
PAF Physiology in Target Organ Systems—A Deep Dive to Understand the PAF Mystery in Pathogenesis of Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
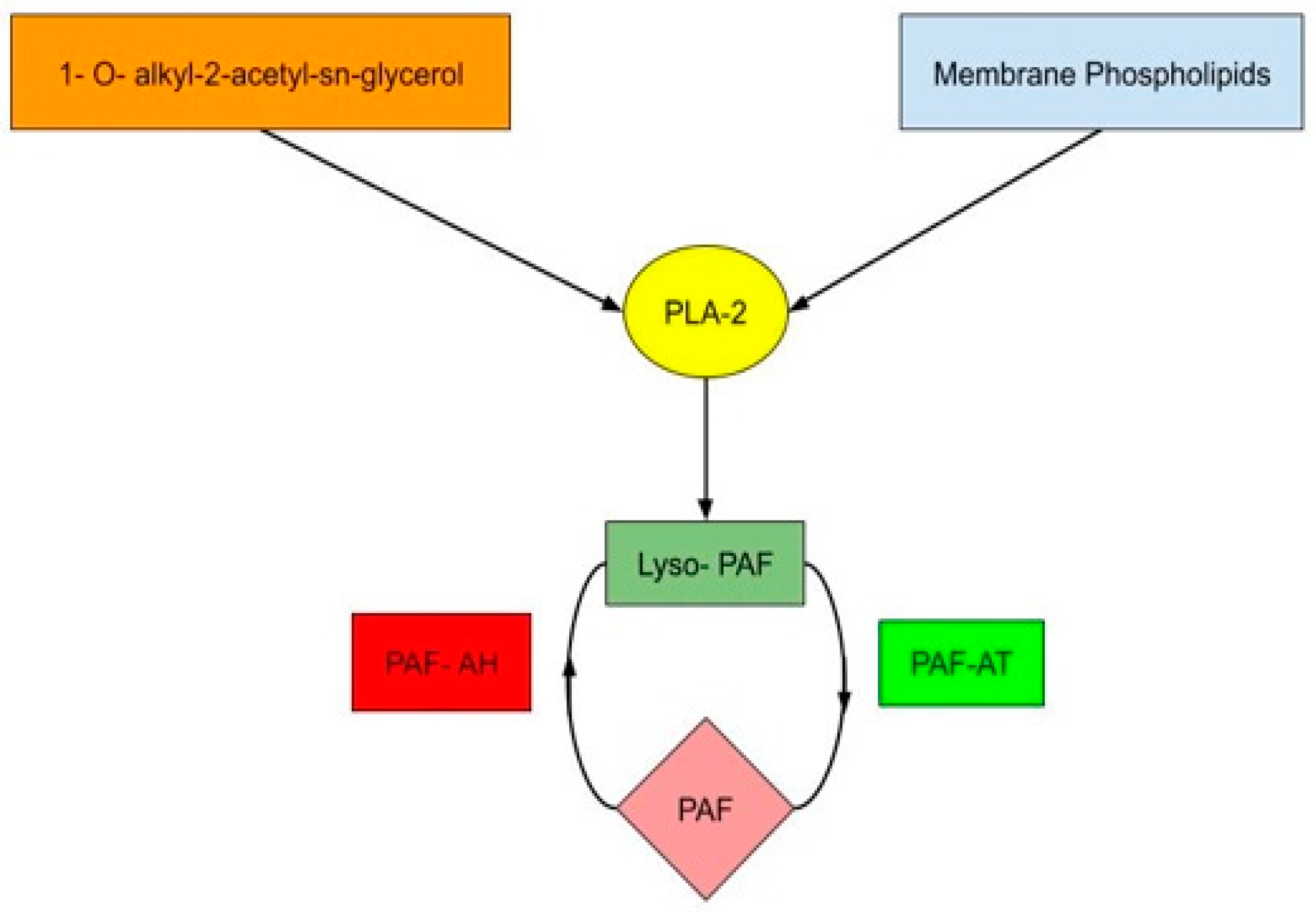
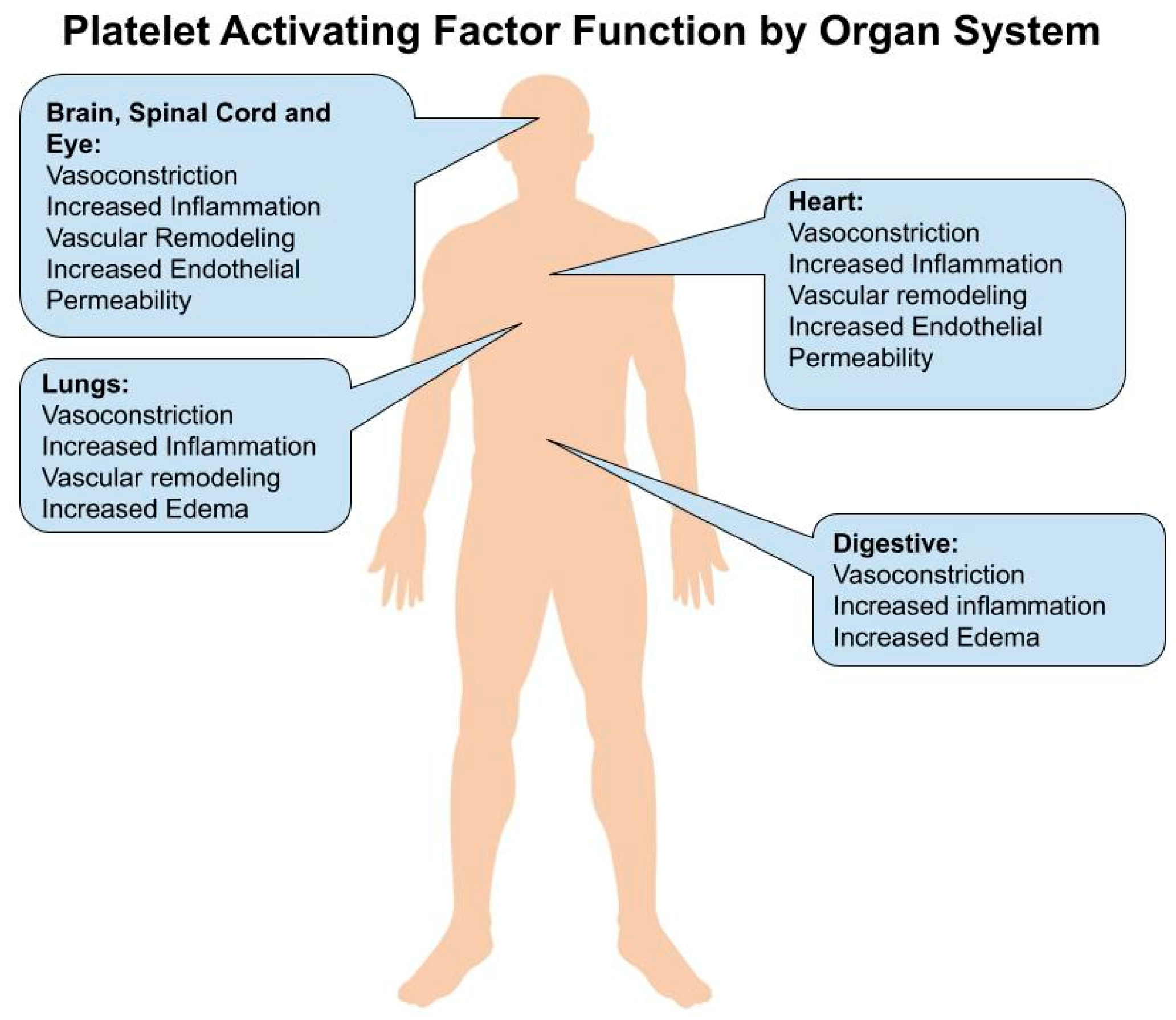
1. Introduction
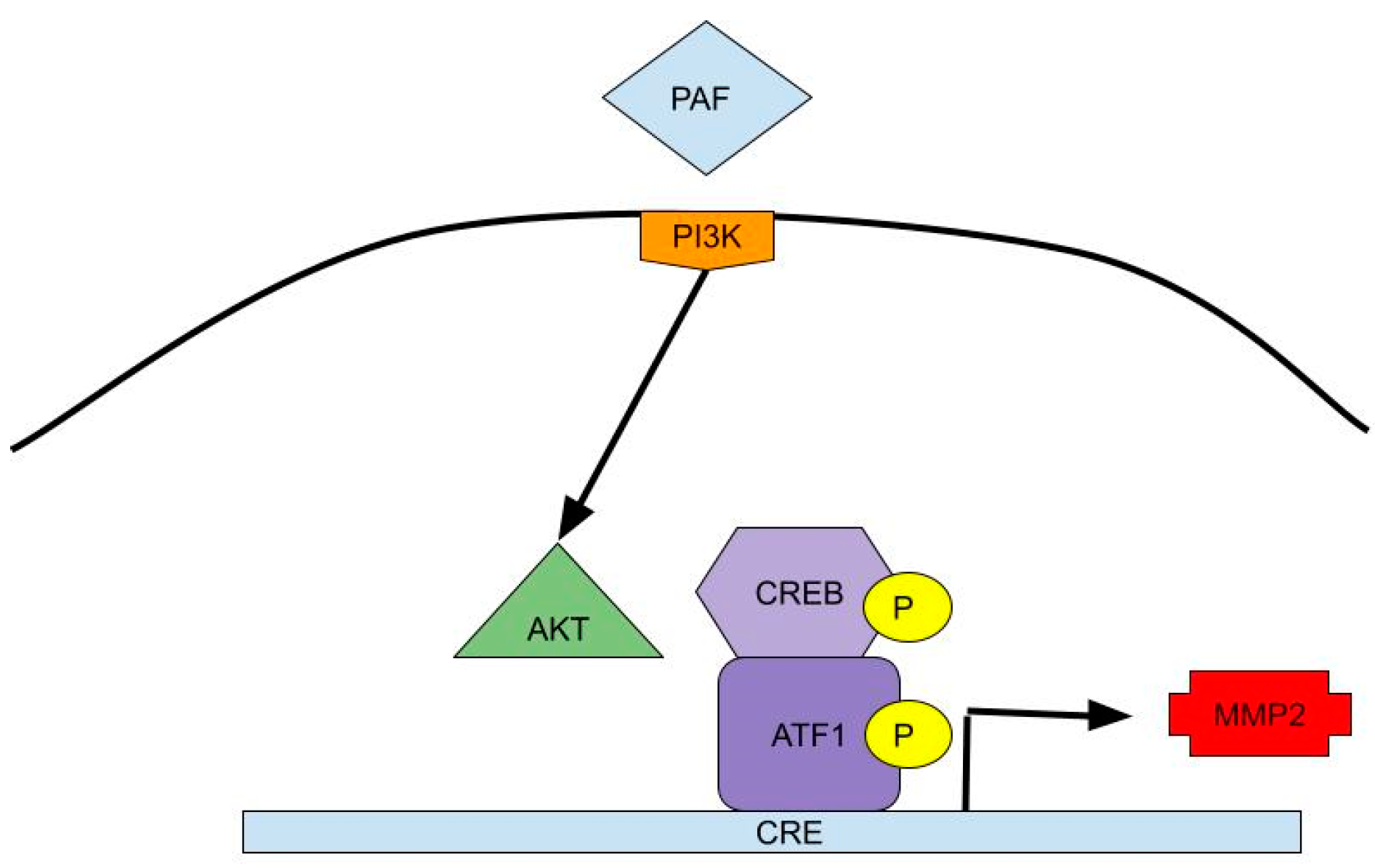
2. Brain and Spinal Cord
3. Eye
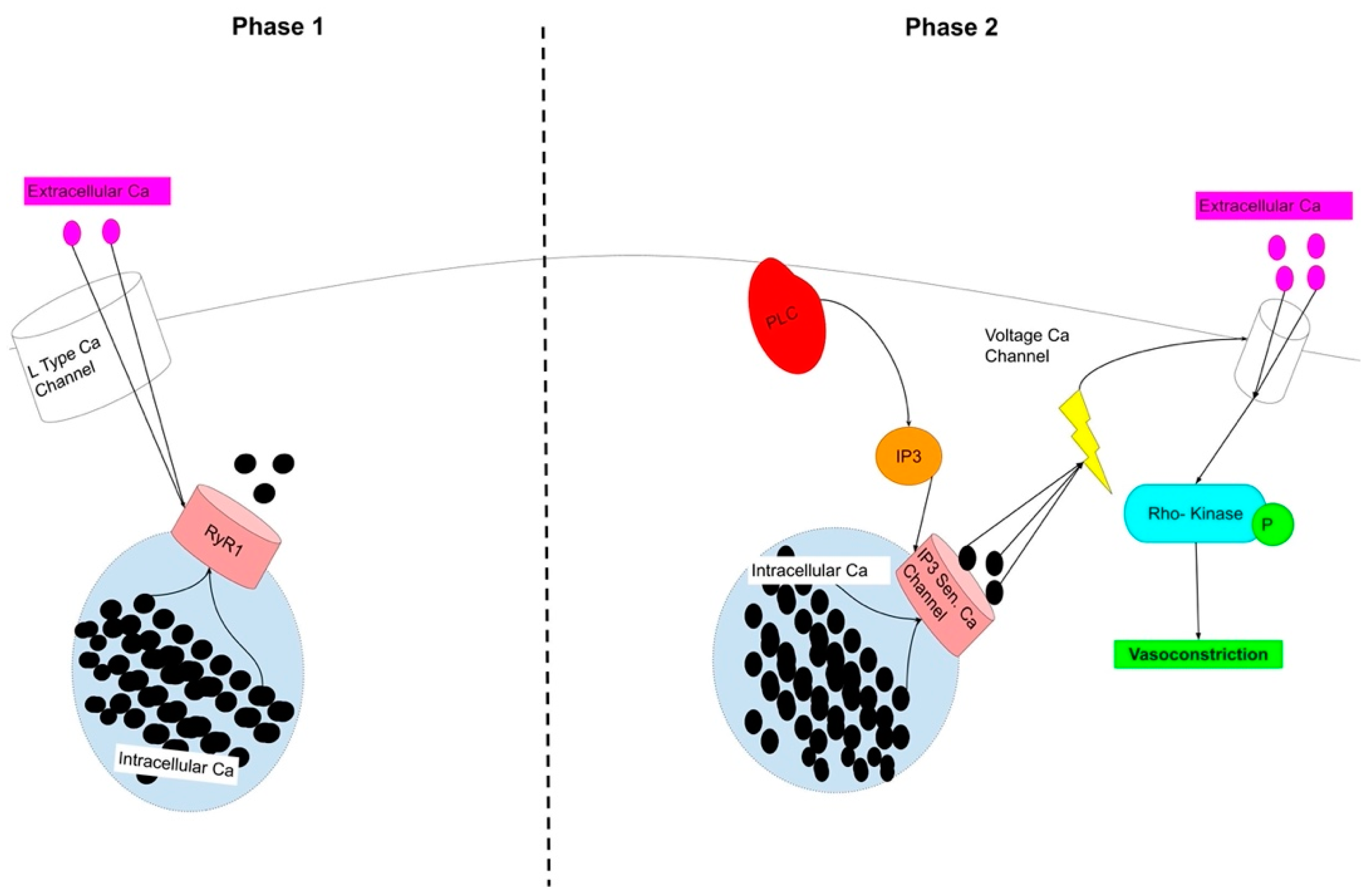
4. Heart
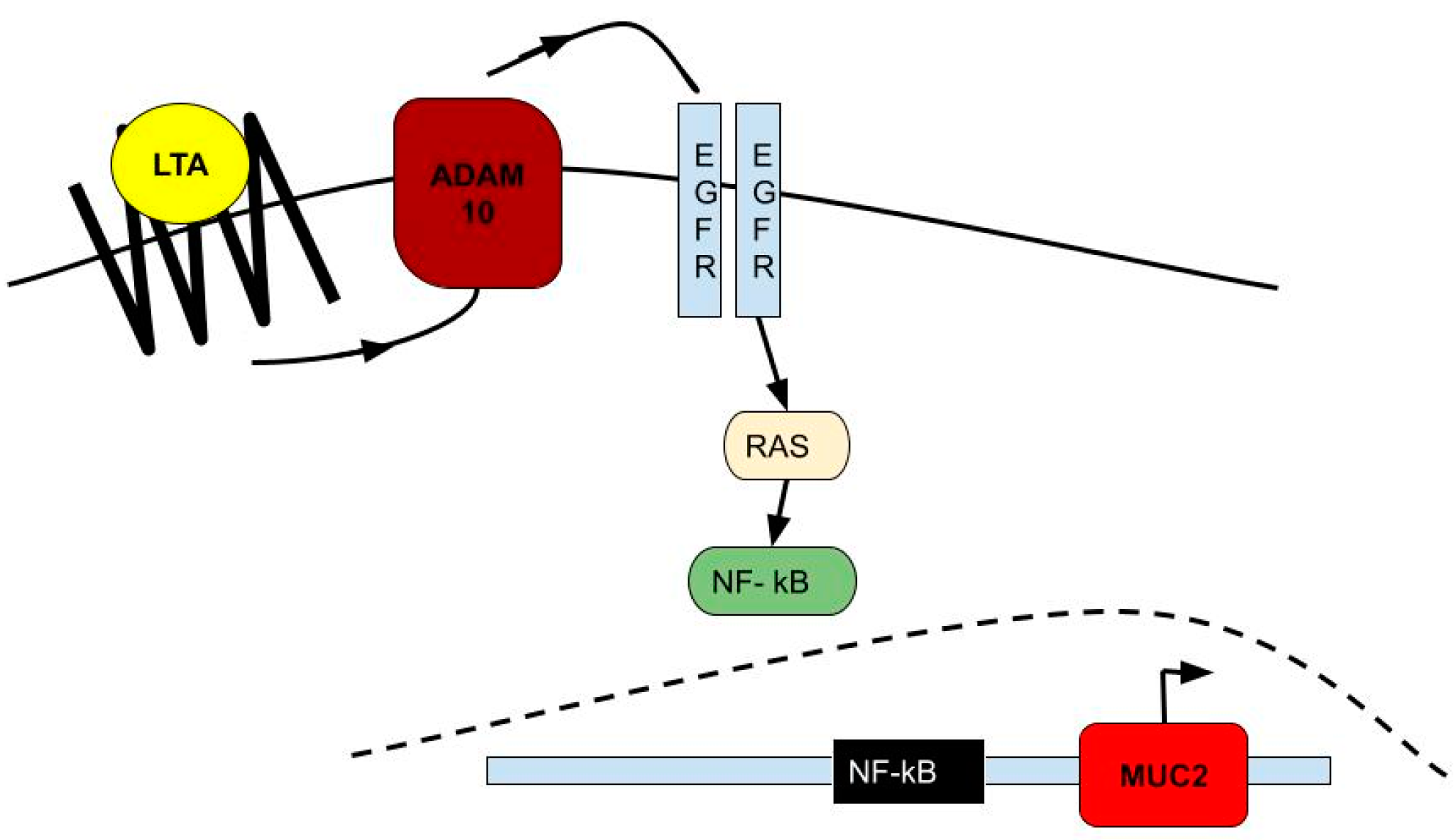
5. Lungs
6. Gastrointestinal
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Palur Ramakrishnan, A.V.K.; Varghese, T.P.; Vanapalli, S.; Nair, N.K.; Mingate, M.D. Platelet activating factor: A potential biomarker in acute coronary syndrome? Cardiovasc. Ther. 2017, 35, 64–70. [Google Scholar] [CrossRef]
- Reznichenko, A.; Korstanje, R. The Role of Platelet-Activating Factor in Mesangial Pathophysiology. Am. J. Pathol. 2015, 185, 888–896. [Google Scholar] [CrossRef]
- Gill, P.; Jindal, N.L.; Jagdis, A.; Vadas, P. Platelets in the immune response: Revisiting platelet-activating factor in anaphylaxis. J. Allergy Clin. Immunol. 2015, 135, 1424–1432. [Google Scholar] [CrossRef]
- Kelesidis, T.; Papakonstantinou, V.; Detopoulou, P.; Fragopoulou, E.; Chini, M.; Lazanas, M.C.; Antonopoulou, S. The Role of Platelet-Activating Factor in Chronic Inflammation, Immune Activation, and Comorbidities Associated with HIV Infection. AIDS Rev. 2015, 17, 191–201. [Google Scholar]
- Papakonstantinou, V.D.; Lagopati, N.; Tsilibary, E.C.; Demopoulos, C.A.; Philippopoulos, A.I. A Review on Platelet Activating Factor Inhibitors: Could a New Class of Potent Metal-Based Anti-Inflammatory Drugs Induce Anticancer Properties? Bioinorg. Chem. Appl. 2017, 2017, 6947034. [Google Scholar] [CrossRef]
- Hostettler, M.E.; Knapp, P.E.; Carlson, S.L. Platelet-activating factor induces cell death in cultured astrocytes and oligodendrocytes: Involvement of caspase-3. Glia 2002, 38, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.; West, E.; Bate, C. The phospholipase A2 pathway controls a synaptic cholesterol ester cycle and synapse damage. J. Cell Sci. 2018, 131, jcs211789. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, L.; Dai, C.; Ma, G.; Zhang, Y.; Zhang, X.; Wu, Z. Neuroprotection by platelet-activating factor acetylhydrolase in a mouse model of transient cerebral ischemia. Neurosci. Lett. 2014, 558, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Brailoiu, E.; Barlow, C.L.; Ramirez, S.H.; Abood, M.E.; Brailoiu, G.C. Effects of Platelet-Activating Factor on Brain Microvascular Endothelial Cells. Neuroscience 2018, 377, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, Z.; Zhang, Y.; Feng, S.-Q.; Liu, Y.; Shields, L.B.E.; Zhao, Y.-Z.; Zhu, Q.; Gozal, D.; Shields, C.B.; et al. Attenuated Reactive Gliosis and Enhanced Functional Recovery Following Spinal Cord Injury in Null Mutant Mice of Platelet-Activating Factor Receptor. Mol. Neurobiol. 2016, 53, 3448–3461. [Google Scholar] [CrossRef] [PubMed]
- Codeluppi, S.; Svensson, C.; Hefferan, M.P.; Valencia, F.; Silldorff, M.D.; Oshiro, M.; Marsala, M.; Pasquale, E.B. The Rheb-mTOR Pathway Is Upregulated in Reactive Astrocytes of the Injured Spinal Cord. J. Neurosci. 2009, 29, 1093–1104. [Google Scholar] [CrossRef]
- He, J. Alkali-Induced Corneal Stromal Melting Prevention by a Novel Platelet-Activating Factor Receptor Antagonist. Arch. Ophthalmol. 2006, 124, 70. [Google Scholar] [CrossRef][Green Version]
- Bazan, H.E.P.; Ottino, P. The role of platelet-activating factor in the corneal response to injury. Prog. Retin Eye Res. 2002, 21, 449–464. [Google Scholar] [CrossRef]
- Sharif, N.A.; Xu, S.; Hellberg, P.E.; Pang, I.-H.; Gamache, D.A.; Yanni, J.M. Human conjunctival epithelial cell responses to platelet-activating factor (PAF): Signal transduction and release of proinflammatory cytokines. Mol. Vis. 2009, 15, 1153–1161. [Google Scholar] [PubMed]
- Penna, C.; Alloatti, G.; Cappello, S.; Gattullo, D.; Berta, G.N.; Mognetti, B.; Losano, G.; Pagliaro, P. Platelet-activating factor induces cardioprotection in isolated rat heart akin to ischemic preconditioning: Role of phosphoinositide 3-kinase and protein kinase C activation. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2512–H2520. [Google Scholar] [CrossRef] [PubMed]
- Penna, C.; Bassino, E.; Alloatti, G. Platelet activating factor: The good and the bad in the ischemic/reperfused heart. Exp. Biol. Med. 2011, 236, 390–401. [Google Scholar] [CrossRef]
- Montrucchio, G.; Alloatti, G.; Camussi, G. Role of Platelet-Activating Factor in Cardiovascular Pathophysiology. Physiol. Rev. 2000, 80, 1669–1699. [Google Scholar] [CrossRef]
- Zheng, G.-H.; Xiong, S.-Q.; Mei, L.-J.; Chen, H.-Y.; Wang, T.; Chu, J.-F. Elevated Plasma Platelet Activating Factor, Platelet Activating Factor Acetylhydrolase Levels and Risk of Coronary Heart Disease or Blood Stasis Syndrome of Coronary Heart Disease in Chinese: A Case Control Study: A Case–Control Study. Inflammation 2012, 35, 1419–1428. [Google Scholar] [CrossRef]
- Quarck, R.; De Geest, B.; Stengel, D.; Mertens, A.; Lox, M.; Theilmeier, G.; Michiels, C.; Raes, M.; Bult, H.; Collen, D.; et al. Adenovirus-Mediated Gene Transfer of Human Platelet-Activating Factor–Acetylhydrolase Prevents Injury-Induced Neointima Formation and Reduces Spontaneous Atherosclerosis in Apolipoprotein E–Deficient Mice. Circulation 2001, 103, 2495–2500. [Google Scholar] [CrossRef]
- Lemjabbar, H.; Basbaum, C. Platelet-activating factor receptor and ADAM10 mediate responses to Staphylococcus aureus in epithelial cells. Nat. Med. 2002, 8, 41–46. [Google Scholar] [CrossRef]
- Altura, B.; Shah, N.; Shah, G.; Perez-Albela, J.L.; Altura, B. Insights into the possible mechanisms by which platelet-activating factor and PAF-receptors function in vascular smooth muscle in magnesium deficiency and vascular remodeling: Possible links to atherogenesis, hypertension and cardiac failure. Int. J. Cardiol. Res. 2016, 3, 1–3. [Google Scholar]
- Uhlig, S.; Göggel, R.; Engel, S. Mechanisms of platelet-activating factor (PAF)-mediated responses in the lung. Pharmacol. Rep. PR 2005, 57, S206–S221. [Google Scholar]
- Martin, C.; Göggel, R.; Ressmeyer, A.-R.; Uhlig, S. Pressor responses to platelet-activating factor and thromboxane are mediated by Rho-kinase. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 287, L250–L257. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, M.; Zhang, X.; Cai, Q.; Zhu, Z.; Jiang, W.; Xu, C. Transactivation of epidermal growth factor receptor through platelet-activating factor/receptor in ovarian cancer cells. J. Exp. Clin. Cancer Res. 2014, 33, 85. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Burgel, P.-R. Roles of epidermal growth factor receptor activation in epithelial cell repair and mucin production in airway epithelium. Thorax 2004, 59, 992–996. [Google Scholar] [CrossRef]
- LeMessurier, K.S.; Häcker, H.; Chi, L.; Tuomanen, E.; Redecke, V. Type I Interferon Protects against Pneumococcal Invasive Disease by Inhibiting Bacterial Transmigration across the Lung. PLoS Pathog. 2013, 9, e1003727. [Google Scholar] [CrossRef]
- Shukla, S.D.; Sohal, S.S.; O’Toole, R.F.; Eapen, M.S.; Walters, E.H. Platelet activating factor receptor: Gateway for bacterial chronic airway infection in chronic obstructive pulmonary disease and potential therapeutic target. Expert Rev. Respir. Med. 2015, 9, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Tejera Alvarez, P.; Zhang, R.; Su, L.; Christiani, D. Decreased Expression in Platelet-Activating Factor Acetylhydrolase (PAF-AH) Gene Is Associated with ARDS. Am. J. Respir. Crit. Care Med. 2018, 197, A7530. [Google Scholar]
- Garcia, C.C.; Russo, R.C.; Guabiraba, R.; Fagundes, C.T.; Polidoro, R.B.; Tavares, L.P.; Salgado, A.P.C.; Cassali, G.D.; Sousa, L.P.; Machado, A.V.; et al. Platelet-Activating Factor Receptor Plays a Role in Lung Injury and Death Caused by Influenza A in Mice. PLoS Pathog. 2010, 6, e1001171. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.-X.; Liu, S.-S.; Li, K.; Cui, B.; Liu, C.; Hu, Z.-W. Cigarette smoke promotes COPD by activating platelet-activating factor receptor and inducing neutrophil autophagic death in mice. Oncotarget 2017, 8, 74720–74735. [Google Scholar] [CrossRef]
- Sharma, J.; Young, D.M.; Marentette, J.O.; Rastogi, P.; Turk, J.; McHowat, J. Lung endothelial cell platelet-activating factor production and inflammatory cell adherence are increased in response to cigarette smoke component exposure. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 302, L47–L55. [Google Scholar] [CrossRef]
- Shukla, S.D.; Muller, H.K.; Latham, R.; Sohal, S.S.; Walters, E.H. Platelet-activating factor receptor (PAFr) is upregulated in small airways and alveoli of smokers and COPD patients: Small airway PAFr expression in COPD. Respirology 2016, 21, 504–510. [Google Scholar] [CrossRef]
- Shukla, S.; Sohal, S.; Mahmood, M.; Reid, D.; Muller, H.; Walters, E. Airway epithelial platelet-activating factor receptor expression is markedly upregulated in chronic obstructive pulmonary disease. Int. J. Chron. Obstruct. Pulmon. Dis. 2014, 9, 853–861. [Google Scholar] [CrossRef]
- Riaz, M.S.; Kaur, A.; Shwayat, S.N.; Behboudi, S.; Kishore, U.; Pathan, A.A. Direct Growth Inhibitory Effect of Platelet Activating Factor C-16 and Its Structural Analogs on Mycobacteria. Front. Microbiol. 2018, 9, 1903. [Google Scholar] [CrossRef]
- Lautenschläger, I.; Wong, Y.L.; Sarau, J.; Goldmann, T.; Zitta, K.; Albrecht, M.; Frerichs, I.; Weiler, N.; Uhlig, S. Signalling mechanisms in PAF-induced intestinal failure. Sci. Rep. 2017, 7, 13382. [Google Scholar] [CrossRef] [PubMed]
- Souza, D.G.; Pinho, V.; Soares, A.C.; Shimizu, T.; Ishii, S.; Teixeira, M.M. Role of PAF receptors during intestinal ischemia and reperfusion injury. A comparative study between PAF receptor-deficient mice and PAF receptor antagonist treatment: PAF receptor and reperfusion injury. Br. J. Pharmacol. 2003, 139, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.L.; Caplan, M.S. Necrotizing enterocolitis: Pathophysiology, platelet-activating factor, and probiotics. Semin. Pediatr. Surg. 2013, 22, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Pierce, M.; Franklin, A.; Jilling, T.; Stafforini, D.M.; Caplan, M. Dual Roles of Endogenous Platelet-Activating Factor Acetylhydrolase in a Murine Model of Necrotizing Enterocolitis. Pediatr. Res. 2010, 68, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Caplan, M.S.; Hedlund, E.; Adler, L.; Lickerman, M.; Hsueh, W. The Platelet-Activating Factor Receptor Antagonist WEB 2170 Prevents Neonatal Necrotizing Enterocolitis in Rats. J. Pediatr. Gastroenterol. Amp. Nutr. 1997, 24, 296–301. [Google Scholar] [CrossRef]
- Caplan, M.S.; Lickerman, M.; Adler, L.; Dietsch, G.N.; Yu, A. The Role of Recombinant Platelet-Activating Factor Acetylhydrolase in a Neonatal Rat Model of Necrotizing Enterocolitis. Pediatr. Res. 1997, 42, 779–783. [Google Scholar] [CrossRef]
- Yu, P.-W.; Xiao, G.X.; Qin Xj, X.; Zhou, L.-X.; Wang, Z.-Q. The effects of PAF antagonist on intestinal mucosal microcirculation after burn in rats. World J. Gastroenterol. 2000, 6, 906–908. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cicchitti, L.; Martelli, M.; Cerritelli, F. Chronic Inflammatory Disease and Osteopathy: A Systematic Review. PLoS ONE 2015, 10, e0121327. [Google Scholar] [CrossRef] [PubMed]
- Perez, L.L.; Sneed, J.A.; Eland, D. Evidence-based osteopathic manipulative treatment for common conditions. Osteopath Fam. Physician 2012, 4, 8–12. [Google Scholar] [CrossRef]
- Hibler, J.; Perkins, J.; Eland, D.; Sammons, D. Osteopathic Manipulative Medicine for Inflammatory Skin Diseases. Available online: https://cdn.ymaws.com/www.aocd.org/resource/resmgr/jaocd/contents/volume31/31-01.pdf (accessed on 1 November 2021).





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shah, N.; Kumar, K.; Shah, N. PAF Physiology in Target Organ Systems—A Deep Dive to Understand the PAF Mystery in Pathogenesis of Disease. Hearts 2021, 2, 551-560. https://doi.org/10.3390/hearts2040042
Shah N, Kumar K, Shah N. PAF Physiology in Target Organ Systems—A Deep Dive to Understand the PAF Mystery in Pathogenesis of Disease. Hearts. 2021; 2(4):551-560. https://doi.org/10.3390/hearts2040042
Chicago/Turabian StyleShah, Nilank, Karan Kumar, and Nikeith Shah. 2021. "PAF Physiology in Target Organ Systems—A Deep Dive to Understand the PAF Mystery in Pathogenesis of Disease" Hearts 2, no. 4: 551-560. https://doi.org/10.3390/hearts2040042
APA StyleShah, N., Kumar, K., & Shah, N. (2021). PAF Physiology in Target Organ Systems—A Deep Dive to Understand the PAF Mystery in Pathogenesis of Disease. Hearts, 2(4), 551-560. https://doi.org/10.3390/hearts2040042