
Surfactant Temperature-Dependent Critical Micelle Concentration Prediction with Uncertainty-Aware Graph Neural Network

Abstract
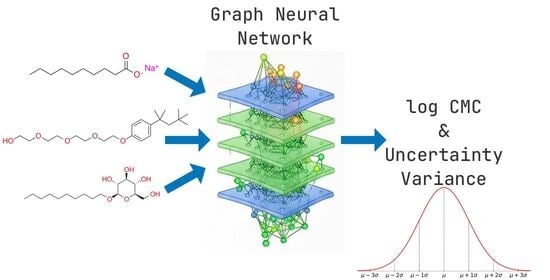
1. Introduction
2. Materials and Methods
2.1. Fingerprint-Based Machine Learning Models
2.2. Graph Neural Network
- L represents the base loss function (MSE in the current implementation);
- σ2 denotes the predicted variance;
- ∈ [0, 1] is a hyperparameter that balances the standard prediction loss and the uncertainty calibration term;
- denotes the expectation over the training samples.
2.3. Performance Metrics
- is the number of observations;
- represents the observed value for the i-th sample;
- represents the predicted value for the i-th sample;
- is the mean of the observed values: .
3. Results and Discussion
3.1. Dataset Preparation
3.1.1. General
- Hödl et al.’s test set [25] comprised 140 data points (for assessing CMC prediction accuracy across all surfactant types, including gemini surfactants, log CMC values range from −0.5 to −6.0 and molecular masses range between 174 and 1196 g/mol);
- Brozos et al’s. test set [24] comprised 218 data points (for assessing CMC’s temperature dependency across multiple surfactant types, log CMC values range from 0.2 to −4.7 and molecular masses are between 90 and 663 g/mol).
- Chemical structure canonicalization: SMILES were canonicalized using the canonicalize method of MoleculeContainer from Chython library. Separately canonicalization using “canonicalize” and “clean_stereo” methods was performed for inspecting different stereoisomers. Stereoisomers without provided anomeric configuration or stereo features were dropped.
- Exact duplicates—defined as identical SMILES, temperature and log CMC values—were removed to prevent overrepresentation in the dataset. Generally, primary sources were prioritized over aggregated literature sources and datasets. Surfactants with multiple measurements at the same temperature were manually examined, and the log CMC values were averaged (“CMC_average”) if the range (Δlog CMC) between the maximum and minimum values was ≤0.01; in other cases, values from less trusted sources were deleted.
- Temperature dependency of each surfactant was manually analyzed by plotting log CMC [mol/L] against temperature [°C]. Measurements significantly deviating from monotonic or U-shaped relationships were removed from the dataset (temperature dependency plots of surfactants in the final dataset are presented in Section 3.1.2).
- Verification of data accuracy through original source: For duplicate entries—defined as identical SMILES and temperature values—present across multiple datasets with conflicting log CMC values, the original cited sources were consulted to verify and select the most accurate and consistent values, ensuring fidelity to the primary experimental data.
- Manual inspection of molecular structures: A final visual audit of all structures was conducted to detect anomalies; any suspicious entries were verified and corrected by consulting the original source literature. Unresolved cases were excluded to ensure structural accuracy and dataset integrity.
3.1.2. Temperature
3.1.3. Molecular Mass
3.1.4. Surfactant Type
3.2. Training and Validation Set Preparation
3.3. Modeling
3.3.1. Predictive Performance of RF and XGB Models
- max_features explored fractional (0.1–0.5) values and heuristics (“sqrt”, “log2”);
- max_depth explicitly included None (unconstrained trees) and values between 10 and 50 to evaluate depth necessity;
- min_samples_split (2–20);
- min_samples_leaf (1–15);
- n_estimators used log-scale sampling (100–1000);
- bootstrap parameters (max_samples, 0.5–1.0) were conditionally optimized only with enabled bootstrap.
- max_depth was restricted to values between 3 and 10 to model chemical relationships without overfitting;
- colsample_bytree (0.3–0.7) and subsample (0.6–0.95) mitigated feature redundancy;
- reg_alpha and reg_lambda were explored over 10−8 to 10.0 with log-uniform sampling, ensuring robust exploration of orders-of-magnitude effects;
- tree_method “hist” ensured computational efficiency for large feature spaces.
3.3.2. Predictive Performance of GNN Models and Comparison with Previous Studies
4. GNN Model Uncertainty Prediction Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CMC | Critical micelle concentration |
| GNN | Graph neural network |
| RMSE | Root mean squared error |
| MSE | Mean squared error |
| MAE | Mean absolute error |
| MD | Molecular dynamics |
| NLL | Negative log-likelihood |
| QSPR | Quantitative structure–property relationship |
| COSMO-RS | Conductor-like screening model for realistic solvation |
| ML | Machine learning |
| MLR | Multiple linear regression |
| PLS | Partial least squares |
| ANN | Artificial neural network |
| SVR | Support vector regression |
| GNN/GP | GNN model augmented with Gaussian processes |
| FP | (Molecular) Fingerprint |
| RF | Random forest regressor |
| XGB | XGBoost regressor |
| MLP | Multilayer perceptron |
| SMILES | Simplified molecular input line entry system |
| CV | Cross-validation |
| NIST | National Institute of Standards and Technology |
References
- Turchi, M.; Karcz, A.P.; Andersson, M.P. First-Principles Prediction of Critical Micellar Concentrations for Ionic and Nonionic Surfactants. J. Colloid Interface Sci. 2022, 606, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas, H.; Kamrul-Bahrin, M.A.H.; Seddon, D.; Othman, J.; Cabral, J.T.; Mejía, A.; Shahruddin, S.; Matar, O.K.; Müller, E.A. Determining Interfacial Tension and Critical Micelle Concentrations of Surfactants from Atomistic Molecular Simulations. J. Colloid Interface Sci. 2024, 674, 1071–1082. [Google Scholar] [CrossRef]
- Mattei, M.; Kontogeorgis, G.M.; Gani, R. Modeling of the Critical Micelle Concentration (CMC) of Nonionic Surfactants with an Extended Group-Contribution Method. Ind. Eng. Chem. Res. 2013, 52, 12236–12246. [Google Scholar] [CrossRef]
- Smith, C.; Lu, J.R.; Thomas, R.K.; Tucker, I.M.; Webster, J.R.P.; Campana, M. Markov Chain Modeling of Surfactant Critical Micelle Concentration and Surface Composition. Langmuir 2019, 35, 561–569. [Google Scholar] [CrossRef]
- Sanchez-Lengeling, B.; Aspuru-Guzik, A. Inverse Molecular Design Using Machine Learning: Generative Models for Matter Engineering. Science 2018, 361, 360–365. [Google Scholar] [CrossRef]
- Afonina, V.A.; Mazitov, D.A.; Nurmukhametova, A.; Shevelev, M.D.; Khasanova, D.A.; Nugmanov, R.I.; Burilov, V.A.; Madzhidov, T.I.; Varnek, A. Prediction of Optimal Conditions of Hydrogenation Reaction Using the Likelihood Ranking Approach. Int. J. Mol. Sci. 2021, 23, 248. [Google Scholar] [CrossRef]
- Albrijawi, M.T.; Alhajj, R. LSTM-Driven Drug Design Using SELFIES for Target-Focused de Novo Generation of HIV-1 Protease Inhibitor Candidates for AIDS Treatment. PLoS ONE 2024, 19, e0303597. [Google Scholar] [CrossRef]
- Bushuev, K.R.; Lobanov, I.S. Machine Learning Method for Computation of Optimal Transitions in Magnetic Nanosystems. Nanosyst. Physics Chem. Math. 2020, 11, 642–650. [Google Scholar] [CrossRef]
- Huibers, P.D.T.; Lobanov, V.S.; Katritzky, A.R.; Shah, D.O.; Karelson, M. Prediction of Critical Micelle Concentration Using a Quantitative Structure-Property Relationship Approach. 1. Nonionic Surfactants. Langmuir 1996, 12, 1462–1470. [Google Scholar] [CrossRef]
- Huibers, P.D.T.; Lobanov, V.S.; Katritzky, A.R.; Shah, D.O.; Karelson, M. Prediction of Critical Micelle Concentration Using a Quantitative Structure–Property Relationship Approach. 2. Anionic Surfactants. J. Colloid Interface Sci. 1997, 187, 113–120. [Google Scholar] [CrossRef]
- Li, X.; Zhang, G.; Dong, J.; Zhou, X.; Yan, X.; Luo, M. Estimation of Critical Micelle Concentration of Anionic Surfactants with QSPR Approach. J. Mol. Struct. THEOCHEM 2004, 710, 119–126. [Google Scholar] [CrossRef]
- Wang, Z.; Huang, D.; Gong, S.; Li, G. Prediction on Critical Micelle Concentration of Nonionic Surfactants in Aqueous Solution: Quantitative Structure-Property Relationship Approach. Chin. J. Chem. 2003, 21, 1573–1579. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Pacureanu, L.; Dobchev, D.; Karelson, M. QSPR Study of Critical Micelle Concentration of Anionic Surfactants Using Computational Molecular Descriptors. J. Chem. Inf. Model. 2007, 47, 782–793. [Google Scholar] [CrossRef]
- Jiao, L.; Wang, Y.; Qu, L.; Xue, Z.; Ge, Y.; Liu, H.; Lei, B.; Gao, Q.; Li, M. Hologram QSAR Study on the Critical Micelle Concentration of Gemini Surfactants. Colloids Surf. A Physicochem. Eng. Asp. 2020, 586, 124226. [Google Scholar] [CrossRef]
- Creton, B.; Barraud, E.; Nieto-Draghi, C. Prediction of Critical Micelle Concentration for Per- and Polyfluoroalkyl Substances. SAR QSAR Environ. Res. 2024, 35, 309–324. [Google Scholar] [CrossRef]
- Anoune, N.; Nouiri, M.; Berrah, Y.; Gauvrit, J.; Lanteri, P. Critical Micelle Concentrations of Different Classes of Surfactants: A Quantitative Structure Property Relationship Study. J. Surfactants Deterg. 2002, 5, 45–53. [Google Scholar] [CrossRef]
- Rahal, S.; Hadidi, N.; Hamadache, M. In Silico Prediction of Critical Micelle Concentration (CMC) of Classic and Extended Anionic Surfactants from Their Molecular Structural Descriptors. Arab. J. Sci. Eng. 2020, 45, 7445–7454. [Google Scholar] [CrossRef]
- Laidi, M.; Abdallah, E.; Si-Moussa, C.; Benkortebi, O.; Hentabli, M.; Hanini, S. CMC of Diverse Gemini Surfactants Modelling Using a Hybrid Approach Combining SVR-DA. Chem. Ind. Chem. Eng. Q. 2021, 27, 299–312. [Google Scholar] [CrossRef]
- Soria-Lopez, A.; García-Martí, M.; Barreiro, E.; Mejuto, J.C. Ionic Surfactants Critical Micelle Concentration Prediction in Water/Organic Solvent Mixtures by Artificial Neural Network. Tenside Surfactants Deterg. 2024, 61, 519–529. [Google Scholar] [CrossRef]
- Boukelkal, N.; Rahal, S.; Rebhi, R.; Hamadache, M. QSPR for the Prediction of Critical Micelle Concentration of Different Classes of Surfactants Using Machine Learning Algorithms. J. Mol. Graph. Model. 2024, 129, 108757. [Google Scholar] [CrossRef]
- Qin, S.; Jin, T.; Lehn, R.C.V.; Zavala, V.M. Predicting Critical Micelle Concentrations for Surfactants Using Graph Convolutional Neural Networks. J. Phys. Chem. B 2021, 125, 10610–10620. [Google Scholar] [CrossRef]
- Moriarty, A.; Kobayashi, T.; Salvalaglio, M.; Striolo, A.; McRobbie, I. Analyzing the Accuracy of Critical Micelle Concentration Predictions Using Deep Learning. J. Chem. Theory Comput. 2023, 19, 7371–7386. [Google Scholar] [CrossRef] [PubMed]
- Theis Marchan, G.; Balogun, T.O.; Territo, K.; Das, D.; Olayiwola, T.; Kumar, R.; Romagnoli, J.A. Toward Harnessing AI for Surfactant Chemistry: Prediction of Critical Micelle Concentration. Comput. Mater. Sci. 2026, 265, 114548. [Google Scholar] [CrossRef]
- Brozos, C.; Rittig, J.G.; Bhattacharya, S.; Akanny, E.; Kohlmann, C.; Mitsos, A. Predicting the Temperature Dependence of Surfactant CMCs Using Graph Neural Networks. J. Chem. Theory Comput. 2024, 20, 5695–5707. [Google Scholar] [CrossRef]
- Hödl, S.L.; Hermans, L.; Dankloff, P.F.J.; Piruska, A.; Huck, W.T.S.; Robinson, W.E. SurfPro—A Curated Database and Predictive Model of Experimental Properties of Surfactants. Digit. Discov. 2025, 4, 1176–1187. [Google Scholar] [CrossRef]
- Chen, J.; Hou, L.; Nan, J.; Ni, B.; Dai, W.; Ge, X. Prediction of Critical Micelle Concentration (CMC) of Surfactants Based on Structural Differentiation Using Machine Learning. Colloids Surf. A Physicochem. Eng. Asp. 2024, 703, 135276. [Google Scholar] [CrossRef]
- Barbosa, G.D.; Striolo, A. Machine Learning Prediction of Critical Micellar Concentration Using Electrostatic and Structural Properties as Descriptors. J. Chem. Eng. Data 2025, 70, 4019–4030. [Google Scholar] [CrossRef]
- Nugmanov, R.; Dyubankova, N.; Gedich, A.; Wegner, J.K. Bidirectional Graphormer for Reactivity Understanding: Neural Network Trained to Reaction Atom-to-Atom Mapping Task. J. Chem. Inf. Model. 2022, 62, 3307–3315. [Google Scholar] [CrossRef] [PubMed]
- Fallani, A.; Nugmanov, R.; Arjona-Medina, J.; Wegner, J.K.; Tkatchenko, A.; Chernichenko, K. Pretraining Graph Transformers with Atom-in-a-Molecule Quantum Properties for Improved ADMET Modeling. J. Cheminform. 2025, 17, 25. [Google Scholar] [CrossRef]
- Arjona-Medina, J.; Nugmanov, R. Analysis of Atom-Level Pretraining with Quantum Mechanics (QM) Data for Graph Neural Networks Molecular Property Models. arXiv 2024, arXiv:2405.14837. [Google Scholar] [CrossRef]
- Saifullin, E.R.; Gimadiev, T.R.; Khakimova, A.A.; Varfolomeev, M.A. Game Changer in Chemical Reagents Design for Upstream Applications: From Long-Term Laboratory Studies to Digital Factory Based On AI. In Proceedings of the Abu Dhabi International Petroleum Exhibition and Conference (ADIPEC), Abu Dhabi, United Arab Emirates, 4–7 November 2024; p. D031S088R006. [Google Scholar]
- RDKit. Available online: https://www.rdkit.org/ (accessed on 18 November 2025).
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- XGBoost Python Package—Xgboost 3.0.5 Documentation. Available online: https://xgboost.readthedocs.io/en/release_3.0.0/python/index.html (accessed on 18 November 2025).
- Kwon, Y. Uncertainty-Aware Prediction of Chemical Reaction Yields with Graph Neural Networks. J. Cheminform. 2022, 14, 2. [Google Scholar] [CrossRef]
- Chython/Chython. Available online: https://github.com/chython/chython (accessed on 18 November 2025).
- Chython/Chytorch. Available online: https://github.com/chython/chytorch (accessed on 18 November 2025).
- Chython/Chytorch-Rxnmap. Available online: https://github.com/chython/chytorch-rxnmap (accessed on 18 November 2025).
- Scholz, N.; Behnke, T.; Resch-Genger, U. Determination of the Critical Micelle Concentration of Neutral and Ionic Surfactants with Fluorometry, Conductometry, and Surface Tension—A Method Comparison. J. Fluoresc. 2018, 28, 465–476. [Google Scholar] [CrossRef]
- Mukerjee, P.; Mysels, K. Critical Micelle Concentrations of Aqueous Surfactant Systems; National Bureau of Standards: Gaithersburg, MD, USA, 1971; p. NBS NSRDS 36. [Google Scholar]
- Frey, J.G.; Pearman-Kanza, S.; Munday, S. Critical Micelle Concentration (CMC) Data Collection. Available online: https://resources.psdi.ac.uk/data/a7c82670-d2e2-46c6-920a-74294289aa34 (accessed on 30 December 2025).
- Perger, T.-M.; Bešter-Rogač, M. Thermodynamics of Micelle Formation of Alkyltrimethylammonium Chlorides from High Performance Electric Conductivity Measurements. J. Colloid Interface Sci. 2007, 313, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Galgano, P.D.; El Seoud, O.A. Micellar Properties of Surface Active Ionic Liquids: A Comparison of 1-Hexadecyl-3-Methylimidazolium Chloride with Structurally Related Cationic Surfactants. J. Colloid Interface Sci. 2010, 345, 1–11. [Google Scholar] [CrossRef]
- Angarten, R.G.; Loh, W. Thermodynamics of Micellization of Homologous Series of Alkyl Mono and Di-Glucosides in Water and in Heavy Water. J. Chem. Thermodyn. 2014, 73, 218–223. [Google Scholar] [CrossRef]
- Cheng, C.; Qu, G.; Wei, J.; Yu, T.; Ding, W. Thermodynamics of Micellization of Sulfobetaine Surfactants in Aqueous Solution. J. Surfactants Deterg. 2012, 15, 757–763. [Google Scholar] [CrossRef]
- González-Pérez, A.; Ruso, J.M.; Romero, M.J.; Blanco, E.; Prieto, G.; Sarmiento, F. Application of Thermodynamic Models to Study Micellar Properties of Sodium Perfluoroalkyl Carboxylates in Aqueous Solutions. Chem. Phys. 2005, 313, 245–259. [Google Scholar] [CrossRef]
- Blanco, E.; González-Pérez, A.; Ruso, J.M.; Pedrido, R.; Prieto, G.; Sarmiento, F. A Comparative Study of the Physicochemical Properties of Perfluorinated and Hydrogenated Amphiphiles. J. Colloid Interface Sci. 2005, 288, 247–260. [Google Scholar] [CrossRef]
- Meguro, K.; Takasawa, Y.; Kawahashi, N.; Tabata, Y.; Ueno, M. Micellar Properties of a Series of Octaethyleneglycol-n-Alkyl Ethers with Homogeneous Ethylene Oxide Chain and Their Temperature Dependence. J. Colloid Interface Sci. 1981, 83, 50–56. [Google Scholar] [CrossRef]
- Castro, G.; Garrido, P.F.; Amigo, A.; Brocos, P. Boosting the Use of Thermoacoustimetry in Micellization Thermodynamics Studies by Easing an Objective Determination of the Cmc. Fluid Phase Equilibria 2018, 478, 1–13. [Google Scholar] [CrossRef]
- Eggenberger, D.N.; Harwood, H.J. Conductometric Studies of Solubility and Micelle Formation1. J. Am. Chem. Soc. 1951, 73, 3353–3355. [Google Scholar] [CrossRef]
- Akiba, T.; Sano, S.; Yanase, T.; Ohta, T.; Koyama, M. Optuna: A Next-Generation Hyperparameter Optimization Framework. In Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, Anchorage, AK, USA, 4–8 August 2019; Association for Computing Machinery: New York, NY, USA, 2019. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models | Test Set | R2 | MAE | RMSE |
|---|---|---|---|---|
| RF | Hödl et al. Test Set | 0.695 | 0.393 | 0.592 |
| XGB | Hödl et al. Test Set | 0.677 | 0.390 | 0.608 |
| RF | Brozos et al. Test Set | 0.727 | 0.402 | 0.541 |
| XGB | Brozos et al. Test Set | 0.778 | 0.372 | 0.488 |
| Models | Hödl et al. Test Set | Brozos et al. Test Set | ||||||
|---|---|---|---|---|---|---|---|---|
| R2 | MAE | RMSE | Spearman ρ | R2 | MAE | RMSE | Spearman ρ | |
| GNN Architecture № 1 | 0.890 | 0.230 | 0.355 | 0.238 | 0.868 | 0.255 | 0.375 | 0.227 |
| GNN Architecture № 2 | 0.882 | 0.241 | 0.367 | 0.177 | 0.850 | 0.278 | 0.401 | 0.377 |
| GNN Architecture № 3 | 0.877 | 0.240 | 0.376 | 0.230 | 0.892 | 0.225 | 0.340 | 0.345 |
| Hödl et al. (2025) single-property model * | – | 0.241 | 0.365 | – | – | – | – | – |
| Brozos et al. (2024) model | – | – | – | – | 0.95 | 0.15 | 0.24 | – |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Adygamov, M.S.; Saifullin, E.R.; Gimadiev, T.R.; Serov, N.Y. Surfactant Temperature-Dependent Critical Micelle Concentration Prediction with Uncertainty-Aware Graph Neural Network. Chemistry 2026, 8, 26. https://doi.org/10.3390/chemistry8020026
Adygamov MS, Saifullin ER, Gimadiev TR, Serov NY. Surfactant Temperature-Dependent Critical Micelle Concentration Prediction with Uncertainty-Aware Graph Neural Network. Chemistry. 2026; 8(2):26. https://doi.org/10.3390/chemistry8020026
Chicago/Turabian StyleAdygamov, Musa Sh., Emil R. Saifullin, Timur R. Gimadiev, and Nikita Yu. Serov. 2026. "Surfactant Temperature-Dependent Critical Micelle Concentration Prediction with Uncertainty-Aware Graph Neural Network" Chemistry 8, no. 2: 26. https://doi.org/10.3390/chemistry8020026
APA StyleAdygamov, M. S., Saifullin, E. R., Gimadiev, T. R., & Serov, N. Y. (2026). Surfactant Temperature-Dependent Critical Micelle Concentration Prediction with Uncertainty-Aware Graph Neural Network. Chemistry, 8(2), 26. https://doi.org/10.3390/chemistry8020026