiparametric Echocardiography Scores for the Diagnosis of Cardiac Amyloidosis. Cardiovasc. Imaging 2019, 13, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Tsuruda, T.; Nakada, H.; Yamamura, Y.; Matsuura, Y.; Ogata, M.; Tanaka, M.; Suiko, Y.; Komaki, S.; Tanaka, H.; Moribayashi, K.; et al. Basal inferoseptal segment is highly susceptible to deformation in the clinical spectrum of transthyretin-derived amyloid cardiomyopathy. Eur. Heart J. Open 2024, 4, oeae076. [Google Scholar] [CrossRef]
- Knight, D.S.; Zumbo, G.; Barcella, W.; Steeden, J.A.; Muthurangu, V.; Martinez-Naharro, A.; Treibel, T.A.; Abdel-Gadir, A.; Bulluck, H.; Kotecha, T.; et al. Cardiac Structural and Functional Consequences of Amyloid Deposition by Cardiac Magnetic Resonance and Echocardiography and Their Prognostic Roles. JACC Cardiovasc. Imaging 2018, 12, 823–833. [Google Scholar] [CrossRef]
- de Carvalho, F.P.; Erthal, F.; Azevedo, C.F. The Role of Cardiac MR Imaging in the Assessment of Patients with Cardiac Amyloidosis. Magn. Reson. Imaging Clin. N.Am. 2019, 27, 453–463. [Google Scholar] [CrossRef]
- Fontana, M.; Martinez-Naharro, A.; Hawkins, P.N. Staging Cardiac Amyloidosis With CMR: Understanding the Different Phenotypes. JACC Cardiovasc. Imaging 2016, 9, 1278–1279. [Google Scholar] [CrossRef]
- Fontana, M.; Pica, S.; Reant, P.; Abdel-Gadir, A.; Treibel, T.A.; Banypersad, S.M.; Maestrini, V.; Barcella, W.; Rosmini, S.; Bulluck, H.; et al. Prognostic Value of Late Gadolinium Enhancement Cardiovascular Magnetic Resonance in Cardiac Amyloidosis. Circulation 2015, 132, 1570–1579. [Google Scholar] [CrossRef]
- Zhao, L.; Tian, Z.; Fang, Q. Diagnostic accuracy of cardiovascular magnetic resonance for patients with suspected cardiac amyloidosis: A systematic review and meta-analysis. BMC Cardiovasc. Disord. 2016, 16, 129. [Google Scholar] [CrossRef]
- Starekova, J.; Pirasteh, A.; Reeder, S.B. Update on Gadolinium-Based Contrast Agent Safety, From the AJR Special Series on Contrast Media. Am. J. Roentgenol. 2024, 223, e2330036. [Google Scholar] [CrossRef] [PubMed]
- Banypersad, S.M.; Sado, D.M.; Flett, A.S.; Gibbs, S.D.; Pinney, J.H.; Maestrini, V.; Cox, A.T.; Fontana, M.; Whelan, C.J.; Wechalekar, A.D.; et al. Quantification of myocardial extracellular volume fraction in systemic AL amyloidosis: An equilibrium contrast cardiovascular magnetic resonance study. Circ. Cardiovasc. Imaging 2013, 6, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Messroghli, D.R.; Moon, J.C.; Ferreira, V.M.; Grosse-Wortmann, L.; He, T.; Kellman, P.; Mascherbauer, J.; Nezafat, R.; Salerno, M.; Schelbert, E.B.; et al. Clinical recommendations for cardiovascular magnetic resonance mapping of T1, T2, T2* and extracellular volume: A consensus statement by the Society for Cardiovascular Magnetic Resonance (SCMR) endorsed by the European Association for Cardiovascular Imaging (EACVI). J. Cardiovasc. Magn. Reson. 2017, 19, 75, Erratum in J. Cardiovasc. Magn. Reson. 2018, 20, 9. [Google Scholar]
- Fontana, M.; Chung, R.; Hawkins, P.N.; Moon, J.C. Cardiovascular magnetic resonance for amyloidosis. Heart Fail. Rev. 2015, 20, 133–144. [Google Scholar] [CrossRef]
- Tahara, N.; Lairez, O.; Endo, J.; Okada, A.; Ueda, M.; Ishii, T.; Kitano, Y.; Lee, H.; Russo, E.; Kubo, T. 99mTechnetium-pyrophosphate scintigraphy: A practical guide for early diagnosis of transthyretin amyloid cardiomyopathy. ESC Heart Fail. 2021, 9, 251–262. [Google Scholar] [CrossRef]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2022, 145, e895–e1032. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Judge, D.P.; Cappelli, F.; Fontana, M.; Garcia-Pavia, P.; Gibbs, S.; Grogan, M.; Hanna, M.; Hoffman, J.; Masri, A.; et al. Efficacy and Safety of Acoramidis in Transthyretin Amyloid Cardiomyopathy. N. Engl. J. Med. 2024, 390, 132–142. [Google Scholar] [CrossRef]
- Judge, D.P.; Gillmore, J.D.; Alexander, K.M.; Ambardekar, A.V.; Cappelli, F.; Fontana, M.; García-Pavía, P.; Grodin, J.L.; Grogan, M.; Hanna, M.; et al. Long-Term Efficacy and Safety of Acoramidis in ATTR-CM: Initial Report From the Open-Label Extension of the ATTRibute-CM Trial. Circulation 2024. Advance online publication. [Google Scholar] [CrossRef]
- Maurer, M.S.; Bokhari, S.; Damy, T.; Dorbala, S.; Drachman, B.M.; Fontana, M.; Grogan, M.; Kristen, A.V.; Lousada, I.; Nativi-Nicolau, J.; et al. Expert Consensus Recommendations for the Suspicion and Diagnosis of Transthyretin Cardiac Amyloidosis. Circ. Heart Fail. 2019, 12, e006075. [Google Scholar] [CrossRef]
- Obici, L.; Kuks, J.B.; Buades, J.; Adams, D.; Suhr, O.B.; Coelho, T.; Kyriakides, T. Recommendations for presymptomatic genetic testing and management of individuals at risk for hereditary transthyretin amyloidosis. Curr. Opin. Neurol. 2016, 29, S27–S35. [Google Scholar] [CrossRef]
- Alreshq, R.; Ruberg, F.L. Clinical approach to genetic testing in amyloid cardiomyopathy: From mechanism to effective therapies. Curr. Opin. Cardiol. 2021, 36, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.F.; Miller, E.J. Can We Manage Presymptomatic TTR V142I Related Risk? Heart Fail. 2022, 10, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Conceição, I.; Damy, T.; Romero, M.; Galán, L.; Attarian, S.; Luigetti, M.; Sadeh, M.; Sarafov, S.; Tournev, I.; Ueda, M. Early diagnosis of ATTR amyloidosis through targeted follow-up of identified carriers of TTR gene mutations. Amyloid 2019, 26, 3–9. [Google Scholar] [CrossRef] [PubMed]
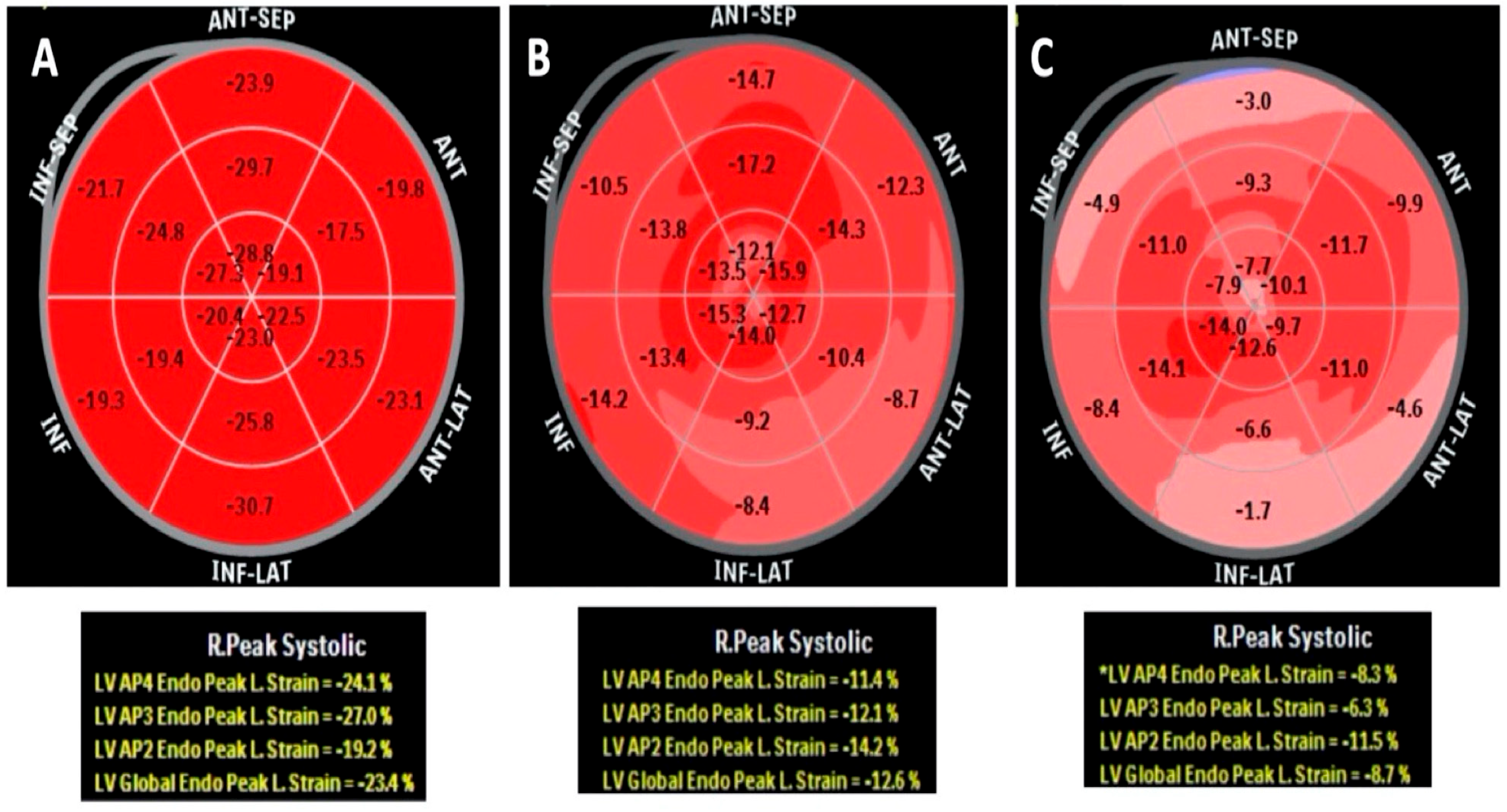
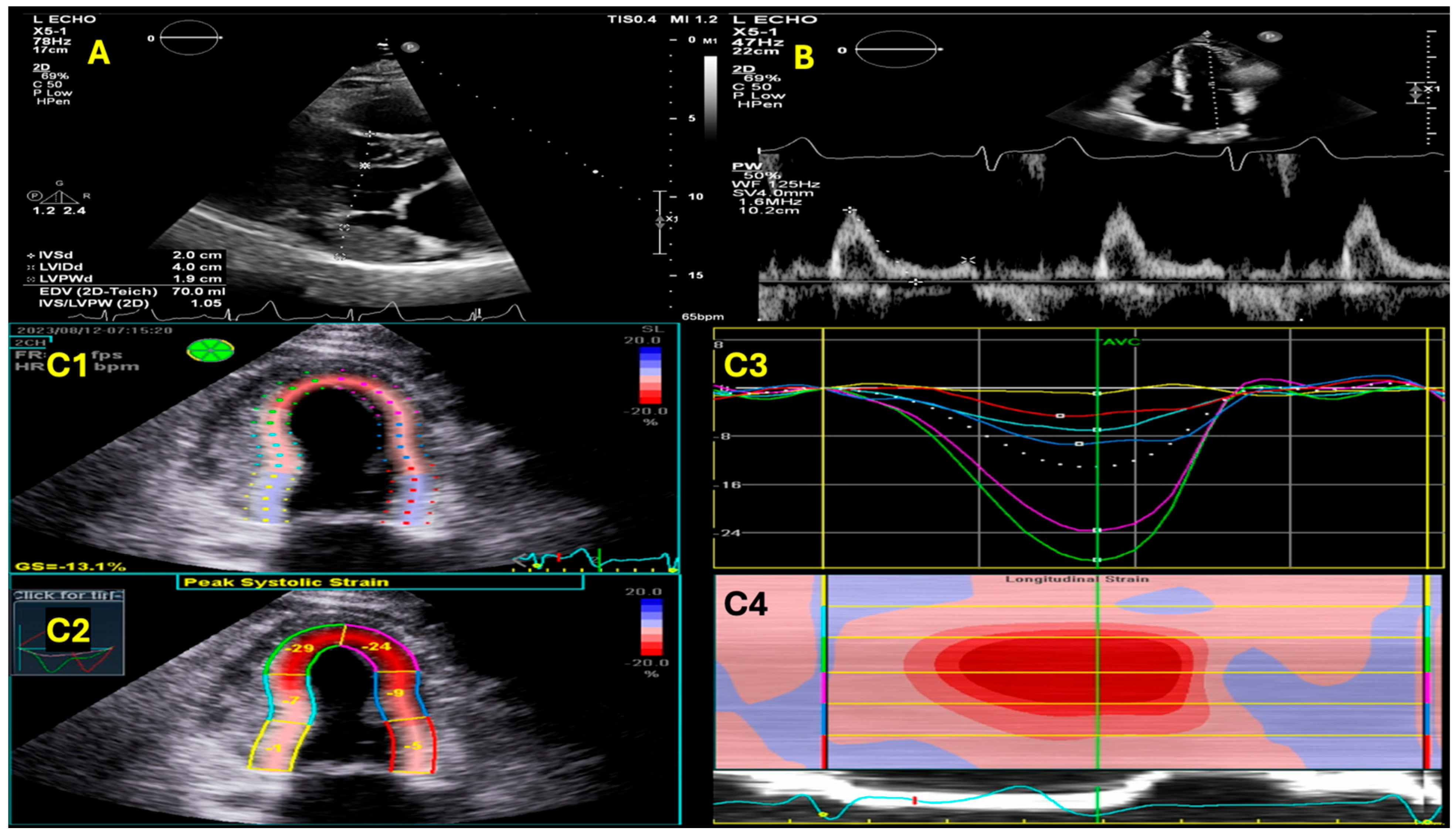
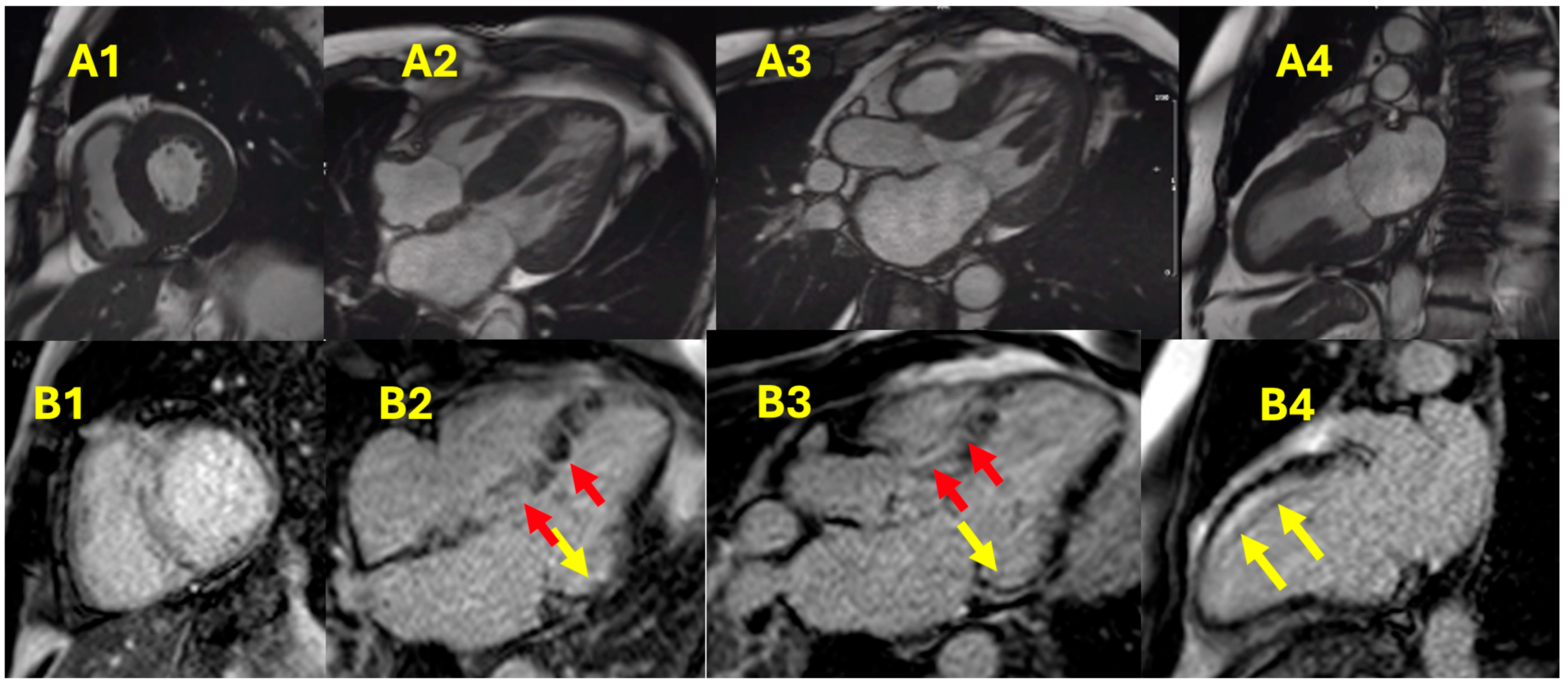
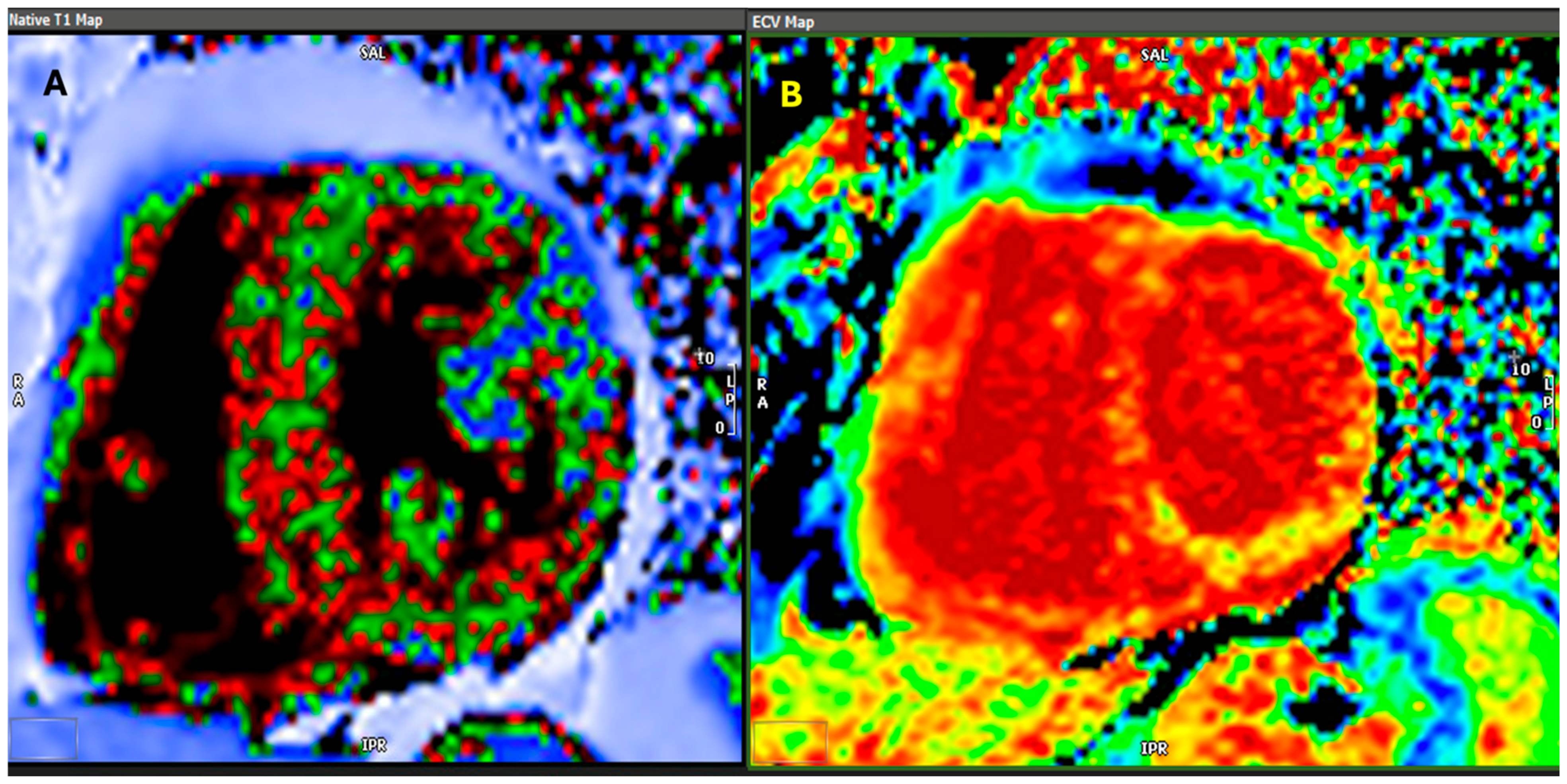

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Precursor Protein | Primary Organ Involvement | Cardiac Involvement |
|---|---|---|---|
| AL (Primary) Amyloidosis | Immunoglobulin Light Chains | Heart, Kidney, Nervous System | Frequent and severe |
| ATTR (Transthyretin) Amyloidosis | Transthyretin (TTR) | Heart, Nervous System | Common, particularly in hereditary forms |
| AA (Secondary) Amyloidosis | Serum Amyloid A | Kidney, Liver, Spleen | Rare |
| Aβ2M (Dialysis-related) | β2-microglobulin | Joints, Bones | Rare |
| Hereditary Non-TTR Amyloidosis | Fibrinogen Apolipoproteins | Kidney, Liver, Nerves | Variable |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sawalha, K.; Alkhatib, D.A. A General Overview of Transthyretin Cardiac Amyloidosis and Summary of Expert Opinions on Pre-Symptomatic Testing and Management of Asymptomatic Patients with a Focus on Transthyretin V122I. Hearts 2025, 6, 6. https://doi.org/10.3390/hearts6010006
Sawalha K, Alkhatib DA. A General Overview of Transthyretin Cardiac Amyloidosis and Summary of Expert Opinions on Pre-Symptomatic Testing and Management of Asymptomatic Patients with a Focus on Transthyretin V122I. Hearts. 2025; 6(1):6. https://doi.org/10.3390/hearts6010006
Chicago/Turabian StyleSawalha, Khalid, and Deya A. Alkhatib. 2025. "A General Overview of Transthyretin Cardiac Amyloidosis and Summary of Expert Opinions on Pre-Symptomatic Testing and Management of Asymptomatic Patients with a Focus on Transthyretin V122I" Hearts 6, no. 1: 6. https://doi.org/10.3390/hearts6010006
APA StyleSawalha, K., & Alkhatib, D. A. (2025). A General Overview of Transthyretin Cardiac Amyloidosis and Summary of Expert Opinions on Pre-Symptomatic Testing and Management of Asymptomatic Patients with a Focus on Transthyretin V122I. Hearts, 6(1), 6. https://doi.org/10.3390/hearts6010006