Abstract
Protein-DNA interactions are the core of the cell’s molecular machinery. For a long time, conventional biochemical methods served as a powerful investigatory basis of protein-DNA interactions and target search mechanisms. Currently single-molecule (SM) techniques have emerged as a complementary tool for studying these interactions and have revealed plenty of previously obscured mechanistic details. In comparison to the traditional ones, SM methods allow direct monitoring of individual biomolecules. Therefore, SM methods reveal reactions that are otherwise hidden by the ensemble averaging observed in conventional bulk-type methods. SM biophysical techniques employing various nanobiotechnology methods for immobilization of studied molecules grant the possibility to monitor individual reaction trajectories of biomolecules. Next-generation in vitro SM biophysics approaches enabling high-throughput studies are characterized by much greater complexity than the ones developed previously. Currently, several high-throughput DNA flow-stretch assays have been published and have shown many benefits for mechanistic target search studies of various DNA-binding proteins, such as CRISPR-Cas, Argonaute, various ATP-fueled helicases and translocases, and others. This review focuses on SM techniques employing surface-immobilized and relatively long DNA molecules for studying protein-DNA interaction mechanisms.
1. Introduction
Protein-DNA interactions are fundamental to the existence of all living organisms. At the cellular level, these interactions govern different biomolecular processes, such as compaction and recombination of genetic material [1,2], DNA repair [3,4] and replication [5], and transcription and its regulation [6,7]. In order to combat bacteriophage infections, bacteria and archaea employ diverse protein-based defense systems-restriction-modification [8], CRISPR-Cas [9], and others. All of them are based on the ability of proteins to specifically interact with their invading DNA target sequences. Therefore, the extensive studies of protein-DNA interactions are of exceptional importance for a better understanding of the aforementioned biological processes and for a deeper insight into the essential components that drive the antiviral protection mechanisms in certain microorganisms.
There are numerous traditional biochemical methods that allow one to identify and analyze protein-DNA interactions: electrophoretic mobility shift assay, cross-linking, footprinting, systematic evolution of ligands by exponential enrichment, chromatin immunoprecipitation, and others [10]. All these techniques are accompanied by execution of the bulk experiments, where the properties of billions of molecules present in a solution are measured simultaneously. However, each studied molecule is unique and changing at any given time because of its dynamics and constant interaction with the other such molecules as well as with a solvent. Ensemble measurements usually mask these processes of molecular dynamics, since at ensemble level, a certain measured characteristic is the asynchronous average of the contribution of each separate molecule in the sample [11]. In contrast, SM methods provide an opportunity to trace the reaction trajectories of individual molecules and thus reveal even the smallest details of multistep processes. Therefore, SM techniques open a path to probe the real-time dynamics of single molecules and assess the heterogeneity of the entire population of investigatory molecules [12].
2. Main Single-Molecule Techniques for Studying Protein-DNA Interactions
SM-level studies of protein-DNA interactions are usually conducted by employing distinct SM biophysical methods and by combining them with each other. Currently, tethered particle motion (TPM), optical tweezers (OTs), magnetic tweezers (MTs), atomic force microscopy (AFM), single-molecule Förster resonance energy transfer (smFRET), and DNA flow-stretch assays mainly prevail in this research field. Some other techniques can be used as well for this purpose. For instance, nanopore sensing enables the investigation of protein-DNA interactions at the level of a single molecule, since nanopores are capable of capturing and manipulating individual protein-DNA complexes [13,14,15]. It is also possible to capture discrete stretched DNA molecules in nanopores by the means of an electroosmotic flow, where this hydrodynamic flow-driven process can be exploited for various applications [16,17]. At the level of functionality, each of these SM approaches fundamentally differs. Therefore, they can provide unique mechanistic insights into the dynamic interactions happening between various proteins and DNA, or even reveal valuable information about the mechanophysical and structural properties of these biomolecular complexes.
TPM analysis allows detecting the position of a metallic or polystyrene bead attached to one end of DNA, when the other end of DNA is immobilized on the surface (Figure 1A) [18]. Surface tethering of an individual DNA molecule restricts the particle-specific Brownian motion in aqueous solution. This constraint is proportional to the length of the DNA molecule, so the amount of the bead’s Brownian motion conveys the information about the ongoing DNA-shortening events [19,20]. TPM has sensitivity to DNA length changes of at least 10–15 nm (60–70 bp) when the size of the immobilized DNA molecules ranges from 100 to 4000 bp [21]. Therefore, TPM is ideal for studying the dynamics of proteins that are able to shorten the DNA molecule by bending, looping, or translocating along it.
OTs, or optical traps, are one of the most versatile techniques of SM manipulation as they allow both real-time force and motion measurements. OTs are established by focusing the laser beam to a diffraction-limited spot with a high numerical aperture microscope objective [22]. An extremely potent electric field gradient is then generated next to the focus area. Due to the transmission of momentum from the absorption or scattering of incident photons, dielectric beads, such as polystyrene or quartz microspheres, located near the beam focus are subjected to an optical force, which is usually composed of two components—scattering and gradient forces [23]. In the case of the SM-level studies of protein-DNA interactions, a typical OTs experiment involves the tethering of one end of an individual DNA molecule to a dielectric bead trapped by a powerful laser beam. The remaining DNA end is immobilized on the glass surface (single OTs) (Figure 1B) or attached to another dielectric bead held by a micropipette (also single OTs) or by a second laser beam (double OTs) (Figure 1C). By gradually moving the surface (in the case of single OTs) or one of the trapped beads (in the double OTs configuration) away from the other captured bead, it becomes possible to apply increasing tension to the nucleoprotein complex [21]. Hence, OTs grant the possibility to confer variable pN loads to the single interacting DNA and protein molecules and thus to investigate force-dependent kinetic reaction rates, whereas double OTs can also be employed for the measurement of sub-nanometer displacements induced by protein-DNA interactions.
Typical MTs experiments utilize tethering of one DNA molecule end to a surface and conjugation of the second end with a micron-sized paramagnetic bead (Figure 1D) [24]. A magnetic field is created by placing two permanent magnets or electromagnets above such a surface at a distance of a few millimeters. This field generates forces that vertically attract the magnetic bead and causes stretching of the DNA molecule. By altering the magnets’ distance from the surface, the produced forces can be varied in the range of 0.001–100 pN to obtain the desired DNA extension [11]. A torque can also be induced on the DNA molecule by rotating the magnets on the x-y plane [25,26]. In general, MTs enable the measurement of slow molecular processes, in which both torque and certain forces are involved. Due to the provided ability to precisely control the resulting tension and the accumulation of positive and negative twists on the DNA molecule, this biophysical approach is well suited for studying DNA topology and enzyme-catalyzed mechanisms, which modulate DNA supercoiling, at the SM level [27].
One of the SM techniques belonging to the class of scanning probe microscopy is AFM, in which surface properties are examined by using an atomically sharp tip (Figure 1E). This permits one to map the surface characteristics of a sample at sub-nanometer resolution [22]. The tip attached to the AFM cantilever interacts with the sample surface under the action of diverse non-covalent forces, such as electrostatic interaction, van der Waals attraction, and Pauli or Coulomb repulsion. Attractive forces pull the probe towards the sample surface, while repulsion forces deflect the probe from the surface [28]. The nanometer resolution of individual molecules and molecular aggregates is one of the main reasons that make AFM imaging an appealing choice for SM-level studies of protein-DNA interactions. This technique allows direct visualization of individual protein-DNA complexes under different conditions and in the context of protein-specific DNA targets [29]. Images acquired during AFM imaging provide structural information about transient complexes of proteins bound to non-specific DNA sites. With the help of AFM, it is also possible to detect protein-induced DNA structural changes, such as compaction or looping of the DNA [30]. Moreover, the 3D resolution ensured by this SM method provides an opportunity to investigate the stoichiometric and conformational properties of protein-DNA complexes [31].
FRET is a distance-dependent non-radiative energy transfer from the donor fluorophore to the acceptor through the dipole-dipole interaction [32]. smFRET experiments usually rely on the excitation and detection of a pair of donor and acceptor fluorophores located on the same individual molecule or two individual interacting molecules (Figure 1F) [33]. Since FRET efficiency is a function of the distance between the donor and the acceptor, smFRET allows one to perform 1–10 nm distance measurements at the SM level. For this reason, it becomes possible to observe nanometer-level conformational changes or interactions, when two molecules come into close proximity. Commonly, DNA sensors with a FRET pair are employed to study its interaction with a protein that changes the distance between the pair, for example, by bending DNA molecules [34,35]. Another approach—attaching FRET acceptor to a DNA molecule and donor to a DNA-interacting protein—allows measuring the interaction of DNA and protein and even performing FRET-restrained modeling of structures, [36,37,38]. As this technique provides dynamic temporal resolution at the SM level, it grants a possibility to not only study the dynamic processes of individual molecules, but also to investigate the heterogeneity of sample molecules, which can be either static or dynamic [23,39].

Figure 1.
Major techniques for investigating protein-DNA interactions at the single-molecule (SM) level. (A) Tethered particle motion. Here, one end of the DNA molecule is immobilized on the surface, while the other end is tethered to a bead (grey). Protein (dark blue) binding to its target sequences (red) causes corresponding changes in the DNA length, thus affecting the allowed bead’s diffusion range (dashed line). (B) Single optical tweezers. A dielectric bead (grey) attached to one of the DNA ends is optically trapped by a diffraction-limited spot-focused laser beam (orange). Gradual movement of the surface can be employed to apply a tension force both to the DNA molecule and surface-immobilized DNA-bound protein (dark blue). (C) Double optical tweezers. Both DNA ends are tethered to the separate dielectric beads (grey) optically trapped by two powerful laser beams (orange). By moving one of the beads away from the other, an increasing tension can be conferred to the DNA and the protein molecule (red) interacting with it. (D) Magnetic tweezers. The DNA molecule is anchored on the surface by one of its ends, whilst another end is attached to a paramagnetic bead (grey). Two permanent magnets (yellow triangles) are then used to create a magnetic field and generate forces enabling the vertical extension of the immobilized DNA. Rotation of the magnets on the x-y plane also allows one to induce torque τ (red arrow) on the DNA molecule. (E) Atomic force microscopy. The surface properties of individual nucleoprotein complexes are probed by the atomically sharp head. The cantilever displacements caused by the combination of attraction and repulsion forces acting during the scanning of the surface are detected by the optical system, in which a laser beam reflected from the back of a cantilever is projected onto a position-sensitive quadrant photodetector. (F) Single-molecule Förster resonance energy transfer (smFRET). Conformational changes in a single protein-DNA complex (grey) result in the distance alteration between the donor (green) and acceptor (red) fluorophores, which is represented by the different FRET efficiency values. (G) DNA flow-stretch assay. The DNA molecule is initially immobilized on the surface through one of its ends and then is extended along the surface by applying a buffer flow. Fluorescent labeling of such a DNA molecule and the investigatory protein (red) enables the direct visualization of discrete protein-DNA interactions by the means of total internal reflection fluorescence microscopy, as both these biomolecules are located in the evanescent field (green) that decays exponentially when moving further away from the surface. (A–D) and (G) are adapted from open-access source [27]. (E) is adapted from [31], with permission from Elsevier. (F) is adapted from open-access source [39].
3. DNA Flow-Stretch Assays
3.1. Traditional Approach
Traditional DNA flow-stretch assays are based on non-predefined position immobilization of typically one of the individual DNA molecule ends on the coverslip surface (Figure 1G) [40,41,42]. In most cases, surface tethering of DNA is achieved through the use of high-affinity non-covalent interactions that ensure a straightforward and specific DNA anchoring on the surface. Streptavidin (sAv)-biotin interaction is commonly employed for such a purpose, since it is one of the strongest naturally occurring non-covalent interactions with a characteristic KD of ~10−15 M [43]. In DNA flow-stretch assays, the surface of a glass coverslip can be coated with various non-specific blocking proteins, such as bovine serum albumin (BSA), a lipid bilayer, or chemically modified with polyethylene glycol (PEG) molecules [44]. All of these approaches not only enable the surface immobilization of individual DNA molecules, but they also passivate the exposed surface of a glass coverslip against non-specific biomolecules adsorption. PEGylation of the surface is the most widely utilized procedure in these kinds of assays, as the glass surface is at first functionalized with methoxy-PEG and biotin-PEG molecules and then streptavidin is deposited on such a surface to allow the immobilization of a biotinylated DNA [45,46]. Upon the application of a buffer flow, single-tethered DNA molecules are stretched along the surface due to the hydrodynamic force. The flexibility and elastic behavior of DNA can be mathematically characterized by the worm-like chain model, which is able to accurately predict the deformation of a linear long-chain polymer under the applied external force [47,48]. Using this model, it has been theoretically estimated that in order to extend a 97 kb bacteriophage λ DNA (λ-DNA) dimer, entropic forces in the range of a few pN are required [49]. Since shear flow of 0.5–2.0 mL/min can induce forces up to 4 pN on surface-immobilized λ-DNA molecules, buffer flow of such velocities is more than sufficient to break the entropic barrier and stretch the single-tethered DNA to ~90% of its full contour length [50]. Thus, fluid flow also reduces the lateral movement of the immobilized DNA molecules [51]. If the second end of surface-immobilized DNA is functionalized, for example, by biotin, a buffer flow can also be used to specifically attach the second end of the DNA molecule on the surface [52].
Due to the flow-stretched DNA molecules being in the vicinity of the glass surface, DNA flow-stretch assays are perfectly compatible with total internal reflection fluorescence (TIRF) microscopy. This assay enables the selective visualization of protein-DNA interactions at the SM level with a low background even when fluorescently labelled protein is diffusing in a solution [53]. Individual DNA molecules are usually labeled with fluorescent intercalating dyes, as this makes it possible to observe the full contour length of the extended DNA. The proteins of interest are typically labeled with bright organic fluorophores or quantum dots. In TIRF microscopy, the intensity of the produced evanescent field decays exponentially, while moving away from the surface [54]. Thus, the illumination is generated only at the glass surface, whereas the background signal produced by the fluorophores diffusing in the solution is greatly reduced. By eliminating the background fluorescence, high signal-to-noise ratios are achieved and that allows easy detecting of individual DNA-interacting molecules in a solution [55].
Although DNA flow-stretch assays enable the immobilization and hydrodynamic flow-directed extension of both relatively short and long DNA molecules, all of this method’s potential is best uncovered when using relatively long DNA substrates. Given that long DNA molecules are tethered and stretched along the surface, it then becomes really convenient to visualize and track the movement of fluorescently labeled proteins of interest along the DNA at the SM level in real time [56]. This also allows one to determine protein-binding profiles on DNA molecules and dwell times, assess the preferential binding sites of the protein, and thoroughly characterize the real-time dynamics of the ongoing individual protein-DNA interactions [57]. Nevertheless, long DNA molecules are more prone to self-entanglement into knots, especially under the conditions of a rapid linearization force, which can induce the formation of DNA knots [58]. In this case, the real binding landscapes of DNA-interacting proteins would then be somewhat distorted, as by reducing the overall length of the DNA molecule, DNA knots could alter the exact positioning of a protein on the DNA or even render some DNA sequences inaccessible for the binding of various proteins. There is a possibility that the hydrodynamic force of a buffer flow could very well trigger the knotting of surface-immobilized DNA molecules. However, the supporting evidence of DNA knots caused solely by fluid-flow stretching of the DNA is currently lacking since in the majority of corresponding studies, the formation of DNA knots is typically induced purposely by mainly using strong electric fields or OTs [59,60]. λ-DNA, which is 48,502 bp long and has a B-form contour length of ~16.5 µm, is currently the most widely employed DNA substrate in such SM experiments [61,62,63,64]. Not only is it relatively long and commercially available, but it can also be easily functionalized with distinct heterologous labels, as λ-DNA possesses 12 bp long cos overhangs at both of its ends, all of which make λ-DNA a particularly attractive option in the DNA flow-stretch-assay-based studies of individual protein-DNA interactions.
All of this information gathered during the SM experiments of DNA flow-stretch assays greatly contributes to the fundamental studies of target search mechanisms employed by different DNA-binding proteins [65,66,67]. It is still not particularly clear how diverse proteins are able to exploit these target search mechanisms for their own benefit to selectively and rapidly locate cognate DNA among a multitude of other non-specific sequences. SM-level mechanistic studies can definitely provide some detailed answers to this question, and here, DNA flow-stretch assays serve as an excellent approach in order to fulfill this aim.
A diverse set of DNA-binding proteins was investigated at the SM level using conventional DNA flow-stretch assays. They allowed for studying the real-time dynamics of single protein-binding events [68,69,70], DNA unwinding [71], individual DNA-interacting protein sliding, and jumping events along the double-tethered elongated non-cognate DNA molecules [72,73]. This biophysical method was also applied in a few studies of DNA repair proteins [74,75]. Moreover, smFRET was combined with the immobilized flow-stretched DNA and revealed binding and diffusion dynamics of discrete mismatch repair initiation protein MutS [76].
3.2. Magnetic Bead-Based Approach
In traditional DNA flow-stretch assays, DNA is usually immobilized on the surface by only one of its ends, as the remaining DNA end stays completely free. An extension of this SM method is occasionally implemented by attaching a paramagnetic micro bead to that available end of a surface-tethered DNA molecule [77,78]. This DNA-bead conjugate is then subjected to a magnetic force that is generated by a permanent magnet placed above the flow cell. The levitation of the magnetic bead allows one to reduce non-specific interactions between the surface and the bead [23]. When the laminar flow is applied, a controllable pN (0.1–10 pN) tension force, which is proportional to the flow rate and the diameter of a magnetic bead, starts to act on the bead [79]. Subjection of a magnetic bead to this force also results in the stretching of the attached DNA molecule. A 15–50 kb long double-stranded DNA (dsDNA) is typically used in such experiments and the imaging of the bead is usually performed by dark field microscopy, allowing one to achieve high signal-to-noise ratios [80]. Due to the bead‘s spherical symmetry, its centroid position can be easily tracked with a precision of 1–10 nm using a sub-pixel fitting [81].
In comparison to the traditional ones, this type of DNA flow-stretch assay can provide more detailed insights into diverse biological reactions such as protein-induced DNA compaction, the formation and disassembly of different DNA topological features, and interconversion between dsDNA and single-stranded DNA (ssDNA) [82,83]. All of these processes trigger changes in DNA length which in turn affect the motion of the magnetic bead, so these bead-motility fluctuations serve as an excellent information source about the ongoing protein-DNA interactions [84,85]. In order to mimic the physiological growth conditions of the microorganisms with respect to the temperature, this kind of DNA flow-stretch assay was performed at a high temperature (65 °C). Furthermore, hydrodynamic flow-induced stretching of surface-immobilized and bead-tethered DNA molecules was extensively applied for the SM-level studies of DNA replication dynamics [86,87,88].
3.3. DNA Tightropes
DNA tightropes, otherwise known as elevated DNA platforms, also belong to the extensive group of DNA flow-stretch assays. In this kind of SM approach, the ends of the individual DNA molecule are suspended on the top of two micron-sized beads by the means of a hydrodynamic flow [89]. The beads are typically coated with poly-L-lysine, which ensures strong binding of the DNA molecule to the bead. Such an experimental design eliminates the necessity for different immobilization strategies that are based solely on the surface chemistry, and it also rules out the requirement for a continuous buffer flow after the DNA tightropes are formed [20]. Because individual DNA molecules are elevated above the glass surface, any other molecules that enter the focal plane at this height must then be bound to the DNA, whilst all loose molecules only contribute to the total background noise. In this case, proteins that interact with such suspended DNA molecules are located further away from any non-specifically surface-bound fluorophores, so the latter no longer interfere with the discrimination of fluorescent spots during the analysis of recorded images [90,91].
DNA tightropes proved to be beneficial in the SM-level studies of various DNA-binding proteins, especially of these participating in DNA base and nucleotide excision repair or in the maintenance of human telomeres. For these studies, DNA tightropes consisted of phage λ or plasmid concatemer DNA and fluorescently labelled protein binding or movement on the DNA substrate was observed in real time [92,93,94,95,96]. DNA tightropes assembled using either λ-DNA or DNA substrates consisting of alternating regions of telomeric and non-telomeric sequences were employed for probing DNA-interaction dynamics of quantum-dot-labeled proteins [97]. Another study combined oblique angle TIRF-microscopy-based DNA tightrope assay with AFM and prism-type TIRF microscopy imaging to observe DNA-interacting protein-promoted DNA compaction and DNA-DNA bridging [98].
The vast majority of previously described SM biophysical techniques that can be applied for exploring protein and DNA interactions allow one to manipulate only one investigatory biomolecule at a time. The experimental design of DNA flow-stretch assays enables the observation of tens of individual fluid flow-extended DNA molecules in the same field of view of the fluorescence microscope [32]. In comparison to the others, the increased throughput of this method makes it feasible to conduct more efficient SM-level studies of dynamic protein-DNA interactions. Nevertheless, DNA flow-stretch assays are accompanied by several substantial drawbacks that restrict their application in these kinds of studies. As in this approach, the individual DNA molecules are randomly immobilized on the coverslip surface, and the number of DNA molecules per single field of view of the microscope must remain small enough (no more than a few dozens of molecules) in order to avoid overlapping DNA molecules [99]. Such surface-fixed DNA molecules also have varying degrees of extension. Ultimately, the orientation of the double-tethered DNA molecules, in terms of a nucleotide sequence, is unknown.
4. Traditional DNA Curtains
4.1. Technological Design
A novel next-generation in vitro SM biophysical technique—DNA curtains—is by far the most superior type of DNA flow-stretch assay, as it is capable of imaging hundreds or even thousands of separate fluorescently labeled protein-DNA complexes at once in real time [100]. The high-throughput nature of DNA curtains determines one of its fundamental advantages over the other similar approaches—in conjunction with TIRF microscopy, this technology enables the collection of large amounts of statistical data during a single experiment [101]. As well as the other types of DNA flow-stretch assays, this experimental platform can provide a whole set of information regarding the SM dynamics of protein-DNA interactions, in particular, when relatively long DNA substrates are employed. Real-time visualization of protein translocation along the DNA molecule, complete characterization of DNA-binding landscapes, and the assessment of protein dwell times—all of this can be efficiently accomplished by the means of DNA curtains [102].
The technological principle of conventional DNA curtains is implemented through the combination of supported lipid bilayers (SLBs) and lipid diffusion barriers (Figure 2A) [100]. In this approach, the surface of a flow cell is initially passivated with an SLB, which has a fraction of its lipid heads functionalized with biotin. Afterwards, this lipid bilayer is coated either with neutravidin or sAv and the biotinylated DNA end is then immobilized on this protein monolayer. The fluidity of the lipid bilayer ensures the long-range 2D motion of lipid-tethered DNA. By applying a hydrodynamic flow, such DNA molecules can be organized at the lipid diffusion barriers, which are deployed on the flow cell surface and are perpendicular to the buffer flow direction (Figure 2B). These physical barriers disrupt the lateral diffusion of the lipids, as they cannot physically cross such barriers, and that results in the accumulation of lipid-tied DNA molecules along the leading edges of the barriers. Being under the continuous hydrodynamic force, hundreds or even thousands of individual DNA molecules not only align in a parallel manner along these diffusion barriers, but they also stretch along the surface of the flow cell. Such a strategy allows one to visualize these DNA molecules with TIRF microscopy (Figure 2C,D) [99,101].
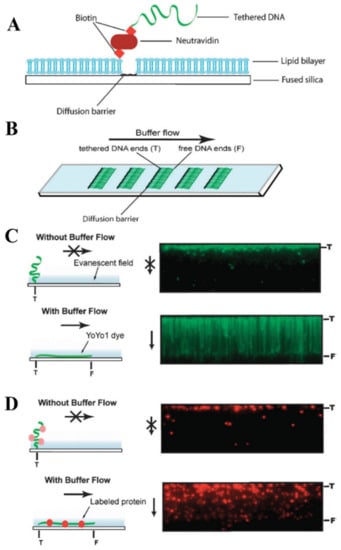
Figure 2.
Technological design of high-throughput traditional DNA curtains. (A) Side view schematically representing the fundamental components that form conventional DNA curtains. (B) Top view of a microscope slide with several arrays of DNA curtains on its surface. (C) Left: schematic representation of surface-tethered DNA molecule response to the buffer flow changes. Right: total internal reflection fluorescence (TIRF) microscopy images showing the response of individual DNA molecules organized in a parallel manner to the application and stopping of a hydrodynamic flow. In the absence of a buffer flow, DNA molecules diffuse away from the glass surface and leave the evanescent field. Here, DNA is stained with the intercalating YOYO1 fluorescent dye. (D) A representative experiment, in which traditional DNA curtains are used to directly observe single protein-DNA interactions. In this case, the DNA molecules are not stained, while the investigatory protein (chromatin remodeling enzyme Rdh54) is fluorescently labeled using a quantum dot. Stopping of the hydrodynamic flow results in the out of evanescent-field diffusion of individual DNA-bound protein molecules. Left: schematic drawing. Right: TIRF microscopy images. Reprinted from [44], with permission from Royal Society of Chemistry.
The employment of SLBs determines another substantial advantage of traditional DNA curtains. Lipid bilayers are splendidly compatible with SM biochemical experiments since in the flow cell they mimic a similar microenvironment that is typically found inside the cell. Lipid bilayers can be readily formed, and they also tolerate a variety of solution conditions [50]. In addition, lipid bilayers can be easily modified by incorporating synthetic lipids with substituted heads, whereas bilayers composed of zwitterionic lipids help to avoid non-specific surface-adsorption of DNA and distinct proteins in the range of near-neutral pH values.
Another essential component of conventional DNA curtains’ technological design—lipid diffusion barriers—can be fabricated by several techniques. The most straightforward approach is the manual etching of diffusion barriers, when the surface of a glass slide is engraved with a diamond-tipped drill [50]. This method does not require any specialized equipment, yet it does not allow control of the width, depth, and arrangement of the etched barriers. For this reason, diffusion barriers are usually formed by the means of nanofabrication which enables surface patterning of a nanometer precision [103]. In electron beam lithography, a microscope slide is at first covered with a thin polymeric film—polymethylmethacrylate (PMMA) bilayer—on top of which a layer of an antistatic agent (Aquasave) is then deployed. Next, the electron beam is used to etch the desired pattern in the polymeric film, thus exposing the underlying glass surface. The metal (typically chromium) is then evaporated in vacuum and deposited on the entire surface including PMMA and the uncovered glass. The remaining polymer is peeled off during the lift-off procedure, as only the metallic structures remain on the microscope slide. These metal assemblies, ultimately, serve as robust lipid diffusion barriers that do not interfere with the fluorescence imaging of the immobilized DNA molecules [100,104,105]. Fabrication of physical barriers can also be implemented through the nanoimprint lithography which is rather similar to the one described above, as this technique conjoins the utilization of electron beam and lift-off with the inductively coupled plasma etching [106]. Although electron beam and nanoimprint lithographies permit the high-resolution patterning of glass substrates, they are restricted by their low-throughput nature, as both these lithographic techniques require the raster scanning of an electron beam along each nanobarrier segment, which limits the overall number of barriers formed on the surface of each slide. Another drawback is the necessity of high-cost specialized equipment. To overcome these shortcomings, a rapid, cost-effective, UV-lithography-based method has been recently developed, enabling the high-throughput manufacturing of chromium-based physical barriers [107].
Lipid diffusion barriers can also be fabricated out of a whole spectrum of different materials. During the production of these nanobarriers by electron beam or nanoimprint lithography, vapor deposition of thin films, composed of such metals or oxides as chromium, titanium oxide, or gold, is usually performed on the surface of a glass slide [50,100,103,108]. Physical barriers impeding the lateral diffusion of lipids can also be made from hydrogen silsesquioxane [104]. This negative resistance eliminates the need for metal evaporation, thus simplifying and shortening the whole process of nanobarrier fabrication. Yet, most of the time, lipid diffusion barriers are formed out of chromium. Since this approach is fairly expensive and time-consuming, chromium-patterned microscope slides are usually reused, which means that such slides must be thoroughly cleaned after each experiment in order to avert the interference of surface-remaining organic matter with the subsequent experiments. However, strong cleaning agents (acetone, acids) and harsh cleaning methods (sonication, boiling) tend to damage chromium barriers; with mild cleaning materials, these barriers still wear out after several reuse cycles, and they also begin to scatter the excitation beam. Recently, it was demonstrated that DNA curtains can be assembled using nanotrenches as barriers to the lateral diffusion of lipids [105]. Nanofabrication of trenches is achieved by the means of electron beam lithography and reactive ion etching, where these physical barriers of different nature are engraved on the glass slide surface. Nanotrenches are robust and do not degrade even under extremely harsh cleaning conditions and that enables the complete removal of any residual surface contaminants. This, in turn, ensures the reliable and repeated reuse of patterned microscope slides and allows one to assemble well-organized high-quality DNA curtains during each successive experiment.
4.2. Single-Tethered DNA Curtains
To address the need for specific conditions dictated by a broad diversity of SM experiments, DNA curtains can be prepared in several different configurations. The most common of them is single-tethered DNA curtains, in which individual DNA molecules are immobilized on a lipid bilayer by one of their ends [109]. TIRF imaging of such DNA molecules is only possible under the conditions of a continuous buffer flow that stretches the DNA along the glass surface. The advantage of this configuration is the ability to temporarily stop the fluid flow, as during the experiment this can be utilized as a standard control to verify if the observed solitary DNA and protein molecules are not non-specifically bound to the flow cell surface. In addition, the constant buffer flow permits one to maintain the same tension for all anchored DNA molecules. Single-tethered DNA curtains can be assembled by employing either linear or zigzag barriers to lipid diffusion (Figure 3A,B) [100,110]. Linear barriers are rather simple, but they can lead to the overlapping of individual surface-tethered DNA molecules, which is undesirable in certain types of experiments. This is typically avoided with the use of zigzag barriers that ensure the spatial separation of each DNA molecule from its proximate neighbor.
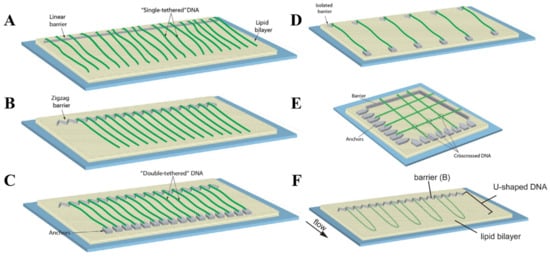
Figure 3.
Configurational diversity of conventional DNA curtains. (A) Single-tethered DNA curtains assembled using a linear barrier to lipid diffusion. (B) Single-tethered DNA curtains assembled utilizing a zigzag barrier to lipid diffusion. The distance between neighboring DNA molecules is determined by a zigzag pattern. (C) Double-tethered DNA curtains. In this configuration, one end of an individual DNA molecule is immobilized on a supported lipid bilayer, while the other DNA end is typically tethered to a downstream pedestal via a specific digoxigenin–anti-digoxigenin interaction. (D) DNA curtains assembled employing a parallel array of double-tethered isolated (PARDI) molecule patterns. Such a configuration of DNA curtains grants the possibility to maintain an extremely low local concentration of surface-immobilized DNA molecules during the experiment. (E) Crisscrossed DNA curtains. Here, the intersections (marked with circles) between the crisscrossed DNA molecules represent the regions of locally high DNA concentration. (F) U-shaped DNA curtains. Since individual DNA molecules in this configuration are anchored on a lipid bilayer by both of their ends, application of a hydrodynamic flow forces the DNA molecules to adopt a “U” shape during their alignment at the lipid diffusion barriers. (A–E) are adapted from [102], with permission from Elsevier. (F) is adapted from open-access source [108].
Single-tethered DNA curtains were utilized for probing the SM dynamics of a broad spectrum of DNA-binding proteins in real time. Abo1 is one of those proteins for which biological functions have been recently uncovered by the use of DNA curtains assembled in a single configuration [111]. Single-tethered DNA curtains were also combined with biochemical assays to gather more molecular details about Abo1-mediated histone loading onto DNA and the ongoing interactions between Abo1 and DNA [112]. Another study combined single-tethered and double-tethered DNA curtains to monitor the motion of individual fluorescently labeled Type I-E CRISPR-Cas proteins on DNA [113]. It revealed that Cas1-Cas2 integrase is able to non-specifically sample the DNA through 3D collisions with a short lifetime, while Cas3 helicase/nuclease translocates on DNA by using two main modes, as at the target sequence it either moves in conjunction with the 11-subunit crRNA-containing Cascade surveillance complex or independently away from Cascade. In the other study, the employment of single-tethered DNA curtains and catalytically dead RNA-programmable DNA endonuclease CRISPR-Cas12a enabled the SM-level real-time fluorescence imaging of artificial virus-like nucleocapsids self-assembly kinetics [114]. Furthermore, single-tethered DNA curtains were applied for directly probing the behavior of individual quantum-dot-labeled DNA translocating proteins [115,116,117]. In these studies, to probe the crowding effect on protein-of-interest translocation, single-tethered DNA curtains were assembled using DNA substrates that were occupied by different proteins, such as EcoRIE111Q, E. coli RNA polymerase holoenzyme, lac repressor, or nucleosomes. Single-tethered DNA curtains were also utilized in the SM-level mechanistic studies of diverse proteins participating in the cellular processes of DNA end resection, such as human resectosome [118], mycobacterial heterodimeric helicase-nuclease AdnAB [119,120], DNA-dependent protein kinase [121], poly(ADP-ribose) polymerase-1 [122], and exonuclease 1 [123]. Moreover, this type of next-generation in vitro DNA flow-stretch assay was proven to be suitable for the efficient studies of discrete interactions between the DNA and Rad51 recombinase, the key component of the eukaryotic HR machinery [124,125,126,127,128,129].
4.3. Double-Tethered DNA Curtains
Certain SM experiments are better performed without a continuous buffer flow, as some of them involve the usage of expensive reagents, while others cover the investigation of individual protein interactions with the surface-immobilized DNA molecules. It is in the latter case that the hydrodynamic force induced by the flow of a buffer can potentially affect the behavior of proteins or their complexes, leading to the disruption of protein-DNA interactions or causing some undesired bias, for instance, during the target search or when a protein is travelling along the DNA. In double-tethered DNA curtains, discrete DNA molecules are immobilized on the surface by both of their extremities, enabling the visualization of the entire DNA contour length in the absence of any fluid flow (Figure 3C) [130]. This configuration is implemented through the use of two structural elements: linear or zigzag barriers and pedestals that are located downstream of these obstructions to lipid diffusion. Single end of the DNA molecule is initially tethered to the lipid bilayer through a specific biotin-sAv interaction. By applying a buffer flow, such DNA molecules are then aligned in a parallel manner at the physical barriers and stretched along the glass surface. Since the other end of the extended DNA molecule is modified with digoxigenin, it is lastly attached to the downstream pedestals coated with digoxigenin antibodies. Therefore, double configuration of DNA curtains ensures that all of the separate surface-fixed DNA molecules have an identical orientation with respect to their nucleotide sequence.
As well as single-tethered ones, DNA curtains assembled in a double configuration were extensively applied in the multitude of mechanistic studies of individual protein-DNA interactions. Single- and double-tethered DNA curtains in tandem with ensemble biochemical measurements were employed to investigate the target search mechanisms of S. pyogenes RNA-programmable DNA endonuclease Cas9 belonging to Type II CRISPR-Cas systems [131]. By utilizing double-tethered DNA curtains, real-time SM fluorescence imaging of target search of E. coli Cascade nucleoprotein complex on λ-DNA was performed, which showed that such a process can be implemented in two distinct pathways—PAM-dependent or PAM-independent [132]. This kind of next-generation in vitro DNA flow-stretch assay was also employed for SM characterization target search mechanisms of various proteins including: Type I-E CRISPR-Cas interference and primed acquisition complexes [133], hexameric DNA translocase FtsK without obstacles on DNA [134], and in the presence of obstacles [135]. Furthermore, DNA curtains assembled in a double configuration were used to observe rapid, highly processive, and ATP-hydrolysis-dependent translocation of S. cerevisiae quantum-dot-labeled condensin along DNA at the SM level [136]. This SM biophysical technique was also utilized to probe the movement of Mlh1-Pms1 along DNA by which this complex locates the lesion-bound Msh2-Msh6 proteins during the eukaryotic postreplicative mismatch repair [137]. Similarly, double-tethered DNA curtains enabled the disclosure of lesion-search mechanism characteristic of Msh2-Msh3 proteins, as it was shown that this eukaryotic mismatch repair complex uses 1D sliding and microscopic hopping to scan individual DNA molecules, which determines its ability to bypass nucleosomes and other protein roadblocks during the lookout for DNA damage [138]. There are several SM-level studies of proteins involved in HR which also make use of DNA curtains assembled in either single or double configuration [139,140,141,142].
4.4. Other Configurations
The configurations described above are the two fundamental types of DNA curtains, as they are typically employed for the efficient mechanistic studies of protein-DNA interactions at the SM level. Still, there are some other occasionally used configurations of traditional DNA curtains which are designed for more specialized SM experiments. DNA curtains can be assembled using parallel array of double-tethered isolated (PARDI) molecule patterns (Figure 3D) [143]. This configuration ensures a 7 µm or even larger spacing between adjacent double-tethered DNA molecules, which means that during the experiment the local concentration of DNA is maintained at an extremely low value. Thus, DNA curtains based on PARDI patterns are suitable for studying individual protein-DNA interactions, in particular protein association kinetics, under the conditions where the effects of locally high DNA concentration are absent. It is the complete opposite with crisscrossed DNA curtains, as in this kind of configuration, high local DNA concentration is established by perpendicularly crossing solitary double-tethered DNA molecules (Figure 3E) [144]. Such DNA molecules are separated one from another at an average of ~106 nm and the intersections between them are considered to be locally high DNA concentration regions, which can be utilized for investigating the capabilities of different proteins to travel between two DNA substrates located in close proximity to each other. There are also U-shaped DNA curtains, in which both biotinylated ends of the individual DNA molecules are initially immobilized on a lipid bilayer (Figure 3F) [108]. The continuous buffer flow is then applied, causing the DNA to adopt a “U” shape, as double-tethered DNA molecules are aligned at the zigzag barriers to lipid diffusion. Such a configuration is especially beneficial for studying distinct mechanisms of DNA compaction, such as loop extrusion induced by condensins, at the SM level, since it enables real-time visualization and tracking of DNA-length and conformational changes during the individual DNA looping events.
4.5. Single-Stranded DNA Curtains
dsDNA is not the only DNA substrate that can be utilized for the assembly of conventional DNA curtains. In most biochemical reactions that comprise the processes of DNA repair and replication, ssDNA plays a key role as an intermediary product. To achieve a deeper understanding of these biomolecular processes, the experimental tool of ssDNA curtains was developed, enabling the fluorescence imaging of surface-tethered stretched ssDNA molecules at the SM level [145,146,147,148]. During the assembly of ssDNA curtains, the synthesis of long ssDNA molecules is at first performed by the means of a rolling circle replication, as φ29 DNA polymerase, a circular ssDNA template and a biotinylated DNA primer are utilized for this reaction (Figure 4A) [149]. The ssDNA products of a rolling circle replication are immobilized on the lipid bilayer through biotin-sAv interaction, and by applying a buffer flow, they are then aligned in a parallel manner along the leading edges of the diffusion barriers. However, optical imaging of such ssDNA molecules is hampered by high ssDNA compactness and the inability to fluorescently label it with intercalating dyes, as they compromise the integrity of the ssDNA molecules. To overcome these limitations, replication protein A (RPA) fused with enhanced green fluorescent protein or mCherry protein is usually employed, as RPA is capable of binding to the ssDNA and removing its secondary structure [146,147]. RPA-ssDNA filaments are much stiffer than the uncovered ssDNA and that allows RPA-bound ssDNA molecules to be stretched under a constant buffer flow and visualized by the means of TIRF microscopy. ssDNA curtains can also be assembled in a double configuration, in which RPA-ssDNA complexes are non-specifically adsorbed on the exposed anchor points located downstream of the lipid diffusion barriers (Figure 4B) [145,147]. Another extension of ssDNA substrate-based DNA curtains is low-complexity ssDNA curtains that allow one to simultaneously synthesize and visualize individual low-complexity ssDNA molecules at the SM level in real time [150]. These DNA curtains are assembled using a phosphorylated template, which is designed in such a manner that standard Watson–Crick base pairing in the generated ssDNA product is non-existent, a biotinylated primer, T4 DNA ligase, and φ29 DNA polymerase. After a primer is annealed with the template, T4 DNA ligase is employed to produce closed minicircles that are then injected into the flow cell. During a rolling circle replication with φ29 DNA polymerase, ssDNA molecules in length of >50,000 nucleotides are synthesized, and then organized at lipid diffusion barriers by employing a buffer flow. Since the produced ssDNA consists of only thymidine and cytidine, the number of possible secondary structures in ssDNA is minimized and the stretching of this molecule can be achieved by applying only minimal force. Such a unique feature of low-complexity DNA curtains makes this kind of experimental platform especially beneficial for investigating the ssDNA physical changes induced by its interactions with other nucleic acids or ssDNA-binding proteins.
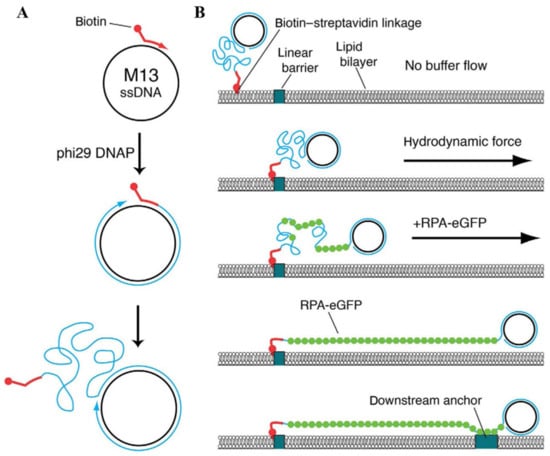
Figure 4.
Assembly of ssDNA curtains. (A) Biotinylated ssDNA substrate is produced by a rolling circle replication. A biotinylated primer, circular ssDNA template such as M13mp18, and φ29 DNA polymerase are employed for this kind of DNA replication. (B) During the assembly of ssDNA curtains, a biotinylated ssDNA substrate is initially immobilized on a supported lipid bilayer and aligned at the lipid diffusion barriers by applying a hydrodynamic flow. Next, RPA-eGFP is introduced into the flow cell. This fusion protein fluorescently labels the ssDNA molecule and removes its secondary structure. As RPA-eGFP enters the channel of a flow cell, ssDNA becomes visible, as it gradually unwinds and stretches along the surface. To assemble double-tethered ssDNA curtains, RPA-ssDNA complex is non-specifically adsorbed on the exposed pedestals deployed downstream of the lipid diffusion barriers. Reprinted from [149], with permission from Elsevier.
ssDNA curtains are most suitable for investigating ssDNA intermediates involving cellular processes, such as DNA replication and HR, at the level of a single molecule. This experimental tool enabled high-throughput SM fluorescent imaging of RADX, a mammalian ssDNA-binding protein that preserves uncapped telomeres and stalled replication forks, and interactions with ssDNA substrates [151]. Another study combined ssDNA curtains with bulk biochemical assays to examine the molecular mechanisms by which RADX regulates RAD51 activity and DNA replication [152]. ssDNA curtains were utilized to probe the mechanism employed by RAD51 paralogs to remodel and stabilize RAD51 filaments [153], investigate the homology search—alignment and pairing of ssDNA with a homologous duplex DNA—of S. cerevisiae RAD51 [154], examine the interplay between S. cerevisiae RPA, RAD52, and RAD51 during the assembly of a presynaptic complex [155], monitor the interactions between human RPA and human RAD51 during presynaptic complex formation [156], and study the properties of presynaptic complexes composed of two S. cerevisiae DNA recombinases—RAD51 and DMC1, of which the latter is a meiosis-specific enzyme [157]. ssDNA curtains in conjunction with the biolayer interferometry measurements allowed one to investigate the behavior of human RECQ5 helicase, which exhibits anti-recombinase activity by negatively regulating RAD51 during HR, loaded on different nucleoprotein complexes at the SM level in real time [158]. Moreover, real-time visualization of individual S. cerevisiae Srs2 superfamily 1 helicase, which is another anti-recombinase maintaining genome integrity through the disassembly of toxic recombination intermediates, complexes acting upon long RAD51-bound ssDNA substrates, was performed using ssDNA curtains [159]. In combination with additional methods, ssDNA curtains were likewise applied in SM-level mechanistic studies of other HR-related proteins, such as RAD52 [160,161], RAD54 [162,163], and RPA [164,165,166].
4.6. Combination with Other Techniques
There are also a few successful examples of traditional DNA curtains being coupled with other manufacturing and biophysical techniques. In order to fully characterize a biochemical reaction, it is commonly necessary to perform multiple long-term experiments of the same nature, in which different reaction conditions related to the changes in protein composition or nucleotide or salt concentration can be thoroughly tested. For this reason, DNA curtains platform was combined with polydimethylsiloxane (PDMS)-based technology of microfluidics [167]. This allowed one to enhance the throughput of DNA curtains tool even further, providing an opportunity to concurrently investigate up to five distinct biochemical reaction conditions at the SM level. Another example is the TIRF-trap microscope—a combination of DNA curtains with OTs—that was developed for investigating the physical properties of separate DNA molecules organized in single-tethered DNA curtains configuration [168]. In this setup, the biotinylated end of a DNA molecule aligned at a nanofabricated diffusion barrier is immobilized on an sAv-covered lipid bilayer, while the other DNA extremity is attached to a fluorescently labeled laser-trapped bead via a digoxigenin–anti-dig linkage. Therefore, this experimental tool not only permits the high-throughput imaging of individual protein-DNA interactions in real time, but it also allows one to simultaneously perform force-based measurements, as the response of these biomolecular complexes to the applied external force can be observed directly.
5. Other Types of DNA Curtains
The technological design of conventional DNA curtains is implemented solely through the use of two fundamental components—SLBs and nanobarriers to the lateral diffusion of lipids. Nevertheless, they likewise confer several shortcomings on this next-generation SM approach. Preparation of lipid-bilayer-based systems may pose some experimental risks related to system stability and defect management. Meanwhile, nanofabrication of physical barriers, which can be executed by the diverse production methods discussed above, is time-consuming, technically challenging, and requires not only specific knowledge in the corresponding field of manufacturing, but also expensive equipment with limited availability. These limiting factors create the need for the development of innovative strategies for assembling DNA curtains in such a way that all the experimental capabilities offered by this platform would be completely retained, whilst banishing the aforementioned drawbacks.
Suspended DNA curtains are somewhat capable of fulfilling such requirements, as this type of DNA curtain provides a few significant advantages over the traditional ones, although it falls short of some basic features. In suspended DNA curtains, individual DNA molecules are, at first, attached to a gold nanowire, which bisects the microfluidic channel of a flow cell, via biotin-sAv interactions, and are then organized in a parallel manner by a buffer-flow-induced stretching [169]. Since this nanowire is elevated, the DNA molecules coupled to it are suspended further away from the flow cell surface, which prevents the DNA molecules from non-specifically interacting with the surface and should, in theory, allow one to only observe those proteins that are bound to DNA. This kind of configuration also determines the overall lower buffer flow rates necessary to achieve the maximum extension of nanowire-tethered DNA molecules, as they experience a more rapid and uniform hydrodynamic flow due to the elevation. However, suspended DNA curtains are still hardly applicable for efficient studies of protein-DNA interactions since the assembling strategy, which this SM platform is based on, results in substantial overlapping of individual nanowire-anchored DNA molecules and, most importantly, it compromises the fundamental feature of DNA curtains—the high-throughput imaging capabilities offered by this technology.
Another type of these next-generation in vitro DNA flow-stretch assays that perfectly meet the criteria stated above is soft DNA curtains [170]. In this recently developed version of DNA curtains, corresponding scanning probe microscopy and soft lithography techniques—AFM and protein lift-off microcontact printing—are combined together, enabling the precise nanopatterning of chemically modified glass coverslip surfaces with protein line-features (Figure 5A). At first, the flat side of PDMS stamp is covered with sAv or traptavidin (tAv) by inking and then the protein ink drop is sucked with the pipette, followed by the washing and drying of the elastomeric stamp. Next, inked PDMS stamp is placed on the Si master, surface of which has inscribed nanometer-sized lines. At this stage, a controllable printing pressure acts on the formed PDMS-master “sandwich”, as this force is generated using a home-built portable printing device. The stamp is then removed from the Si master, determining the selective subtraction of certain protein regions covering the surface of the stamp, and is transferred onto the glass coverslip, the surface of which is coated with a layer of methoxy-PEG and biotin-PEG molecules. At this stage, a controllable printing pressure generated by the home-built portable printing device also acts on the formed PDMS-coverslip “sandwich”. Finally, the stamp is removed from the chemically modified glass coverslip, resulting in the formation of protein line-features on the glass surface.
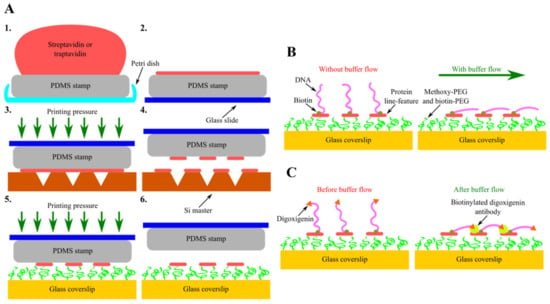
Figure 5.
Assembly and configurational variety of soft DNA curtains. (A) Principal steps of soft DNA curtains assembly—inking of the elastomeric stamp (1, 2) and selective subtraction of proteins by lift-off microcontact printing (3–6). (B) Single-tethered soft DNA curtains. Individual DNA molecules biotinylated only at a single end are immobilized on streptavidin (sAv) or traptavidin (tAv) line-features patterned on the glass coverslip surface. In the absence of a buffer flow, the free end of such DNA molecules is diffused further away from the surface. Upon the application of a hydrodynamic flow, these single-tethered DNA molecules stretch along the glass surface and align in a parallel manner with respect to each other. (C) Double-tethered soft DNA curtains. To assemble double-tethered soft DNA curtains, biotin and digoxigenin-functionalized DNA substrates and biotinylated anti-dig antibodies are employed. These antibodies are initially introduced into the flow cell containing the nanopatterned surface and single-tethered DNA molecules. In the presence of a buffer flow, the digoxigenin end of the stretched DNA molecules then binds to the adjacent anti-dig-coated sAv or tAv line-feature, thus ensuring that such DNA molecules stay aligned in a parallel manner and remain in an extended conformation even when the hydrodynamic flow is halted.
sAv or tAv line-features formed on the PEGylated glass coverslip surface serve as the stable anchor points for the surface immobilization of biotinylated DNA molecules. Moreover, such protein features ensure predefined alignment of the DNA molecules after immobilization. Upon the application of a buffer flow, anchored DNA molecules are stretched along the glass surface, allowing them to be visualized with TIRF microscopy (Figure 5B). Soft DNA curtains can also be assembled in a double configuration, where the digoxigenin-labeled end of the surface-tethered DNA molecule is attached to the adjacent protein line-feature covered in biotinylated digoxigenin antibodies by the means of a hydrodynamic flow (Figure 5C) [171]. Same as the traditional ones, soft DNA curtains enable the high-throughput fluorescence microscopy imaging of individual DNA molecules organized in a massively parallel manner, whereas double-tethered soft DNA curtains also ensure the defined orientation of surface-immobilized DNA. For instance, soft DNA curtains were employed to observe the restriction endonuclease BfiI-catalyzed hydrolysis of discrete λ-DNA molecules and to visualize the dynamics of S. pyogenes Cas9 specific interactions with the double-tethered λ-DNA. Nevertheless, the eliminated need for expensive, specific-handling-knowledge-requiring equipment and the ingenious technological design of soft DNA curtains render them a cost-effective, simple, versatile, and user-friendly platform for efficient SM-level studies of protein-DNA interactions that is worth having in one’s scientific toolbox even for users with a little experience in this research field.
Moreover, an alternative to the whole technology of DNA curtains—DNA skybridge—has been developed recently [172]. As well as DNA curtains, DNA skybridge enables the high-throughput imaging of protein-DNA interactions at the SM level. However, by utilizing some unique strategies, this novel tool fulfills the key concept of DNA curtains somewhat differently. A special 3D structure consisting of 4 µm high thin quartz barriers serves as a basis for the assembly of DNA skybridge. One biotinylated end of an individual DNA molecule is initially immobilized on the sAv-coated apex of the barrier. Upon the application of a buffer flow, this DNA molecule is then stretched, and its other end is tethered to the adjacent quartz ridge via the same biotin-sAv interaction. Ultimately, such DNA molecules are visualized using thin light sheet fluorescence microscopy. These two fundamental differences between DNA curtains and DNA skybridge permit some considerable advantages to the latter platform. The employment of the different optical imaging setup allows one to obtain high signal-to-noise ratios by reducing the background noise caused by the surface-bound fluorophores. Meanwhile, the organization of individual DNA molecules in a manner in which they are located far away from the surface eliminates the impact of false signals coming from non-specifically adsorbed fluorescently labeled proteins on the real-time SM-level visualization of dynamic protein-DNA interactions.
6. Future Perspectives
DNA flow-stretch assays, especially those using relatively long DNA molecules, are a versatile tool for investigating protein-DNA interaction dynamics and real-time protein movement at the SM level, thus revealing the target search mechanisms and many other characteristic features of DNA-binding proteins that are otherwise concealed in traditional bulk-type measurements. In general, DNA flow-stretch assays mostly differ by the surface modification and nanopatterning techniques, DNA immobilization and organization strategies, and the DNA substrates they employ (Table 1). Since protein non-specific adsorption on the surface is still fairly prevalent in most types of DNA flow-stretch assays, novel surface passivation methods can be utilized to create a more inert and biologically compatible environment during the ongoing experiments. It is expected that in future these assays will enable high-throughput imaging of individual protein interactions with other nucleic acid substrates, such as ssDNA-dsDNA, DNA-RNA hybrids, or only RNA. This would greatly expand the capabilities of DNA flow-stretch assays, as new and less characterized proteins such as Cas13 and Cas14 or eukaryotic and prokaryotic Argonautes could be studied at the level of a single molecule. In addition, a common feature of DNA flow-stretch assays is that almost all of them rely on TIRF microscopy imaging. Since novel super-resolution microscopy approaches are constantly emerging and the already existing ones are being rapidly improved, combining them with such assays would provide an opportunity to access currently invisible features of different protein-nucleic acid interactions. Ultimately, DNA flow-stretch assays should become a cost-effective, widely available, and user-friendly set of SM techniques for efficient studies of diverse protein-DNA interactions.

Table 1.
Summary of various techniques used for protein-DNA interaction studies highlighting the technological basis and major benefits and limitations of each method.
Author Contributions
Writing—original draft preparation, review and editing: A.K., M.T., M.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Research Council of Lithuania [S-MIP-20-55 and 09.3.3.-LMT-K-712-19-0113].
Conflicts of Interest
The authors declare no conflict of interest.
References
- Maeshima, K.; Eltsov, M. Packaging the genome: The structure of mitotic chromosomes. J. Biochem. 2008, 143, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Sung, P.; Klein, H. Mechanism of homologous recombination: Mediators and helicases take on regulatory functions. Nat. Rev. Mol. Cell Biol. 2006, 7, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kaçmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef]
- O’Donnell, M.; Langston, L.; Stillman, B. Principles and concepts of DNA Replication in bacteria, archaea, and eukarya. Cold Spring Harb Perspect Biol. 2013, 5, a010108. [Google Scholar] [CrossRef]
- Shandilya, J.; Roberts, S.G.E. The transcription cycle in eukaryotes: From productive initiation to RNA polymerase II recycling. Biochim. Biophys. Acta 2012, 1819, 391–400. [Google Scholar] [CrossRef]
- Spitz, F.; Furlong, E.E.M. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef]
- Tock, M.R.; Dryden, D.T. The biology of restriction and anti-restriction. Curr. Opin. Microbiol. 2005, 8, 466–472. [Google Scholar] [CrossRef]
- Marraffini, L.A. CRISPR-cas immunity in prokaryotes. Nature 2015, 526, 55–61. [Google Scholar] [CrossRef]
- Ferraz, R.A.C.; Lopes, A.L.G.; da Silva, J.A.F.; Moreira, D.F.V.; Ferreira, M.J.N.; de Almeida Coimbra, S.V. DNA–protein interaction studies: A historical and comparative analysis. Plant. Methods 2021, 17, 82. [Google Scholar] [CrossRef]
- Tinoco, I.; Gonzalez, R.L. Biological mechanisms, one molecule at a time. Genes Dev. 2011, 25, 1205–1231. [Google Scholar] [CrossRef]
- Kapanidis, A.N.; Strick, T. Biology, one molecule at a time. Trends Biochem. Sci. 2009, 34, 234–243. [Google Scholar] [CrossRef]
- Yang, W.; Restrepo-Pérez, L.; Bengtson, M.; Heerema, S.J.; Birnie, A.; van der Torre, J.; Dekker, C. Detection of CRISPR-DCas9 on DNA with solid-state nanopores. Nano Lett. 2018, 18, 6469–6474. [Google Scholar] [CrossRef]
- Hornblower, B.; Coombs, A.; Whitaker, R.D.; Kolomeisky, A.; Picone, S.J.; Meller, A.; Akeson, M. Single-molecule analysis of DNA-protein complexes using nanopores. Nat. Methods 2007, 4, 315–317. [Google Scholar] [CrossRef]
- Wanunu, M.; Sutin, J.; Meller, A. DNA profiling using solid-state nanopores: Detection of DNA-binding molecules. Nano Lett. 2009, 9, 3498–3502. [Google Scholar] [CrossRef]
- Wong, C.T.A.; Muthukumar, M. Polymer capture by electro-osmotic flow of oppositely charged nanopores. J. Chem. Phys. 2007, 126, 164903. [Google Scholar] [CrossRef]
- Farajpour, N.; Lastra, L.S.; Sharma, V.; Freedman, K.J. Calibration-less DNA Concentration measurements using EOF volumetric flow and single molecule counting. Front. Nanotechnol. 2021, 3, 58. [Google Scholar] [CrossRef]
- Fan, H.-F.; Ma, C.-H.; Jayaram, M. Single-molecule tethered particle motion: Stepwise Analyses of site-specific DNA recombination. Micromachines 2018, 9, 216. [Google Scholar] [CrossRef]
- Zaremba, M.; Owsicka, A.; Tamulaitis, G.; Sasnauskas, G.; Shlyakhtenko, L.S.; Lushnikov, A.Y.; Lyubchenko, Y.L.; Laurens, N.; van den Broek, B.; Wuite, G.J.L.; et al. DNA synapsis through transient tetramerization triggers cleavage by Ecl18kI restriction enzyme. Nucleic Acids Res. 2010, 38, 7142–7154. [Google Scholar] [CrossRef]
- Hughes, C.D.; Simons, M.; Mackenzie, C.E.; Van Houten, B.; Kad, N.M. Single molecule techniques in DNA repair: A primer. DNA Repair 2014, 20, 2–13. [Google Scholar] [CrossRef][Green Version]
- Mohapatra, S.; Lin, C.-T.; Feng, X.A.; Basu, A.; Ha, T. Single-molecule analysis and engineering of DNA motors. Chem Rev. 2020, 120, 36–78. [Google Scholar] [CrossRef]
- Neuman, K.C.; Nagy, A. Single-molecule force spectroscopy: Optical tweezers, magnetic tweezers and atomic force microscopy. Nat. Methods 2008, 5, 491–505. [Google Scholar] [CrossRef]
- Zhao, D.; Liu, S.; Gao, Y. Single-molecule manipulation and detection. Acta Biochim. Biophys. Sin. 2018, 50, 231–237. [Google Scholar] [CrossRef]
- Hilario, J.; Kowalczykowski, S.C. Visualizing protein–DNA interactions at the single-molecule level. Curr. Opin. Chem. Biol. 2010, 14, 15–22. [Google Scholar] [CrossRef]
- Szczelkun, M.D.; Tikhomirova, M.S.; Sinkunas, T.; Gasiunas, G.; Karvelis, T.; Pschera, P.; Siksnys, V.; Seidel, R. Direct observation of R-loop formation by single RNA-guided Cas9 and cascade effector complexes. Proc. Natl. Acad. Sci. USA 2014, 111, 9798–9803. [Google Scholar] [CrossRef]
- Van Aelst, K.; Martínez-Santiago, C.J.; Cross, S.J.; Szczelkun, M.D. The effect of DNA topology on observed rates of R-loop formation and DNA strand cleavage by CRISPR Cas12a. Genes 2019, 10, 169. [Google Scholar] [CrossRef]
- Monico, C.; Capitanio, M.; Belcastro, G.; Vanzi, F.; Pavone, F.S. Optical methods to study protein-DNA interactions in vitro and in living cells at the single-molecule level. Int. J. Mol. Sci. 2013, 14, 3961–3992. [Google Scholar] [CrossRef]
- Miller, H.; Zhou, Z.; Shepherd, J.; Wollman, A.J.M.; Leake, M.C. Single-molecule techniques in biophysics: A review of the progress in methods and applications. Rep. Prog. Phys. 2017, 81, 024601. [Google Scholar] [CrossRef]
- Bangalore, D.M.; Tessmer, I. Unique insight into protein-DNA interactions from single molecule atomic force microscopy. AIMS Biophys. 2018, 5, 194–216. [Google Scholar] [CrossRef]
- Golovinas, E.; Rutkauskas, D.; Manakova, E.; Jankunec, M.; Silanskas, A.; Sasnauskas, G.; Zaremba, M. Prokaryotic argonaute from archaeoglobus fulgidus interacts with DNA as a homodimer. Sci. Rep. 2021, 11, 4518. [Google Scholar] [CrossRef]
- Beckwitt, E.C.; Kong, M.; Van Houten, B. Studying protein-DNA Interactions using atomic force microscopy. Semin. Cell Dev. Biol. 2018, 73, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Oshige, M.; Katsura, S. DNA manipulation and single-molecule imaging. Molecules 2021, 26, 1050. [Google Scholar] [CrossRef] [PubMed]
- Farooq, S.; Fijen, C.; Hohlbein, J. Studying DNA–protein interactions with single-molecule Förster resonance energy transfer. Protoplasma 2014, 251, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Kozlov, A.G.; Lohman, T.M.; Ha, T. SSB protein diffusion on single-stranded DNA stimulates reca filament formation. Nature 2009, 461, 1092–1097. [Google Scholar] [CrossRef]
- Zhou, R.; Kozlov, A.G.; Roy, R.; Zhang, J.; Korolev, S.; Lohman, T.M.; Ha, T. SSB functions as a sliding platform that migrates on DNA via reptation. Cell 2011, 146, 222–232. [Google Scholar] [CrossRef]
- Singh, D.; Wang, Y.; Mallon, J.; Yang, O.; Fei, J.; Poddar, A.; Ceylan, D.; Bailey, S.; Ha, T. Mechanisms of improved specificity of engineered Cas9s revealed by single-molecule FRET analysis. Nat. Struct Mol. Biol. 2018, 25, 347–354. [Google Scholar] [CrossRef]
- Okafor, I.C.; Singh, D.; Wang, Y.; Jung, M.; Wang, H.; Mallon, J.; Bailey, S.; Lee, J.K.; Ha, T. Single Molecule analysis of effects of non-canonical guide RNAs and specificity-enhancing mutations on Cas9-Induced DNA unwinding. Nucleic Acids Res. 2019, 47, 11880–11888. [Google Scholar] [CrossRef]
- Kalinin, S.; Peulen, T.; Sindbert, S.; Rothwell, P.J.; Berger, S.; Restle, T.; Goody, R.S.; Gohlke, H.; Seidel, C.A.M. A Toolkit and benchmark study for FRET-restrained high-precision structural modeling. Nat. Methods 2012, 9, 1218–1225. [Google Scholar] [CrossRef]
- Senavirathne, G.; Jaszczur, M.; Auerbach, P.A.; Upton, T.G.; Chelico, L.; Goodman, M.F.; Rueda, D. Single-stranded DNA scanning and deamination by APOBEC3G cytidine deaminase at single molecule resolution. J. Biol. Chem. 2012, 287, 15826–15835. [Google Scholar] [CrossRef]
- Lee, S.; Jo, K. Visualization of Surface-tethered large DNA molecules with a fluorescent protein DNA binding peptide. J. Vis. Exp. 2016, 112, e54141. [Google Scholar] [CrossRef]
- Doyle, P.; Ladoux, B.; Viovy, J.-L. Dynamics of a tethered polymer in shear flow. Phys. Rev. Lett. 2000, 84, 4769–4772. [Google Scholar] [CrossRef]
- Van Mameren, J.; Peterman, E.J.G.; Wuite, G.J.L. See me, feel me: Methods to concurrently visualize and manipulate single DNA molecules and associated proteins. Nucleic Acids Res. 2008, 36, 4381–4389. [Google Scholar] [CrossRef]
- Delgadillo, R.F.; Mueser, T.C.; Zaleta-Rivera, K.; Carnes, K.A.; González-Valdez, J.; Parkhurst, L.J. Detailed Characterization of the solution kinetics and thermodynamics of biotin, biocytin and HABA binding to avidin and streptavidin. PLoS ONE 2019, 14, e0204194. [Google Scholar] [CrossRef]
- Visnapuu, M.-L.; Duzdevich, D.; Greene, E.C. The importance of surfaces in single-molecule bioscience. Mol. BioSyst. 2008, 4, 394–403. [Google Scholar] [CrossRef]
- Xiong, K.; Blainey, P.C. Molecular sled sequences are common in mammalian proteins. Nucleic Acids Res. 2016, 44, 2266–2273. [Google Scholar] [CrossRef][Green Version]
- Xue, H.; Zhan, Z.; Zhang, K.; Fu, Y.V. Visualizing the Interaction between the Qdot-labeled protein and site-specifically modified λ DNA at the single molecule level. J. Vis. Exp. 2018, e57967. [Google Scholar] [CrossRef]
- Bouchiat, C.; Wang, M.D.; Allemand, J.-F.; Strick, T.; Block, S.M.; Croquette, V. Estimating the persistence length of a worm-like chain molecule from force-extension measurements. Biophys. J. 1999, 76, 409–413. [Google Scholar] [CrossRef]
- Roy, T.; Szuttor, K.; Smiatek, J.; Holm, C.; Hardt, S. Conformation and dynamics of long-chain end-tethered polymers in microchannels. Polymers 2019, 11, 488. [Google Scholar] [CrossRef]
- Bustamante, C.; Marko, J.F.; Siggia, E.D.; Smith, S. Entropic elasticity of λ-phage DNA. Science 1994, 265, 1599–1600. [Google Scholar] [CrossRef]
- Granéli, A.; Yeykal, C.C.; Prasad, T.K.; Greene, E.C. Organized arrays of individual DNA molecules tethered to supported lipid bilayers. Langmuir 2006, 22, 292–299. [Google Scholar] [CrossRef]
- Smith, D.E.; Babcock, H.P.; Chu, S. Single-polymer dynamics in steady shear flow. Science 1999, 283, 1724–1727. [Google Scholar] [CrossRef]
- Mueller, S.H.; Spenkelink, L.M.; van Oijen, A.M.; Lewis, J.S. Design of customizable long linear DNA substrates with controlled end modifications for single-molecule studies. Anal. Biochem. 2020, 592, 113541. [Google Scholar] [CrossRef]
- Pisapati, A.V.; Wang, Y.; Blauch, M.E.; Wittenberg, N.J.; Cheng, X.; Zhang, X.F. Characterizing single-molecule conformational changes under shear flow with fluorescence microscopy. J. Vis. Exp. 2020, 155, e60784. [Google Scholar] [CrossRef]
- Martin-Fernandez, M.; Tynan, C.; Webb, S. A ‘Pocket Guide’ to total internal reflection fluorescence. J. Microsc. 2013, 252, 16–22. [Google Scholar] [CrossRef]
- Reck-Peterson, S.L.; Derr, N.D.; Stuurman, N. Imaging single molecules using total internal reflection fluorescence microscopy (TIRFM). Cold Spring Harb. Protoc. 2010. [Google Scholar] [CrossRef]
- Kim, H.; Loparo, J.J. Observing bacterial chromatin protein-DNA interactions by combining DNA flow-stretching with single-molecule imaging. In Bacterial Chromatin: Methods and Protocols; Dame, R.T., Ed.; Humana Press: New York, NY, USA, 2018; Volume 1837, pp. 277–299. [Google Scholar] [CrossRef]
- Xiong, K.; Blainey, P.C. A Simple, robust, and high throughput single molecule flow stretching assay implementation for studying transport of molecules along DNA. J. Vis. Exp. 2017, 128, e55923. [Google Scholar] [CrossRef]
- Plesa, C.; Verschueren, D.; Pud, S.; van der Torre, J.; Ruitenberg, J.W.; Witteveen, M.J.; Jonsson, M.P.; Grosberg, A.Y.; Rabin, Y.; Dekker, C. Direct observation of DNA knots using a solid-state nanopore. Nat. Nanotech. 2016, 11, 1093–1097. [Google Scholar] [CrossRef]
- Klotz, A.R.; Soh, B.W.; Doyle, P.S. Motion of knots in DNA stretched by elongational fields. Phys. Rev. Lett. 2018, 120, 188003. [Google Scholar] [CrossRef]
- Bao, X.R.; Lee, H.J.; Quake, S.R. Behavior of comp.plex knots in single DNA molecules. Phys. Rev. Lett. 2003, 91, 265506. [Google Scholar] [CrossRef]
- Wang, W.; Lin, J.; Schwartz, D.C. Scanning force microscopy of DNA molecules elongated by convective fluid flow in an evaporating droplet. Biophys. J. 1998, 75, 513–520. [Google Scholar] [CrossRef]
- Kim, Y.; de la Torre, A.; Leal, A.A.; Finkelstein, I.J. Efficient modification of λ-DNA substrates for single-molecule studies. Sci. Rep. 2017, 7, 2071. [Google Scholar] [CrossRef] [PubMed]
- Gorman, J.; Chowdhury, A.; Surtees, J.A.; Shimada, J.; Reichman, D.R.; Alani, E.; Greene, E.C. Dynamic basis for one-dimensional DNA Scanning by the mismatch repair complex Msh2-Msh6. Mol. Cell 2007, 28, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Komazin-Meredith, G.; Mirchev, R.; Golan, D.E.; van Oijen, A.M.; Coen, D.M. Hopping of a processivity factor on DNA revealed by single-molecule assays of diffusion. Proc. Natl. Acad. Sci. USA 2008, 105, 10721–10726. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Lee, O.; Durang, X.; Jeon, J.-H. A mini-review of the diffusion dynamics of DNA-binding proteins: Experiments and models. J. Korean Phys. Soc. 2021, 78, 408–426. [Google Scholar] [CrossRef]
- Redding, S.; Greene, E.C. How do proteins locate specific targets in DNA? Chem. Phys. Lett. 2013, 570, 1–11. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Halford, S.E.; Marko, J.F. How Do site-specific DNA-binding proteins find their targets? Nucleic Acids Res. 2004, 32, 3040–3052. [Google Scholar] [CrossRef]
- Greene, E.C.; Mizuuchi, K. Direct observation of single MuB polymers: Evidence for a DNA-dependent conformational change for generating an active target complex. Mol. Cell 2002, 9, 1079–1089. [Google Scholar] [CrossRef]
- Greene, E.C.; Mizuuchi, K. Visualizing the assembly and disassembly mechanisms of the MuB transposition targeting complex. J. Biol. Chem. 2004, 279, 16736–16743. [Google Scholar] [CrossRef]
- Tan, X.; Mizuuchi, M.; Mizuuchi, K. DNA Transposition target immunity and the determinants of the MuB distribution patterns on DNA. Proc. Natl. Acad. Sci. USA 2007, 104, 13925–13929. [Google Scholar] [CrossRef]
- Yardimci, H.; Wang, X.; Loveland, A.B.; Tappin, I.; Rudner, D.Z.; Hurwitz, J.; van Oijen, A.M.; Walter, J.C. Bypass of a protein barrier by a replicative DNA helicase. Nature 2012, 492, 205–209. [Google Scholar] [CrossRef]
- Bonnet, I.; Biebricher, A.; Porté, P.-L.; Loverdo, C.; Bénichou, O.; Voituriez, R.; Escudé, C.; Wende, W.; Pingoud, A.; Desbiolles, P. Sliding and jumping of single EcoRV restriction enzymes on non-cognate DNA. Nucleic Acids Res. 2008, 36, 4118–4127. [Google Scholar] [CrossRef]
- Dikić, J.; Menges, C.; Clarke, S.; Kokkinidis, M.; Pingoud, A.; Wende, W.; Desbiolles, P. The rotation-coupled sliding of EcoRV. Nucleic Acids Res. 2012, 40, 4064–4070. [Google Scholar] [CrossRef]
- Blainey, P.C.; van Oijen, A.M.; Banerjee, A.; Verdine, G.L.; Xie, X.S. A Base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proc. Natl. Acad. Sci. USA 2006, 103, 5752–5757. [Google Scholar] [CrossRef]
- Blainey, P.C.; Luo, G.; Kou, S.C.; Mangel, W.F.; Verdine, G.L.; Bagchi, B.; Xie, X.S. Nonspecifically bound proteins spin while diffusing along DNA. Nat. Struct. Mol. Biol. 2009, 16, 1224–1229. [Google Scholar] [CrossRef]
- Cho, W.-K.; Jeong, C.; Kim, D.; Chang, M.; Song, K.-M.; Hanne, J.; Ban, C.; Fishel, R.; Lee, J.-B. ATP Alters the diffusion mechanics of MutS on mismatched DNA. Structure 2012, 20, 1264–1274. [Google Scholar] [CrossRef]
- Williams, K.; Grafe, B.; Burke, K.M.; Tanner, N.; van Oijen, A.M.; Loparo, J.; Price, A.C. A Single molecule DNA Flow stretching microscope for undergraduates. Am. J. Phys. 2011, 79, 1112–1120. [Google Scholar] [CrossRef]
- Wang, J.; Lu, C. Single molecule λ-DNA stretching studied by microfluidics and single particle tracking. J. Appl. Phys. 2007, 102, 074703. [Google Scholar] [CrossRef]
- Smith, S.B.; Finzi, L.; Bustamante, C. Direct mechanical measurements of the elasticity of single DNA molecules by using magnetic beads. Science 1992, 258, 1122–1126. [Google Scholar] [CrossRef]
- Lee, R.; Jeon, Y.; Lee, J.-B. Single-molecule DNA Flow-stretching force spectroscopy with higher resolution using dark-field microscopy. J. Korean Phys. Soc. 2019, 75, 350–354. [Google Scholar] [CrossRef]
- Hill, F.R.; Monachino, E.; van Oijen, A.M. The more the merrier: High-Throughput single-molecule techniques. Biochem Soc. Trans. 2017, 45, 759–769. [Google Scholar] [CrossRef][Green Version]
- Lee, R.; Yang, K.; Lee, J.-B. Multiplexed single-molecule flow-stretching bead assay for DNA enzymology. BMB Rep. 2019, 52, 589–594. [Google Scholar] [CrossRef]
- Kim, S.; Blainey, P.C.; Schroeder, C.M.; Xie, X.S. Multiplexed single-molecule assay for enzymatic activity on flow-stretched DNA. Nat. Methods 2007, 4, 397–399. [Google Scholar] [CrossRef]
- Van Oijen, A.M.; Blainey, P.C.; Crampton, D.J.; Richardson, C.C.; Ellenberger, T.; Xie, X.S. Single-molecule kinetics of lambda exonuclease reveal base dependence and dynamic disorder. Science 2003, 301, 1235–1238. [Google Scholar] [CrossRef]
- Schermerhorn, K.M.; Tanner, N.; Kelman, Z.; Gardner, A.F. High-temperature single-molecule kinetic analysis of thermophilic archaeal MCM helicases. Nucleic Acids Res. 2016, 44, 8764–8771. [Google Scholar] [CrossRef]
- Hamdan, S.M.; Loparo, J.J.; Takahashi, M.; Richardson, C.C.; van Oijen, A.M. Dynamics of DNA replication loops reveal temporal control of lagging-strand synthesis. Nature 2009, 457, 336–339. [Google Scholar] [CrossRef]
- Tanner, A.N.; Hamdan, S.M.; Jergic, S.; Loscha, K.V.; Schaeffer, P.M.; Dixon, N.E.; van Oijen, A.M. Single-molecule studies of fork dynamics of escherichia coli DNA replication. Nat. Struct Mol. Biol. 2008, 15, 170–176. [Google Scholar] [CrossRef]
- Lee, J.-B.; Hite, R.K.; Hamdan, S.M.; Xie, X.S.; Richardson, C.C.; van Oijen, A.M. DNA primase acts as a molecular brake in DNA replication. Nature 2006, 439, 621–624. [Google Scholar] [CrossRef]
- Kad, N.M.; Wang, H.; Kennedy, G.G.; Warshaw, D.M.; van Houten, B. Collaborative dynamic DNA scanning by nucleotide excision repair proteins investigated by single-molecule imaging of quantum-dot-labeled proteins. Mol. Cell 2010, 37, 702–713. [Google Scholar] [CrossRef]
- Springall, L.; Inchingolo, A.V.; Kad, N.M. DNA–protein interactions studied directly using single molecule fluorescence imaging of quantum dot tagged proteins moving on DNA tightropes. In Methods in Molecular Biology: Methods and Protocols; Leake, M.C., Ed.; Humana Press: New York, NY, USA, 2016; Volume 1431, pp. 141–150. [Google Scholar] [CrossRef]
- Wang, J.; Barnett, J.T.; Pollard, M.R.; Kad, N.M. Chapter Seven–Integrating optical tweezers, DNA tightropes, and single-molecule fluorescence imaging: Pitfalls and traps. In Methods in Enzymology; Spies, M., Chemla, Y.R., Eds.; Academic Press: Cambridge, MA, USA, 2017; Volume 582, pp. 171–192. [Google Scholar] [CrossRef]
- Nelson, S.R.; Kathe, S.D.; Hilzinger, T.S.; Averill, A.M.; Warshaw, D.M.; Wallace, S.S.; Lee, A.J. Single molecule glycosylase studies with engineered 8-oxoguanine DNA damage sites show functional defects of a MUTYH polyposis variant. Nucleic Acids Res. 2019, 47, 3058–3071. [Google Scholar] [CrossRef]
- Nelson, S.R.; Dunn, A.R.; Kathe, S.D.; Warshaw, D.M.; Wallace, S.S. Two glycosylase families diffusively scan DNA using a wedge residue to probe for and identify oxidatively damaged bases. Proc. Natl Acad Sci. USA 2014, 111, 2091–2099. [Google Scholar] [CrossRef]
- Barnett, J.T.; Kad, N.M. Understanding the coupling between DNA Damage detection and UvrA’s ATPase using bulk and single molecule kinetics. FASEB J. 2019, 33, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Pollard, M.R.; Hughes, C.D.; Ward, A.D.; Van Houten, B.; Towrie, M.; Botchway, S.W.; Parker, A.W.; Kad, N.M. Directly interrogating single quantum dot labelled UvrA2 molecules on DNA tightropes using an optically trapped nanoprobe. Sci. Rep. 2015, 5, 18486. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.D.; Wang, H.; Ghodke, H.; Simons, M.; Towheed, A.; Peng, Y.; Van Houten, B.; Kad, N.M. Real-Time single-molecule imaging reveals a direct interaction between UvrC and UvrB on DNA tightropes. Nucleic Acids Res. 2013, 41, 4901–4912. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Countryman, P.; Buncher, N.; Kaur, P.; Longjiang, E.; Zhang, Y.; Gibson, G.; You, C.; Watkins, S.C.; Piehler, J.; et al. TRF1 and TRF2 use different mechanisms to find telomeric DNA but share a novel mechanism to search for protein partners at telomeres. Nucleic Acids Res. 2014, 42, 2493–2504. [Google Scholar] [CrossRef]
- Pan, H.; Kaur, P.; Barnes, R.; Detwiler, A.C.; Sanford, S.L.; Liu, M.; Xu, P.; Mahn, C.; Tang, Q.; Hao, P.; et al. Structure, dynamics, and regulation of TRF1-TIN2-mediated trans- and cis-interactions on telomeric DNA. J. Biol. Chem. 2021, 297, 101080. [Google Scholar] [CrossRef]
- Robison, A.D.; Finkelstein, I.J. High-throughput single-molecule studies of protein-DNA interactions. FEBS Lett. 2014, 588, 3539–3546. [Google Scholar] [CrossRef]
- Fazio, T.; Visnapuu, M.-L.; Wind, S.; Greene, E.C. DNA Curtains and nanoscale curtain rods: High-Throughput tools for single molecule imaging. Langmuir 2008, 24, 10524–10531. [Google Scholar] [CrossRef]
- Silverstein, T.D.; Gibb, B.; Greene, E.C. Visualizing protein movement on DNA at the single-molecule level using DNA curtains. DNA Repair 2014, 20, 94–109. [Google Scholar] [CrossRef]
- Collins, B.E.; Ye, L.F.; Duzdevich, D.; Greene, E.C. Chapter 12–DNA curtains: Novel tools for imaging protein–nucleic acid interactions at the single-molecule level. In Quantitative Imaging in Cell Biology; Waters, J.C., Wittman, T., Eds.; Academic Press: Cambridge, MA, USA, 2014; Volume 123, pp. 217–234. [Google Scholar] [CrossRef]
- Greene, E.C.; Wind, S.; Fazio, T.; Gorman, J.; Visnapuu, M.-L. DNA curtains for high-throughput single-molecule optical imaging. Methods Enzym. 2010, 472, 293–315. [Google Scholar] [CrossRef]
- Fazio, T.A.; Lee, J.Y.; Wind, S.J.; Greene, E.C. Assembly of DNA curtains using hydrogen silsesquioxane as a barrier to lipid diffusion. Anal. Chem. 2012, 84, 7613–7617. [Google Scholar] [CrossRef]
- Kang, Y.; Cheon, N.Y.; Cha, J.; Kim, A.; Kim, H.; Lee, L.; Kim, K.O.; Jo, K.; Lee, J.Y. High-throughput single-molecule imaging system using nanofabricated trenches and fluorescent DNA-binding proteins. Biotechnol. Bioeng. 2020, 117, 1640–1648. [Google Scholar] [CrossRef]
- Fazio, T.A.; Visnapuu, M.; Greene, E.C.; Wind, S.J. Fabrication of nanoscale “Curtain Rods” for DNA Curtains using nanoimprint lithography. J. Vac. Sci. Technol. B Nanotechnol. Microelectron. 2009, 27, 3095–3098. [Google Scholar] [CrossRef]
- Gallardo, I.F.; Pasupathy, P.; Brown, M.; Manhart, C.M.; Neikirk, D.P.; Alani, E.; Finkelstein, I.J. High-throughput universal DNA curtain arrays for single-molecule fluorescence imaging. Langmuir 2015, 31, 10310–10317. [Google Scholar] [CrossRef]
- Kong, M.; Cutts, E.E.; Pan, D.; Beuron, F.; Kaliyappan, T.; Xue, C.; Morris, E.P.; Musacchio, A.; Vannini, A.; Greene, E.C. Human condensin I and II drive extensive ATP-dependent compaction of nucleosome-bound DNA. Mol. Cell 2020, 79, 99–114. [Google Scholar] [CrossRef]
- Finkelstein, I.J.; Greene, E.C. Supported lipid bilayers and DNA curtains for high-throughput single-molecule studies. Methods Mol. Biol. 2011, 745, 447–461. [Google Scholar] [CrossRef]
- Visnapuu, M.-L.; Fazio, T.; Wind, S.; Greene, E.C. Parallel arrays of geometric nanowells for assembling curtains of DNA with controlled lateral dispersion. Langmuir 2008, 24, 11293–11299. [Google Scholar] [CrossRef][Green Version]
- Cho, C.; Jang, J.; Kang, Y.; Watanabe, H.; Uchihashi, T.; Kim, S.J.; Kato, K.; Lee, J.Y.; Song, J.-J. Structural basis of nucleosome assembly by the abo1 AAA+ ATPase histone chaperone. Nat. Commun. 2019, 10, 5764. [Google Scholar] [CrossRef]
- Kang, Y.; Cho, C.; Lee, K.S.; Song, J.-J.; Lee, J.Y. Single-molecule imaging reveals the mechanism underlying histone loading of Schizosaccharomyces pombe AAA+ ATPase Abo1. Mol. Cells 2021, 44, 79–87. [Google Scholar] [CrossRef]
- Dillard, K.E.; Schaub, J.M.; Brown, M.W.; Saifuddin, F.A.; Xiao, Y.; Hernandez, E.; Dahlhauser, S.D.; Anslyn, E.V.; Ke, A.; Finkelstein, I.J. Sortase-mediated fluorescent labeling of CRISPR complexes. Methods Enzym. 2019, 616, 43–59. [Google Scholar] [CrossRef]
- Calcines-Cruz, C.; Finkelstein, I.J.; Hernandez-Garcia, A. CRISPR-guided programmable self-assembly of artificial virus-like nucleocapsids. Nano Lett. 2021, 21, 2752–2757. [Google Scholar] [CrossRef]
- Prasad, T.K.; Robertson, R.B.; Visnapuu, M.-L.; Chi, P.; Sung, P.; Greene, E.C. A DNA Translocating Snf2 molecular motor: S. cerevisiae Rdh54 displays processive translocation and can extrude DNA loops. J. Mol. Biol. 2007, 369, 940–953. [Google Scholar] [CrossRef]
- Finkelstein, I.J.; Visnapuu, M.-L.; Greene, E.C. Single-molecule imaging reveals mechanisms of protein disruption by a DNA translocase. Nature 2010, 468, 983–987. [Google Scholar] [CrossRef]
- Terakawa, T.; Redding, S.; Silverstein, T.D.; Greene, E.C. Sequential Eviction of crowded nucleoprotein complexes by the exonuclease RecBCD molecular motor. Proc. Natl. Acad. Sci. USA 2017, 114, 6322–6331. [Google Scholar] [CrossRef]
- Soniat, M.M.; Myler, L.R.; Finkelstein, I.J. Assembling the human resectosome on DNA curtains. Methods Mol. Biol. 2019, 1999, 225–244. [Google Scholar] [CrossRef]
- Jia, N.; Unciuleac, M.C.; Xue, C.; Greene, E.C.; Patel, D.J.; Shuman, S. Structures and Single-molecule analysis of bacterial motor nuclease AdnAB illuminate the mechanism of DNA double-strand break resection. Proc. Natl. Acad. Sci. USA 2019, 116, 24507–24516. [Google Scholar] [CrossRef]
- Unciuleac, M.-C.; Meir, A.; Xue, C.; Warren, G.M.; Greene, E.C.; Shuman, S. Clutch Mechanism of chemomechanical coupling in a DNA resecting motor nuclease. Proc Natl. Acad. Sci. USA 2021, 118, e2023955118. [Google Scholar] [CrossRef]
- Deshpande, R.A.; Myler, L.R.; Soniat, M.M.; Makharashvili, N.; Lee, L.; Lees-Miller, S.P.; Finkelstein, I.J.; Paull, T.T. DNA-dependent protein kinase promotes DNA end processing by MRN and CtIP. Sci. Adv. 2020, 6, eaay0922. [Google Scholar] [CrossRef]
- Caron, M.-C.; Sharma, A.K.; O’Sullivan, J.; Myler, L.R.; Ferreira, M.T.; Rodrigue, A.; Coulombe, Y.; Ethier, C.; Gagné, J.-P.; Langelier, M.-F.; et al. Poly(ADP-Ribose) Polymeras.se-1 antagonizes DNA resection at double-strand breaks. Nat. Commun. 2019, 10, 2954. [Google Scholar] [CrossRef]
- Myler, L.R.; Gallardo, I.F.; Zhou, Y.; Gong, F.; Yang, S.-H.; Wold, M.S.; Miller, K.M.; Paull, T.T.; Finkelstein, I.J. Single-molecule imaging reveals the mechanism of exo1 regulation by single-stranded DNA binding proteins. Proc. Natl. Acad. Sci. USA 2016, 113, 1170–1179. [Google Scholar] [CrossRef]
- Chi, P.; Kwon, Y.; Visnapuu, M.-L.; Lam, I.; Santa Maria, S.R.; Zheng, X.; Epshtein, A.; Greene, E.C.; Sung, P.; Klein, H.L. Analyses of the yeast Rad51 recombinase A265V mutant reveal different in vivo roles of Swi2-like factors. Nucleic Acids Res. 2011, 39, 6511–6522. [Google Scholar] [CrossRef][Green Version]
- Granéli, A.; Yeykal, C.C.; Robertson, R.B.; Greene, E.C. Long-distance lateral diffusion of human Rad51 on double-stranded DNA. Proc. Natl. Acad. Sci. USA 2006, 103, 1221–1226. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.B.; Moses, D.N.; Kwon, Y.; Chan, P.; Chi, P.; Klein, H.; Sung, P.; Greene, E.C. Structural transitions within human Rad51 nucleoprotein filaments. Proc. Natl. Acad. Sci. USA 2009, 106, 12688–12693. [Google Scholar] [CrossRef] [PubMed]
- Prasad, T.K.; Yeykal, C.C.; Greene, E.C. Visualizing the assembly of human Rad51 filaments on double-stranded DNA. J. Mol. Biol. 2006, 363, 713–728. [Google Scholar] [CrossRef] [PubMed]
- Yeykal, C.C.; Greene, E.C. Visualizing the behavior of human Rad51 at the single-molecule level. Cell Cycle 2006, 5, 1033–1038. [Google Scholar] [CrossRef]
- Robertson, R.B.; Moses, D.N.; Kwon, Y.; Chan, P.; Chi, P.; Klein, H.; Sung, P.; Greene, E.C. Visualizing the disassembly of S. cerevisiae Rad51 nucleoprotein filaments. J. Mol. Biol. 2009, 388, 703–720. [Google Scholar] [CrossRef]
- Gorman, J.; Fazio, T.; Wang, F.; Wind, S.; Greene, E.C. Nanofabricated racks of aligned and anchored DNA substrates for single-molecule imaging. Langmuir 2010, 26, 1372–1379. [Google Scholar] [CrossRef]
- Sternberg, S.H.; Redding, S.; Jinek, M.; Greene, E.C.; Doudna, J.A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 2014, 507, 62–67. [Google Scholar] [CrossRef]
- Redding, S.; Sternberg, S.H.; Marshall, M.; Gibb, B.; Bhat, P.; Guegler, C.K.; Wiedenheft, B.; Doudna, J.A.; Greene, E.C. Surveillance and processing of foreign DNA by the escherichia coli CRISPR-Cas system. Cell 2015, 163, 854–865. [Google Scholar] [CrossRef]
- Dillard, K.E.; Brown, M.W.; Johnson, N.V.; Xiao, Y.; Dolan, A.; Hernandez, E.; Dahlhauser, S.D.; Kim, Y.; Myler, L.R.; Anslyn, E.V.; et al. Assembly and translocation of a CRISPR-Cas primed acquisition complex. Cell 2018, 175, 934–946. [Google Scholar] [CrossRef]
- Lee, J.Y.; Finkelstein, I.J.; Crozat, E.; Sherratt, D.J.; Greene, E.C. Single-molecule imaging of DNA curtains reveals mechanisms of KOPS sequence targeting by the DNA translocase FtsK. Proc. Natl. Acad. Sci. USA 2012, 109, 6531–6536. [Google Scholar] [CrossRef]
- Lee, J.Y.; Finkelstein, I.J.; Arciszewska, L.K.; Sherratt, D.J.; Greene, E.C. Single-molecule imaging of FtsK Translocation reveals mechanistic features of protein-protein collisions on DNA. Mol. Cell 2014, 54, 832–843. [Google Scholar] [CrossRef]
- Terakawa, T.; Bisht, S.; Eeftens, J.M.; Dekker, C.; Haering, C.H.; Greene, E.C. The Condensin complex is a mechanochemical motor that translocates along DNA. Science 2017, 358, 672–676. [Google Scholar] [CrossRef]
- Gorman, J.; Plys, A.J.; Visnapuu, M.-L.; Alani, E.; Greene, E.C. Visualizing one-dimensional diffusion of eukaryotic DNA Repair factors along a chromatin lattice. Nat. Struct. Mol. Biol. 2010, 17, 932–938. [Google Scholar] [CrossRef]
- Brown, M.W.; Kim, Y.; Williams, G.M.; Huck, J.D.; Surtees, J.A.; Finkelstein, I.J. Dynamic DNA binding licenses a repair factor to bypass roadblocks in search of DNA lesions. Nat. Commun. 2016, 7, 10607. [Google Scholar] [CrossRef]
- Crickard, J.B.; Moevus, C.J.; Kwon, Y.; Sung, P.; Greene, E.C. Rad54 Drives ATP hydrolysis-dependent DNA sequence alignment during homologous recombination. Cell 2020, 181, 1380–1394. [Google Scholar] [CrossRef]
- Meir, A.; Kong, M.; Xue, C.; Greene, E.C. DNA Curtains shed light on complex molecular systems during homologous recombination. JoVE 2020, 160, e61320. [Google Scholar] [CrossRef]
- Myler, L.R.; Gallardo, I.F.; Soniat, M.M.; Deshpande, R.A.; Gonzalez, X.B.; Kim, Y.; Paull, T.T.; Finkelstein, I.J. Single-Molecule Imaging reveals how Mre11-Rad50-Nbs1 initiates DNA break repair. Mol. Cell 2017, 67, 891–898. [Google Scholar] [CrossRef]
- Myler, L.R.; Soniat, M.M.; Zhang, X.; Deshpande, R.A.; Paull, T.T.; Finkelstein, I.J. Purification and Biophysical characterization of the Mre11-Rad50-Nbs1 complex. Methods Mol. Biol. 2019, 2004, 269–287. [Google Scholar] [CrossRef]
- Wang, F.; Redding, S.; Finkelstein, I.J.; Gorman, J.; Reichman, D.R.; Greene, E.C. The promoter-search mechanism of escherichia coli RNA polymerase is dominated by three-dimensional diffusion. Nat. Struct. Mol. Biol. 2013, 20, 174–181. [Google Scholar] [CrossRef]
- Gorman, J.; Wang, F.; Redding, S.; Plys, A.J.; Fazio, T.; Wind, S.; Alani, E.E.; Greene, E.C. Single-molecule imaging reveals target-search mechanisms during DNA mismatch repair. Proc. Natl. Acad. Sci. USA 2012, 109, 3074–3083. [Google Scholar] [CrossRef]
- Gibb, B.; Silverstein, T.D.; Finkelstein, I.J.; Greene, E.C. Single-stranded DNA curtains for real-time single-molecule visualization of protein–nucleic acid interactions. Anal. Chem. 2012, 84, 7607–7612. [Google Scholar] [CrossRef]
- Ma, C.J.; Steinfeld, J.B.; Greene, E.C. Single-stranded DNA curtains for studying homologous recombination. Methods Enzym. 2017, 582, 193–219. [Google Scholar] [CrossRef]
- Qi, Z.; Greene, E.C. Visualizing recombination intermediates with single-stranded DNA curtains. Methods 2016, 105, 62–74. [Google Scholar] [CrossRef]
- Soniat, M.M.; Myler, L.R.; Schaub, J.M.; Kim, Y.; Gallardo, I.F.; Finkelstein, I.J. Next-generation DNA curtains for single-molecule studies of homologous recombination. Methods Enzym. 2017, 592, 259–281. [Google Scholar] [CrossRef]
- De Tullio, L.; Kaniecki, K.; Greene, E.C. Single-stranded DNA curtains for studying the Srs2 helicase using total internal reflection fluorescence microscopy. Methods Enzym. 2018, 600, 407–437. [Google Scholar] [CrossRef]
- Schaub, J.M.; Zhang, H.; Soniat, M.M.; Finkelstein, I.J. Assessing protein dynamics on low-complexity single-stranded DNA curtains. Langmuir 2018, 34, 14882–14890. [Google Scholar] [CrossRef]
- Zhang, H.; Schaub, J.M.; Finkelstein, I.J. RADX condenses single-stranded DNA to antagonize RAD51 loading. Nucleic Acids Res. 2020, 48, 7834–7843. [Google Scholar] [CrossRef]
- Adolph, M.B.; Mohamed, T.M.; Balakrishnan, S.; Xue, C.; Morati, F.; Modesti, M.; Greene, E.C.; Chazin, W.J.; Cortez, D. RADX controls RAD51 filament dynamics to regulate replication fork stability. Mol. Cell 2021, 81, 1074–1083. [Google Scholar] [CrossRef]
- Taylor, M.R.G.; Špírek, M.; Jian Ma, C.; Carzaniga, R.; Takaki, T.; Collinson, L.M.; Greene, E.C.; Krejci, L.; Boulton, S.J. A polar and nucleotide-dependent mechanism of action for RAD51 paralogs in RAD51 filament remodeling. Mol. Cell 2016, 64, 926–939. [Google Scholar] [CrossRef]
- Qi, Z.; Redding, S.; Lee, J.Y.; Gibb, B.; Kwon, Y.; Niu, H.; Gaines, W.A.; Sung, P.; Greene, E.C. DNA sequence alignment by microhomology sampling during homologous recombination. Cell 2015, 160, 856–869. [Google Scholar] [CrossRef]
- Gibb, B.; Ye, L.F.; Kwon, Y.; Niu, H.; Sung, P.; Greene, E.C. Protein dynamics during presynaptic complex assembly on individual SsDNA molecules. Nat. Struct. Mol. Biol. 2014, 21, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.J.; Gibb, B.; Kwon, Y.; Sung, P.; Greene, E.C. Protein dynamics of human RPA and RAD51 on SsDNA during assembly and disassembly of the RAD51 filament. Nucleic Acids Res. 2017, 45, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Crickard, J.B.; Kaniecki, K.; Kwon, Y.; Sung, P.; Greene, E.C. Spontaneous self-segregation of Rad51 and Dmc1 DNA recombinases within mixed recombinase filaments. J. Biol. Chem. 2018, 293, 4191–4200. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Molnarova, L.; Steinfeld, J.B.; Zhao, W.; Ma, C.; Spirek, M.; Kaniecki, K.; Kwon, Y.; Beláň, O.; Krejci, K.; et al. Single-molecule visualization of human RECQ5 interactions with single-stranded DNA recombination intermediates. Nucleic Acids Res. 2020, 49, 285–305. [Google Scholar] [CrossRef]
- Kaniecki, K.; De Tullio, L.; Gibb, B.; Kwon, Y.; Sung, P.; Greene, E.C. Dissociation of Rad51 presynaptic complexes and heteroduplex DNA joints by tandem assemblies of Srs2. Cell Rep. 2017, 21, 3166–3177. [Google Scholar] [CrossRef]
- Ma, C.J.; Kwon, Y.; Sung, P.; Greene, E.C. Human RAD52 interactions with replication protein A and the RAD51 presynaptic complex. J. Biol. Chem. 2017, 292, 11702. [Google Scholar] [CrossRef]
- Yan, Z.; Xue, C.; Kumar, S.; Crickard, J.B.; Yu, Y.; Wang, W.; Pham, N.; Li, Y.; Niu, H.; Sung, P.; et al. Rad52 restrains resection at DNA double-strand break ends in yeast. Mol. Cell 2019, 76, 699. [Google Scholar] [CrossRef]
- Crickard, J.B.; Kwon, Y.; Sung, P.; Greene, E.C. Rad54 and Rdh54 occupy spatially and functionally distinct sites within the Rad51-SsDNA presynaptic complex. EMBO J. 2020, 39, e105705. [Google Scholar] [CrossRef]
- Crickard, J.B.; Kaniecki, K.; Kwon, Y.; Sung, P.; Lisby, M.; Greene, E.C. Regulation of Hed1 and Rad54 binding during maturation of the meiosis-specific presynaptic complex. EMBO J. 2018, 37, e98728. [Google Scholar] [CrossRef]
- Gibb, B.; Ye, L.F.; Gergoudis, S.C.; Kwon, Y.; Niu, H.; Sung, P.; Greene, E.C. Concentration-dependent exchange of replication protein a on single-stranded DNA revealed by single-molecule imaging. PLoS ONE 2014, 9, e87922. [Google Scholar] [CrossRef]
- Pokhrel, N.; Origanti, S.; Davenport, E.P.; Gandhi, D.; Kaniecki, K.; Mehl, R.A.; Greene, E.C.; Dockendorff, C.; Antony, E. monitoring replication protein A (RPA) dynamics in homologous recombination through site-specific incorporation of non-canonical amino acids. Nucleic Acids Res. 2017, 45, 9413–9426. [Google Scholar] [CrossRef]
- Deng, S.K.; Gibb, B.; de Almeida, M.J.; Greene, E.C.; Symington, L.S. RPA Antagonizes microhomology-mediated repair of DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014, 21, 405–412. [Google Scholar] [CrossRef]
- Robison, A.D.; Finkelstein, I.J. Rapid prototyping of multichannel microfluidic devices for single-molecule DNA curtain imaging. Anal. Chem. 2014, 86, 4157–4163. [Google Scholar] [CrossRef]
- Lee, J.Y.; Wang, F.; Fazio, T.; Wind, S.; Greene, E. Measuring intermolecular rupture forces with a combined TIRF-optical trap microscope and DNA curtains. Biochem. Biophys. Res. Commun. 2012, 426, 565–570. [Google Scholar] [CrossRef]
- Kalkman, G.A.; Zhang, Y.; Monachino, E.; Mathwig, K.; Kamminga, M.E.; Pourhossein, P.; Oomen, P.E.; Stratmann, S.A.; Zhao, Z.; van Oijen, A.M.; et al. Bisecting Microfluidic Channels with Metallic Nanowires Fabricated by Nanoskiving. ACS Nano 2016, 10, 2852–2859. [Google Scholar] [CrossRef]
- Tutkus, M.; Rakickas, T.; Kopustas, A.; Ivanovaitė, Š.; Venckus, O.; Navikas, V.; Zaremba, M.; Manakova, E.; Valiokas, R. Fixed DNA molecule arrays for high-throughput single DNA–protein interaction studies. Langmuir 2019, 35, 5921–5930. [Google Scholar] [CrossRef]
- Kopūstas, A.; Ivanovaitė, Š.; Rakickas, T.; Pocevičiūtė, E.; Paksaitė, J.; Karvelis, T.; Zaremba, M.; Manakova, E.; Tutkus, M. Oriented soft DNA curtains for single-molecule imaging. Langmuir 2021, 37, 3428–3437. [Google Scholar] [CrossRef]
- Kim, D.; Rashid, F.; Cho, Y.; Zaher, M.S.; Cho, I.I.H.; Hamdan, S.M.; Jeong, C.; Lee, J.-B. DNA skybridge: 3D structure producing a light sheet for high-throughput single-molecule imaging. Nucleic Acids Res. 2019, 47, e107. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).