
Copper and Nickel Nanoparticles Prepared by Thermal Treatment of Their Respective Cations Confined in Nanopores through High-Pressure Synthesis

 , and
, and
Abstract
:1. Introduction
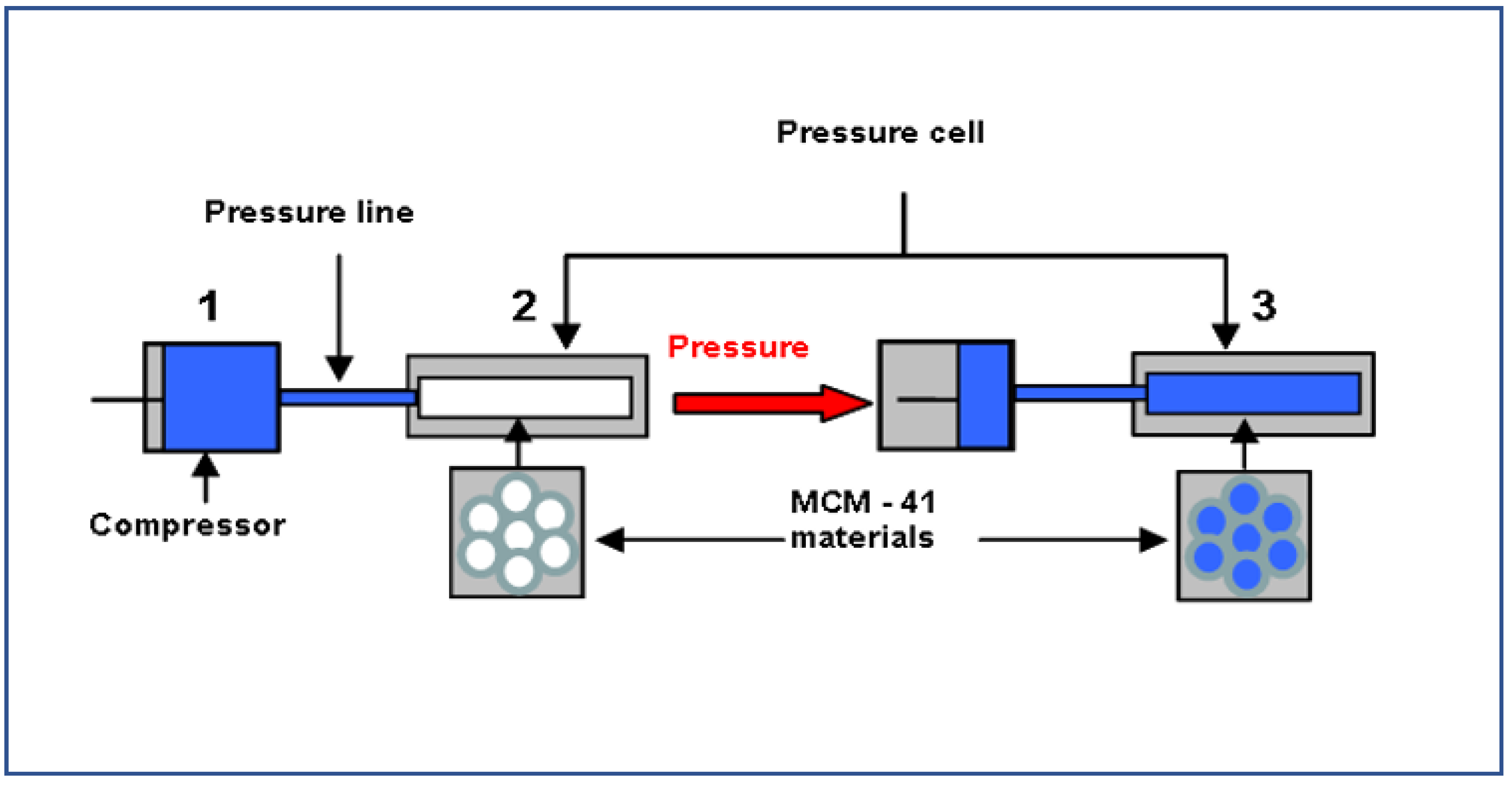
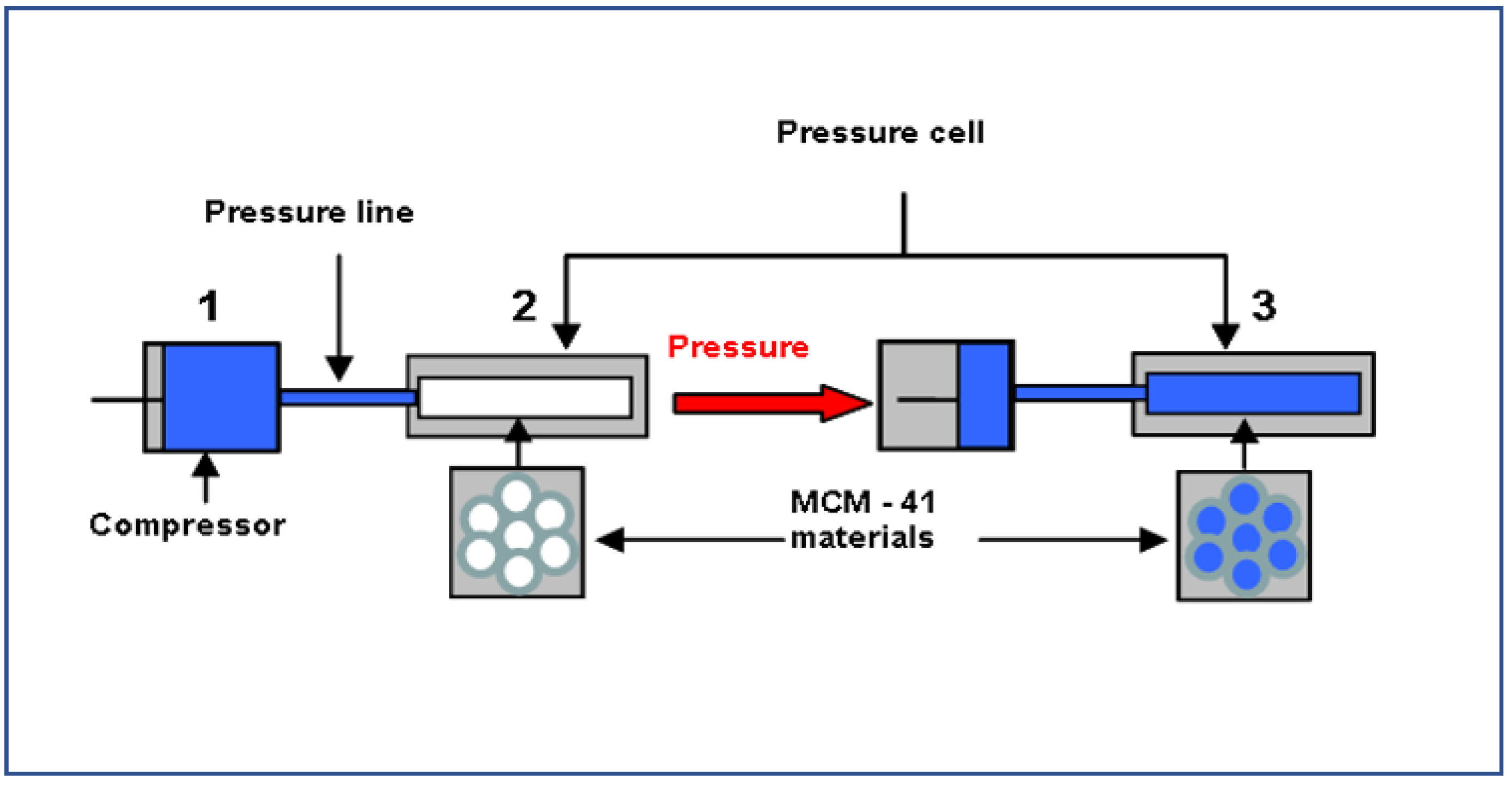
2. Materials and Methods
3. Results and Discussion
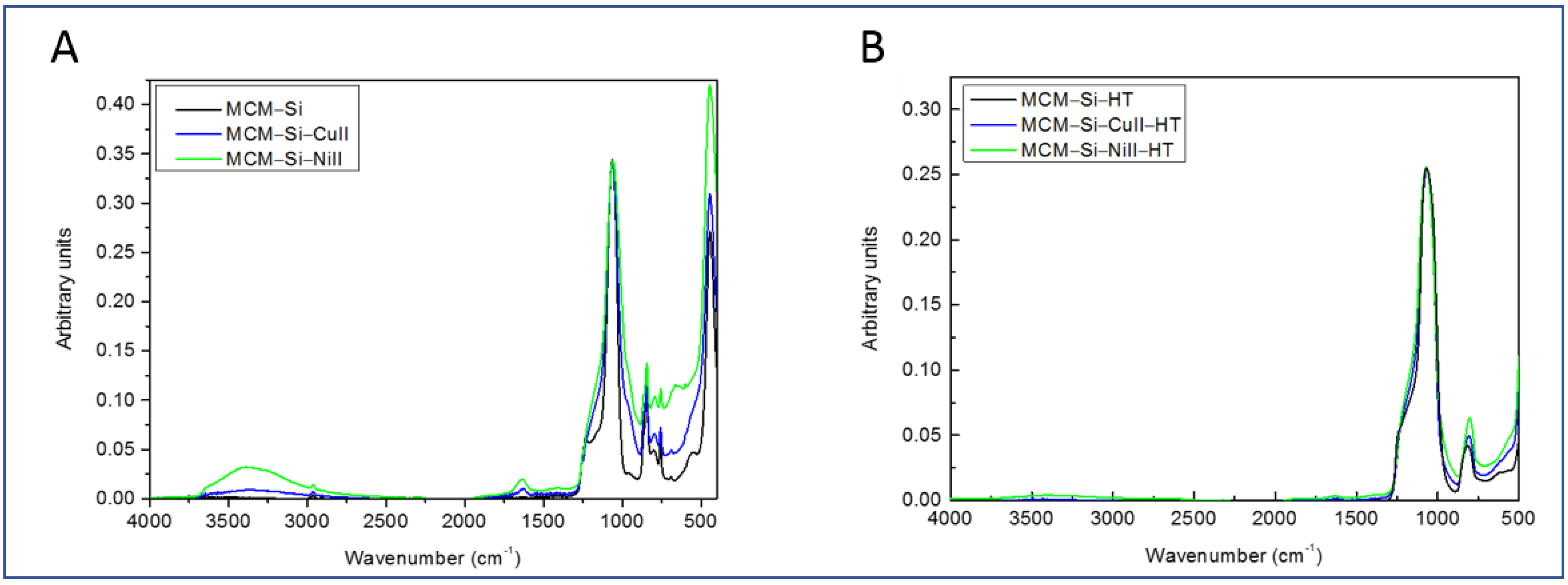
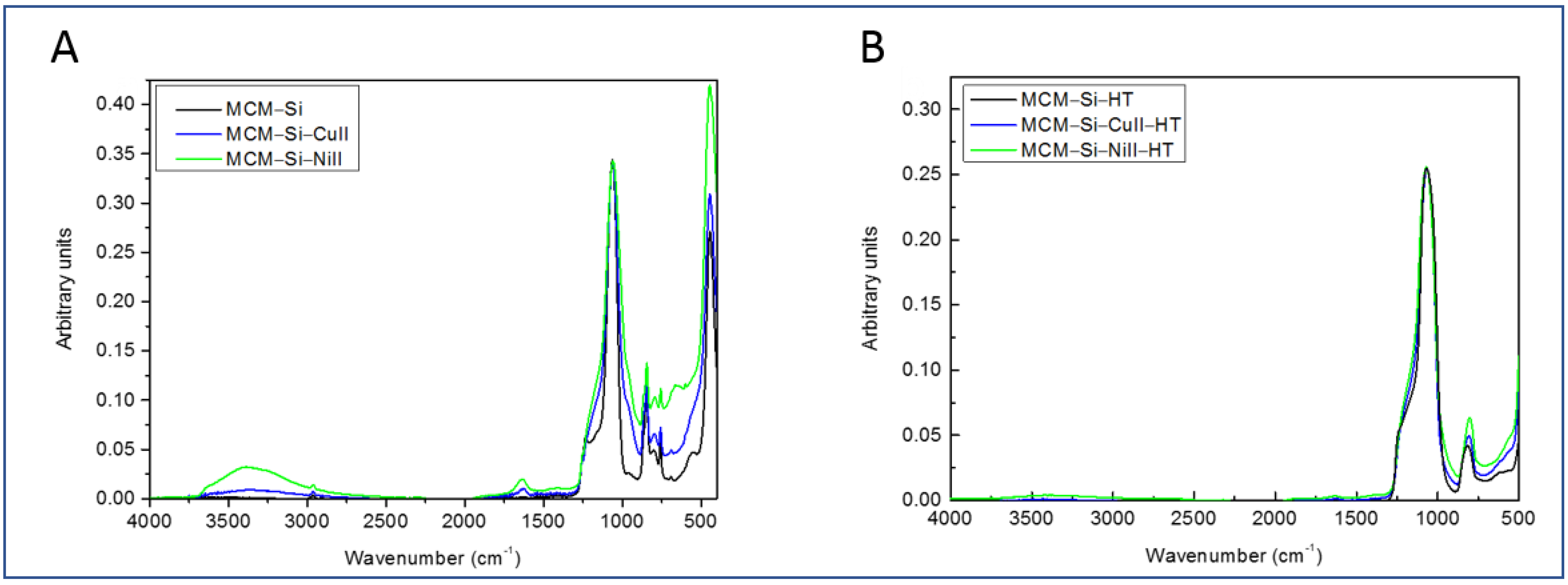
3.1. Fourier Transform Infrared (FTIR) Analysis
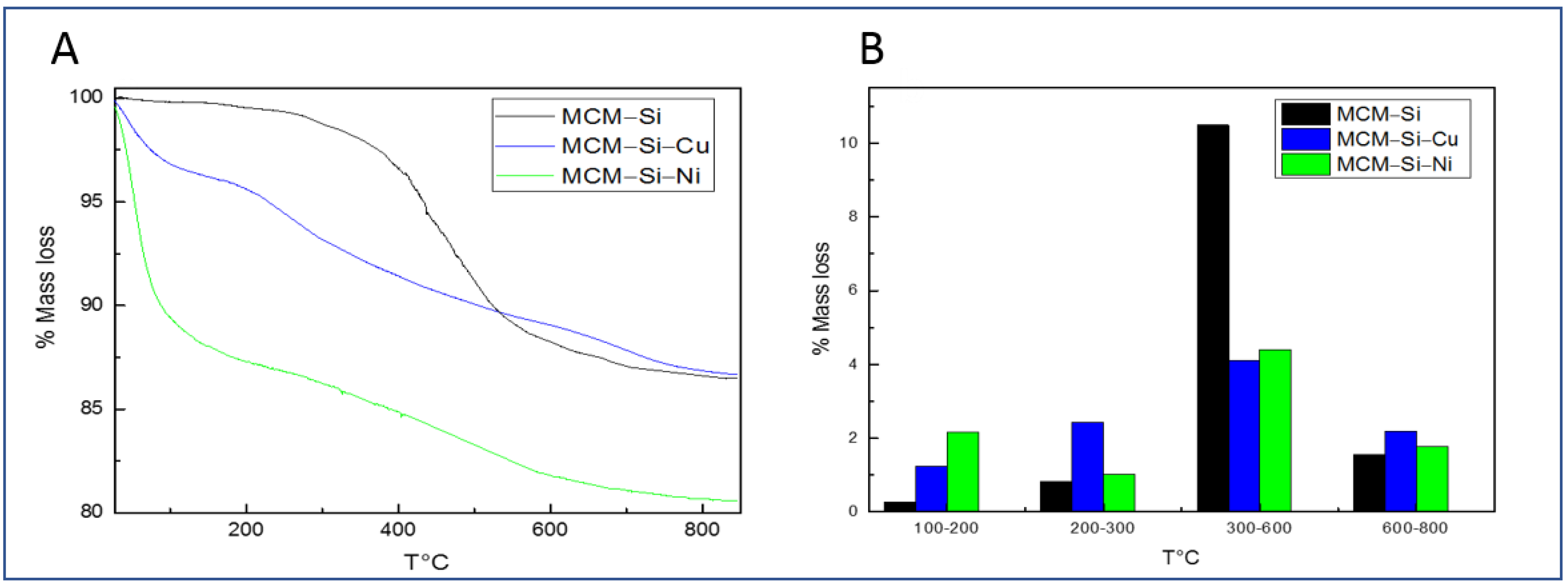
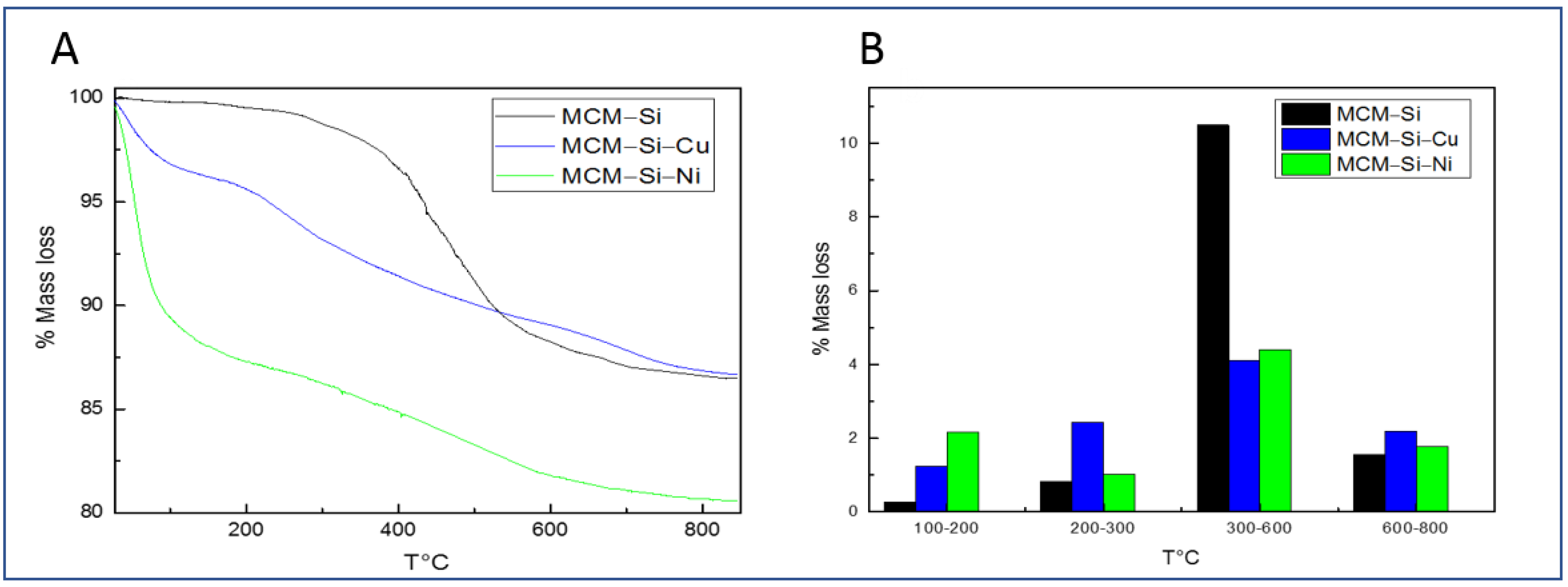
3.2. Thermogravimetric Analysis (TGA)
3.3. Redox Chemistry
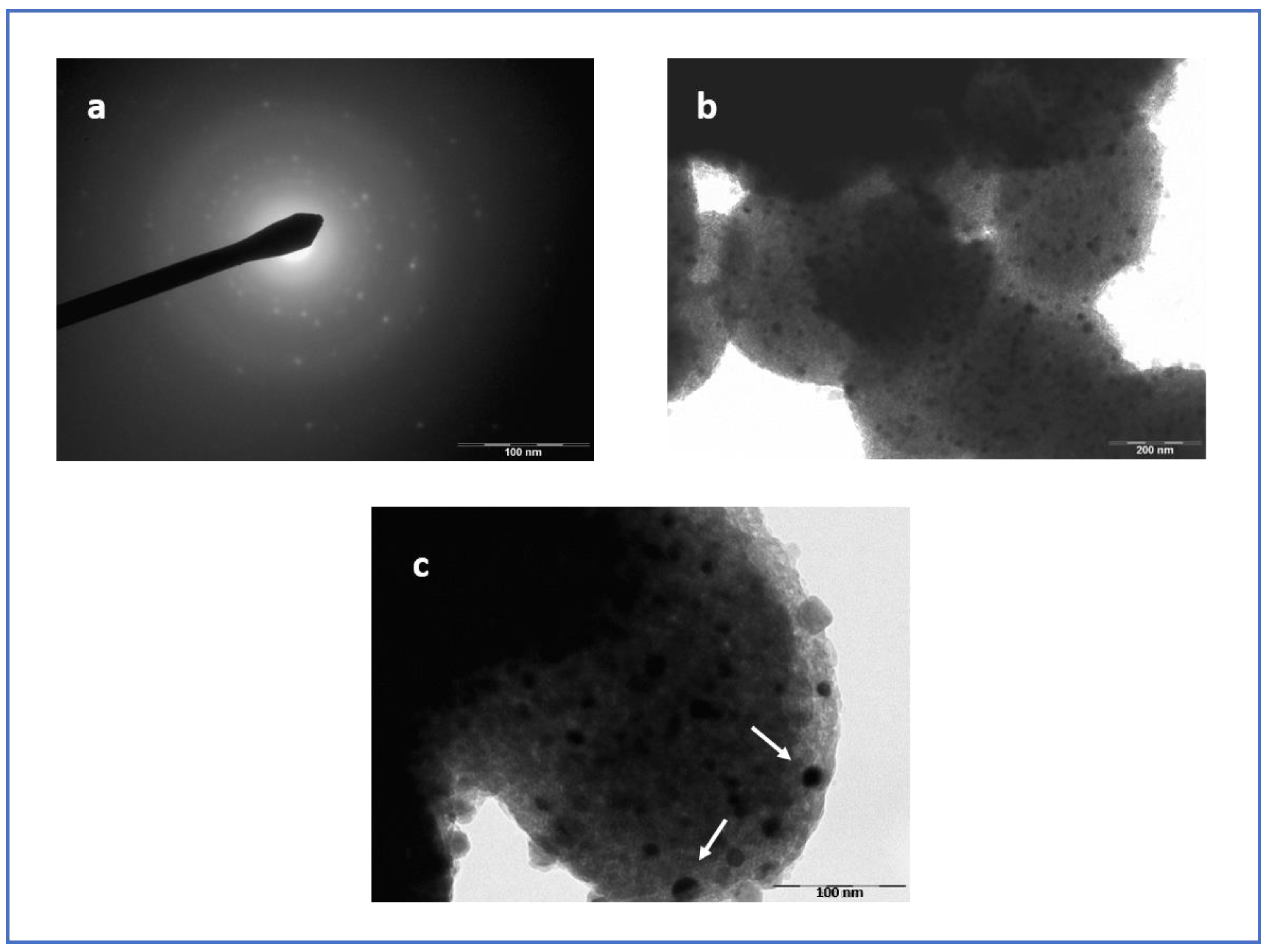
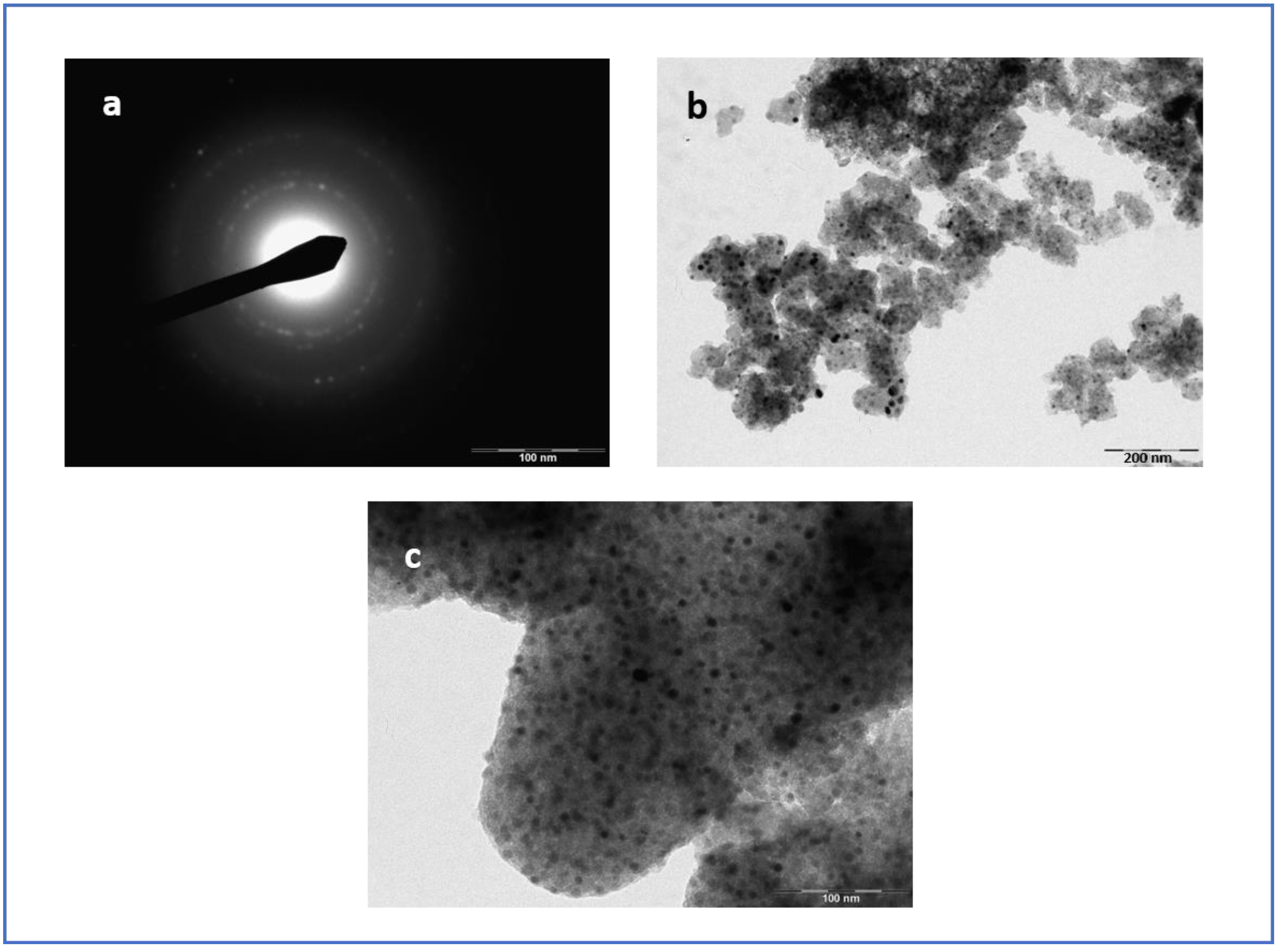
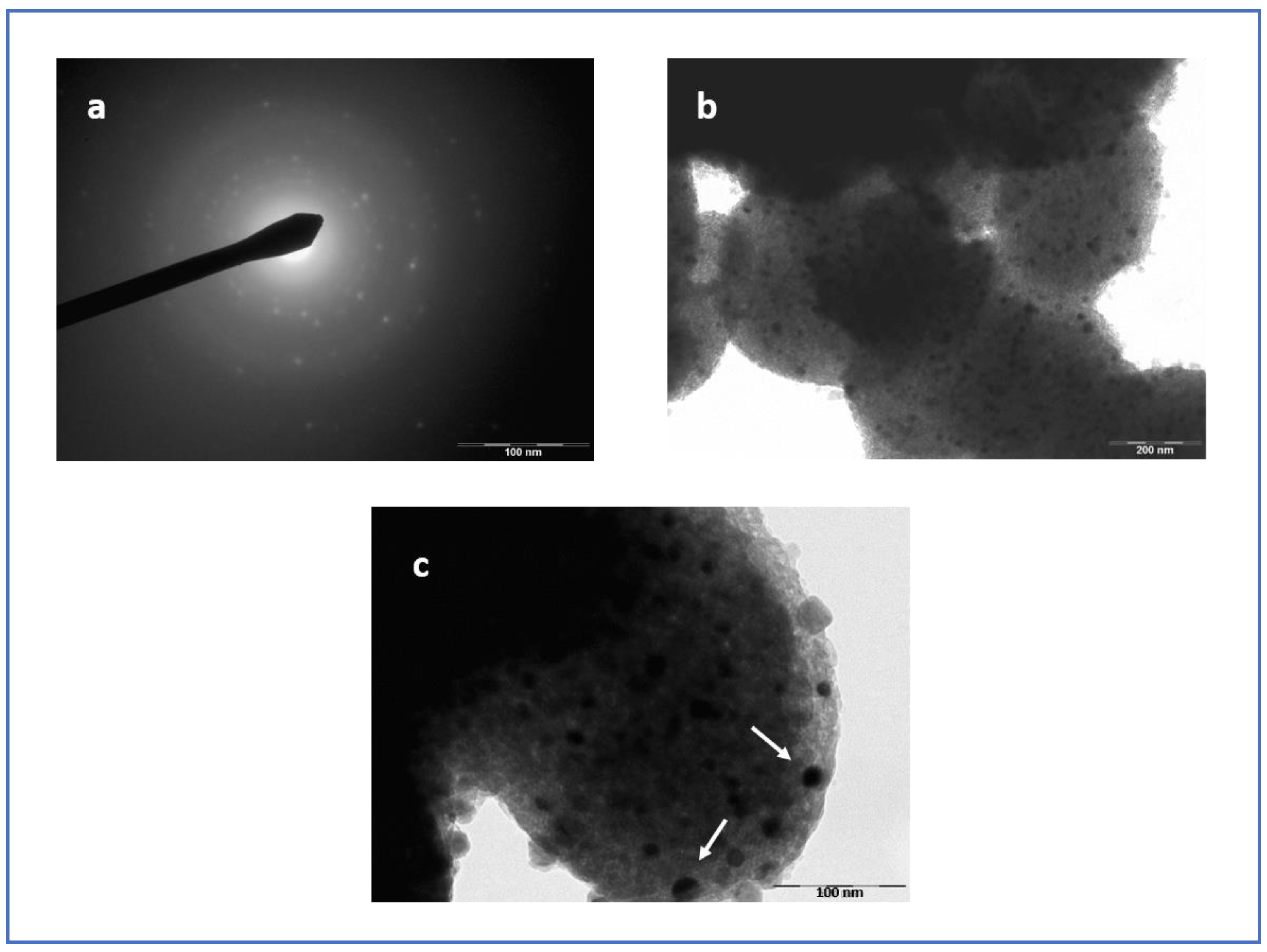
3.4. Micoscopy Analysis
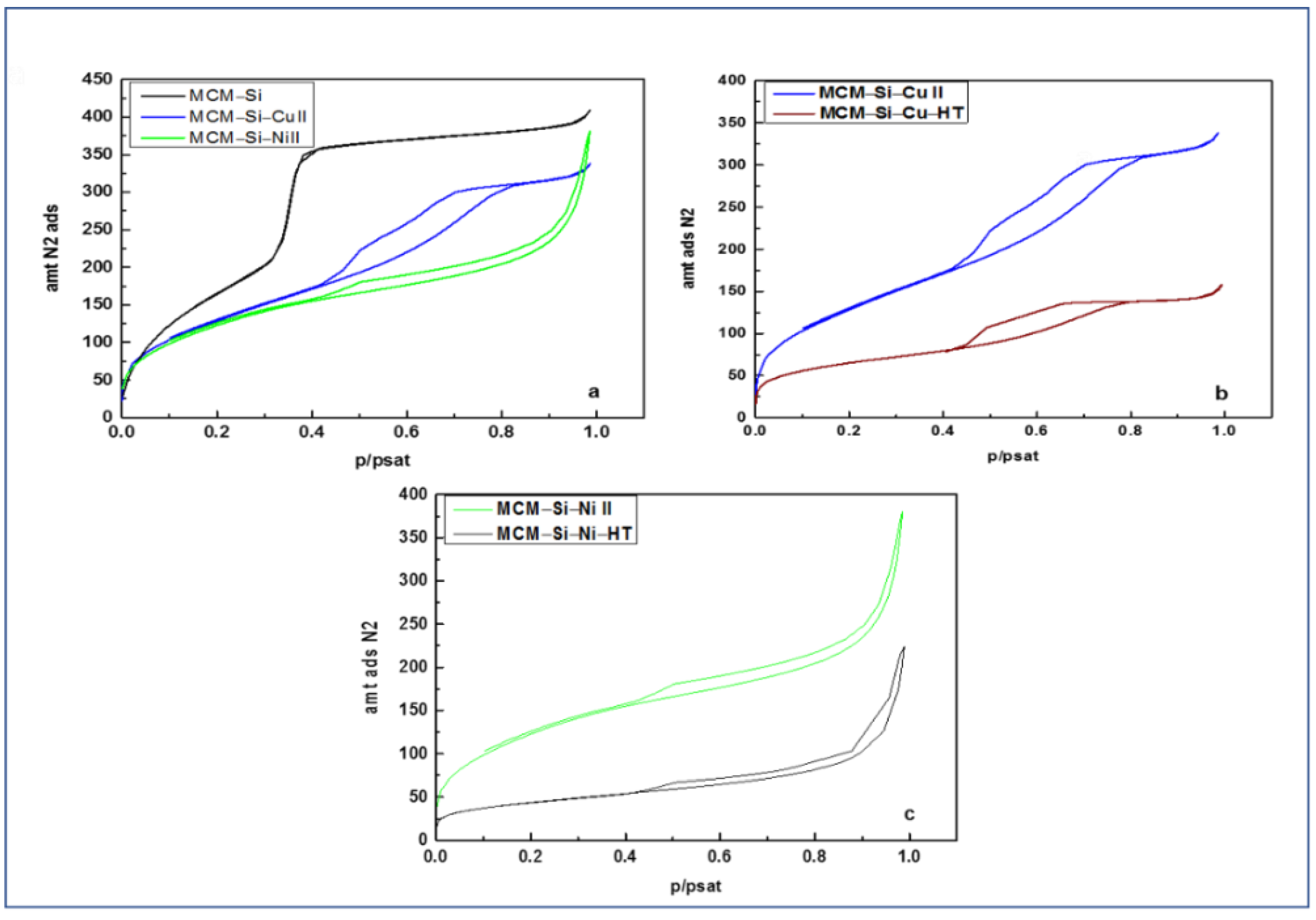
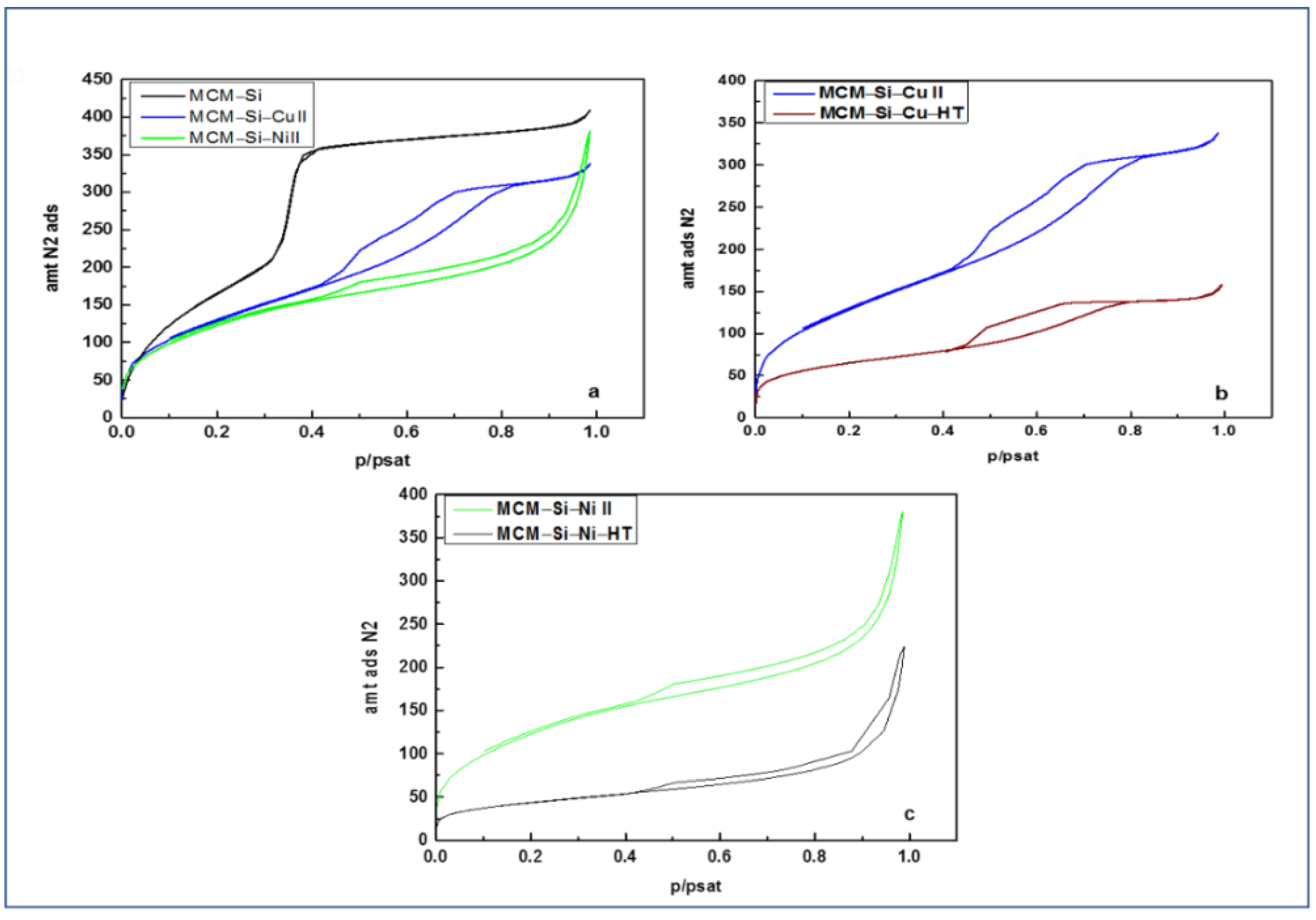
3.5. Porosity Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| MCM–41 | Mobil Composition of Matter 41 |
| TEM | Transmission electron microscopy |
| SAED | Selected area electron diffraction |
| FTIR | Fourier transform infrared |
| TGA | Thermogravimetric analysis |
| NP | Nanoparticle |
| MCM–Si | MCM–41 material, containing O–Si(CH3)3 groups on the surface, with a pore size of 3.82 nm, used for all pressure treatments |
| MCM–Si–M (M = Cu II or Ni II) | MCM–Si material, containing copper II or nickel II, before thermal treatment |
| MCM–Si–M–HT (M = Cu II or Ni II) | MCM–Si–M material, containing copper or nickel NP, after thermal treatment |
| ATR | Attenuated Total Reflectance |
| BET | Brunauer, Emmett and Teller |
| KJS Kruk | Jaroniec and Sayari |
| iKJS | improved KJS |
| BJH | Barrett, Joyner and Halenda |
References
- Baglioni, P.; Carretti, E.; Chelazzi, D. Nanomaterials in art conservation. Nat. Nanotech. 2015, 10, 287–290. [Google Scholar] [CrossRef]
- Tessier, P.M.; Velev, O.D.; Kalambur, A.T.; Rabolt, J.F.; Lenhoff, A.M.; Kaler, E.W. Assembly of gold nanostructured films templated by colloidal crystals and use in surface-enhanced Raman spectroscopy. J. Am. Chem. Soc. 2000, 122, 9554–9555. [Google Scholar] [CrossRef]
- Chaughule, R.S.; Ramanujan, R.V. Nanoparticles: Synthesis, Characterization and Applications; Chaughule, R.S., Ramanujan, R.V., Eds.; American Scientific Publishers: Stevenson Ranch, CA, USA, 2010. [Google Scholar]
- Goodman, D.W. Supported metal clusters: Synthesis, structure, and catalysis. Chem. Rev. 1995, 95, 511–522. [Google Scholar]
- Astruc, D.M.C. Gold nanoparticles: Assembly, supramolecular chemistry, quantum-size-related properties, and applications toward biology, catalysis, and nanotechnology. Chem. Rev. 2004, 104, 293–346. [Google Scholar]
- Prasanth, K.; Catoire, L.; Deschamps, J. Aluminium doping composite metal-organic framework by alane nanoconfinement: Impact on the room temperature hydrogen uptake. Micropor. Mesopor. Mat. 2017, 243, 214–220. [Google Scholar]
- Pansare, A.V.; Amol, A.; Shedge, A.A.; Patil, V.R. Discrete SeNPs-Macromolecule Binding Manipulated by Hydrophilic Interaction. Int. J. Biol. Macromol. 2018, 107, 1982–1987. [Google Scholar] [CrossRef]
- Shedgea, A.A.; Pansare, S.V.; Khairkarc, S.R.; Chatred, S.Y.; Chakrabartib, S.; Nagarkare, A.A.; Pansare, A.V.; Patil, V.R. Nanocomposite of functional silver metal containing curcumin biomolecule model systems: Protein BSA bioavailability. J. Inorg. Biochem. 2020, 212, 111210. [Google Scholar] [CrossRef]
- Kumar, K.H.; Venkatesh, N.; Bhowmik, H.; Kuila, A. Metallic nanoparticle: A review. Biomed. J. Sci. Tech. Res. 2018, 4, 3765–3775. [Google Scholar]
- Roldan Cuenya, B. Metal nanoparticle catalysts beginning to shape-up. Acc. Chem. Res. 2013, 46, 1682–1691. [Google Scholar] [CrossRef]
- Ealias, A.M.; Saravanakumar, M.B. A Review on the classification, characterisation, synthesis of nanoparticles and their application. In Proceedings of the IOP Conference Series: Materials Science and Engineering 2017, Busan, Korea, 25–27 August 2017; p. 032019. [Google Scholar]
- Govindrao Jamkhande, P.; Ghule, N.; Haque Bamer, A.; Kalaskar, M. Metal nanoparticles synthesis: An overview on methods of preparation, advantages and disadvantages, and applications. J. Drug Deliv. Sci. Technol. 2019, 53, 101174–101185. [Google Scholar] [CrossRef]
- Kato, Y.; Suzuki, M. Synthesis of Metal Nanoparticles by Microorganisms. Crystals 2020, 10, 589. [Google Scholar] [CrossRef]
- Campbell, C.T. The Energetics of Supported Metal Nanoparticles: Relationships to sintering rates and catalytic activity. Acc. Chem. Res. 2013, 46, 1712–1719. [Google Scholar] [CrossRef]
- Abrokwah, R.; Deshmane, V.; Kuila, D. Comparative performance of M-MCM-41 (M: Cu, Co, Ni, Pd, Zn and Sn) catalysts for steam reforming of methanol. J. Mol. Cat. A Chem. 2016, 425, 10–20. [Google Scholar] [CrossRef] [Green Version]
- Zienkiewicz-Strzalka, M.; Pasieczna-Patkowska, S.; Kozak, M.; Pikus, S. Silver nanoparticles incorporated onto ordered mesoporous silica from tollen’s reagent. App. Surf. Sci. 2013, 266, 337–343. [Google Scholar] [CrossRef]
- Brodie-Linder, N.; Audonnet, F.; Dosseh, G.; Alba-Simionesco, C.; Imperor, M. SBA-15 synthesis: Are there lasting effects of temperature change within the first 10 minutes of TEOS polymerization? Mater. Chem. Phys. 2008, 108, 73–81. [Google Scholar] [CrossRef]
- Brodie-Linder, N.; Besse, R.; Audonnet, F.; Le Caër, S.; Deschamps, J.; Imperor-Clerc, M.; Alba-Simionesco, C. The key to control Cu II loading in silica based mesoporous materials. Micr. Mes. Mat. 2010, 132, 518–525. [Google Scholar] [CrossRef]
- Brodie-Linder, N.; Le Caër, S.; Alam, M.S.; Renault, J.P.; Alba-Simionesco, C. H2 formation by electron irradiation of SBA-15 materials and the effect of Cu(II) grafting. Phys. Chem. Chem. Phys. 2010, 12, 14188–14195. [Google Scholar] [CrossRef]
- Brodie-Linder, N.; Deschamps, J.; Audonnet, F.; Alba-Simionesco, C. Method to create a hydrophilic environment within hydrophobic nanostructures. Micropor. Mesopor. Mat. 2013, 179, 17–21. [Google Scholar] [CrossRef]
- Schoeffel, M.; Brodie-Linder, N.; Audonnet, F.; Alba-Simionesco, C. Wall thickness determination of hydrophobically functionalized MCM-41 materials. J. Mat. Chem. 2012, 22, 557–567. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, E.; Teller, P.H. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Kruk, M.; Jaroniec, M.; Sayari, A. Application of large pore MCM-41 Molecular sieves to improve pore size analysis using nitrogen adsorption measurements. Langmuir 1997, 13, 6267–6273. [Google Scholar] [CrossRef]
- Jaroniec, M.; Solovyov, L.A. Improvement of the Kruk-Jaroniec-Sayari method for pore size analysis of ordered silicas with cylindrical mesopores. Langmuir 2006, 22, 6757–6760. [Google Scholar] [CrossRef] [PubMed]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. The Determination of pore volume and area distributions in porous substances. I. Computations from Nitrogen Isotherms. J. Am. Chem. Soc. 1951, 73, 373–380. [Google Scholar] [CrossRef]
- Knight, A.; Kalugin, N.; Coker, E.; Iigen, A. Water properties under nano-scale confinement. Sci. Rep. 2019, 9, 8246. [Google Scholar] [CrossRef]
- Zhang, K.; Lee, T.H.; Bubach, B.; Jang, H.W.; Ostadhassan, M.; Choi, J.-W.; Shokouhimehr, M. Graphite carbon-encapsulated metal nanoparticles derived from Prussian blue analogs growing on natural loofa as cathode materials for rechargeable aluminum-ion batteries. Sci. Rep. 2019, 9, 13665–13674. [Google Scholar] [CrossRef]
- Pansare, A.V.; Khairkar, S.R.; Shedge, A.A.; Chhatre, S.Y.; Patil, V.R.; Nagarkar, A.A. In situ nanoparticle embedding for authentication of epoxy composites. Adv. Mater. 2018, 30, 1801523. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.; Oliver, J.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution. Pure Appl. Chem. 2015, 879, 105–1069. [Google Scholar]
- Deschamps, J.; Audonnet, F.; Brodie-Linder, N.; Schoeffel, M.; Alba-Simionesco, C. A thermodynamic limit of the melting/freezing processes of water under strongly hydrophobic nanoscopic confinement. Phys. Chem. Chem. Phys. 2010, 12, 1440–1443. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MCM–Si–M | % C per Weight | % Weight Loss | % N per Weight | % M per Weight |
|---|---|---|---|---|
| MCM–Si | 11.28 | 10.5 | – | – |
| MCM–Si–Cu II | 4.11 | 0.52 | 7.5 | |
| MCM–Si–Ni II | – | 4.40 | 0.32 | 10.0 |
| MCM–Si–Cu–HT | 0.71 (0.70) a | – | – | – |
| MCM–Si–Ni–HT | 1.49 (1.0) a | – | – | – |
| Materials | ABET/(m2·g−1) | Vp/(cm3·g−1) | d iKJS/nm | C |
|---|---|---|---|---|
| MCM–Si | 658.1 | 0.600 | 3.82 | 24 |
| MCM–Si–Cu II | 474.8 | 0.495 | 2.12 | 9 |
| MCM–Si–Ni II | 456.1 | 0.321 | 2.10 | 47 |
| Materials | ABET/(m2·g−1) | Vp/(cm3·g−1) | d iKJS/nm | C |
|---|---|---|---|---|
| MCM–Si–Cu–HT | 234.2 | 0.278 | 1.86 | 129 |
| MCM–Si–Ni–HT | 157.4 | 0.369 | 1.82 | 129 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brodie-Linder, N.; Deschamps, J.; Bombled, M.; Pasternak, N.; Audonnet, F.; Beaunier, P.; Alba-Simionesco, C. Copper and Nickel Nanoparticles Prepared by Thermal Treatment of Their Respective Cations Confined in Nanopores through High-Pressure Synthesis. Appl. Nano 2021, 2, 278-288. https://doi.org/10.3390/applnano2030020
Brodie-Linder N, Deschamps J, Bombled M, Pasternak N, Audonnet F, Beaunier P, Alba-Simionesco C. Copper and Nickel Nanoparticles Prepared by Thermal Treatment of Their Respective Cations Confined in Nanopores through High-Pressure Synthesis. Applied Nano. 2021; 2(3):278-288. https://doi.org/10.3390/applnano2030020
Chicago/Turabian StyleBrodie-Linder, Nancy, Johnny Deschamps, Marianne Bombled, Nicolas Pasternak, Fabrice Audonnet, Patricia Beaunier, and Christiane Alba-Simionesco. 2021. "Copper and Nickel Nanoparticles Prepared by Thermal Treatment of Their Respective Cations Confined in Nanopores through High-Pressure Synthesis" Applied Nano 2, no. 3: 278-288. https://doi.org/10.3390/applnano2030020
APA StyleBrodie-Linder, N., Deschamps, J., Bombled, M., Pasternak, N., Audonnet, F., Beaunier, P., & Alba-Simionesco, C. (2021). Copper and Nickel Nanoparticles Prepared by Thermal Treatment of Their Respective Cations Confined in Nanopores through High-Pressure Synthesis. Applied Nano, 2(3), 278-288. https://doi.org/10.3390/applnano2030020