
Photocyclization of Alkenes and Arenes: Penetrating Through Aromatic Armor with the Help of Excited State Antiaromaticity



Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
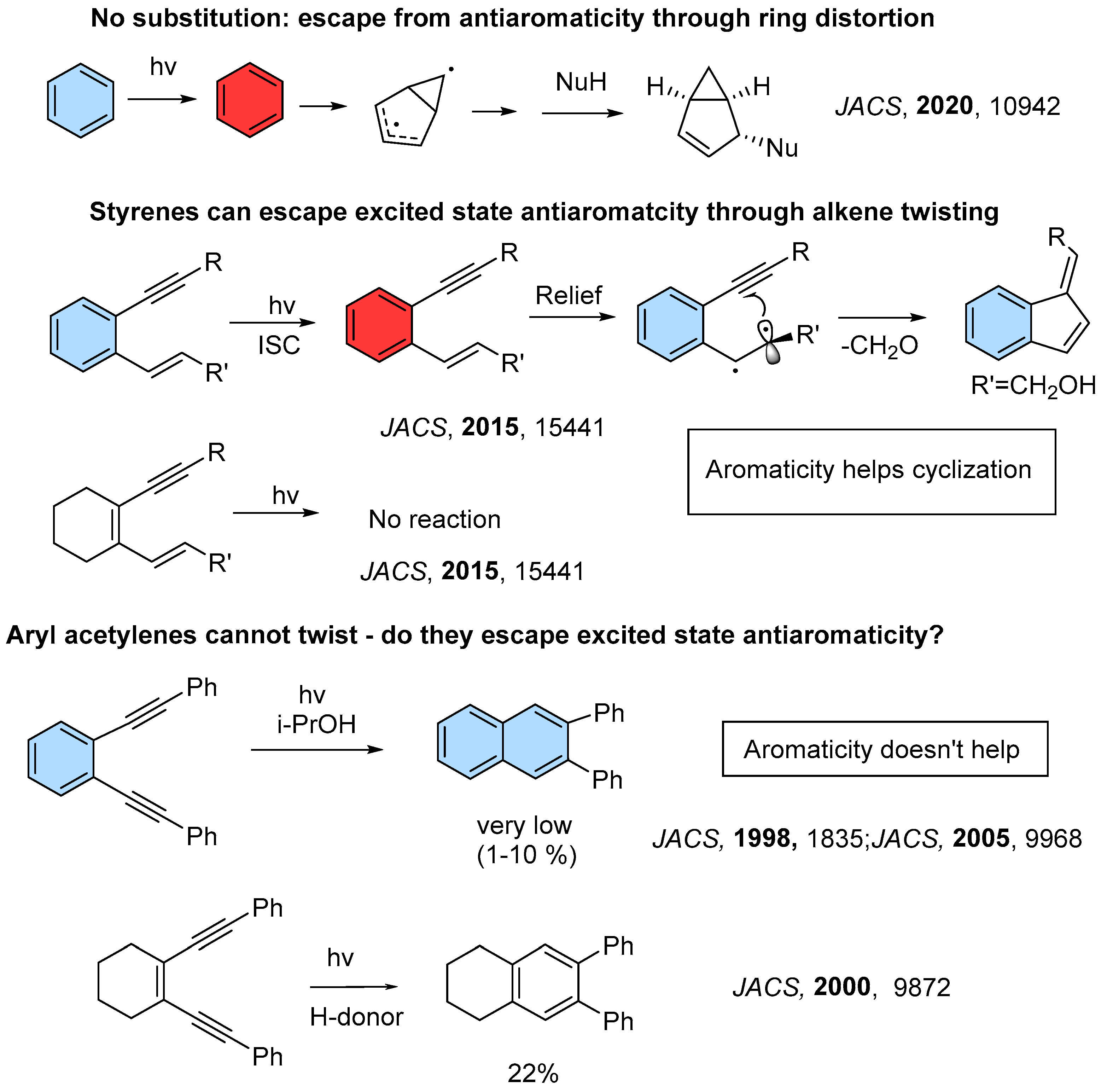
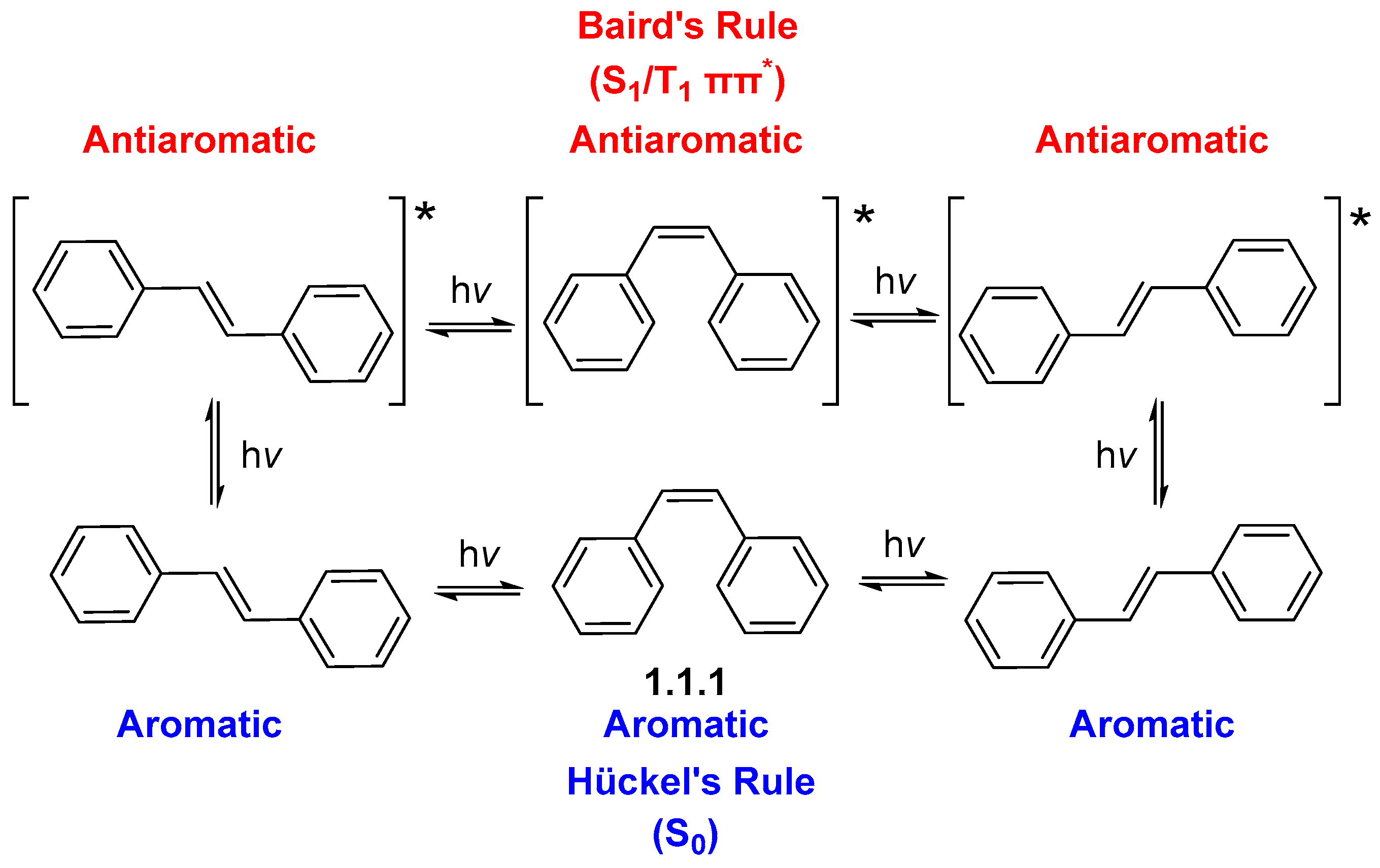
- Escape from Antiaromaticity Converts Benzene into a Highly Reactive Species
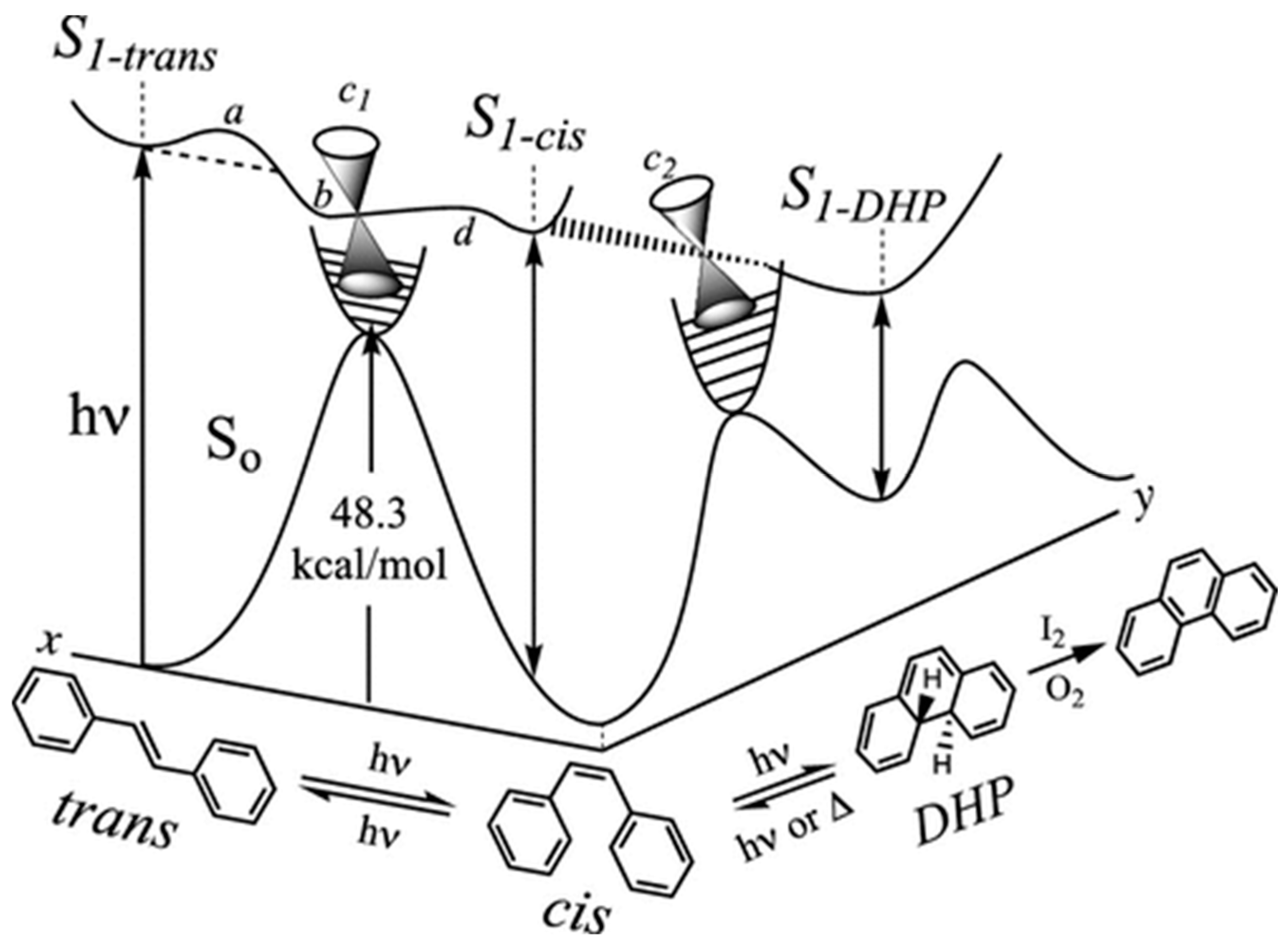
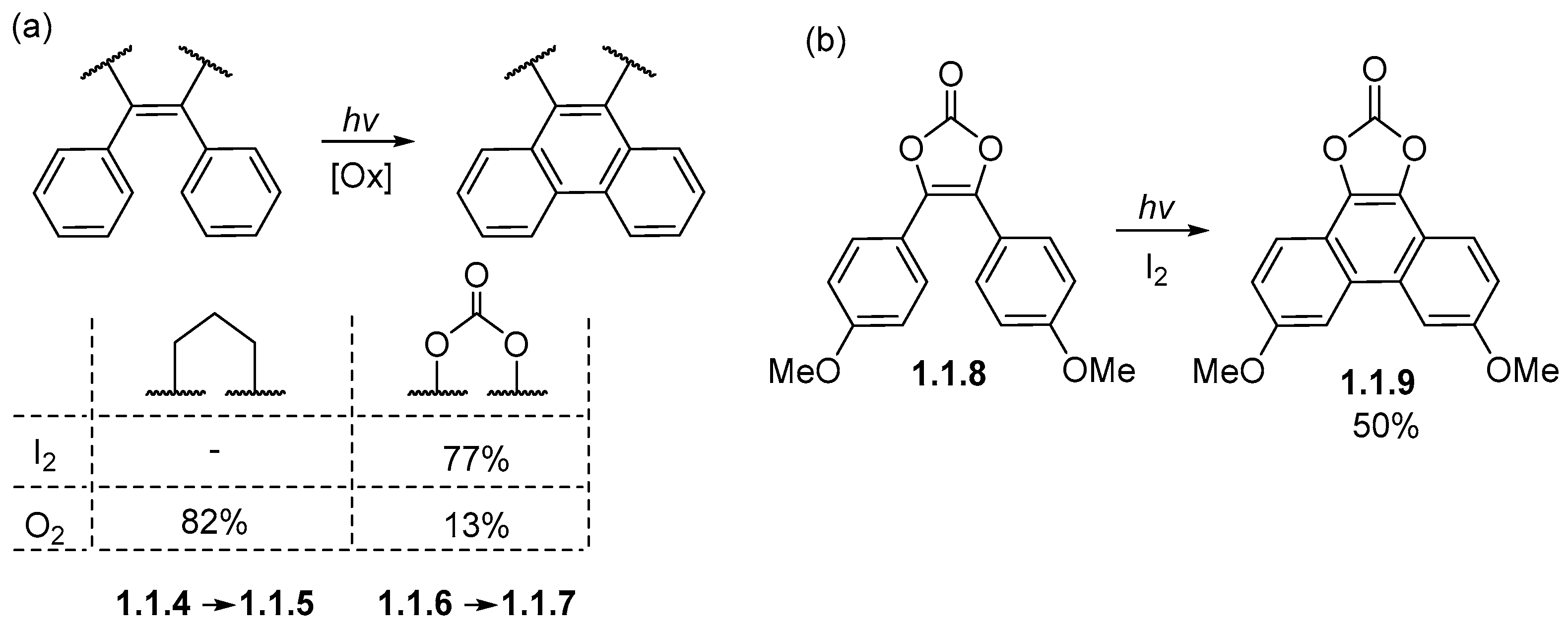
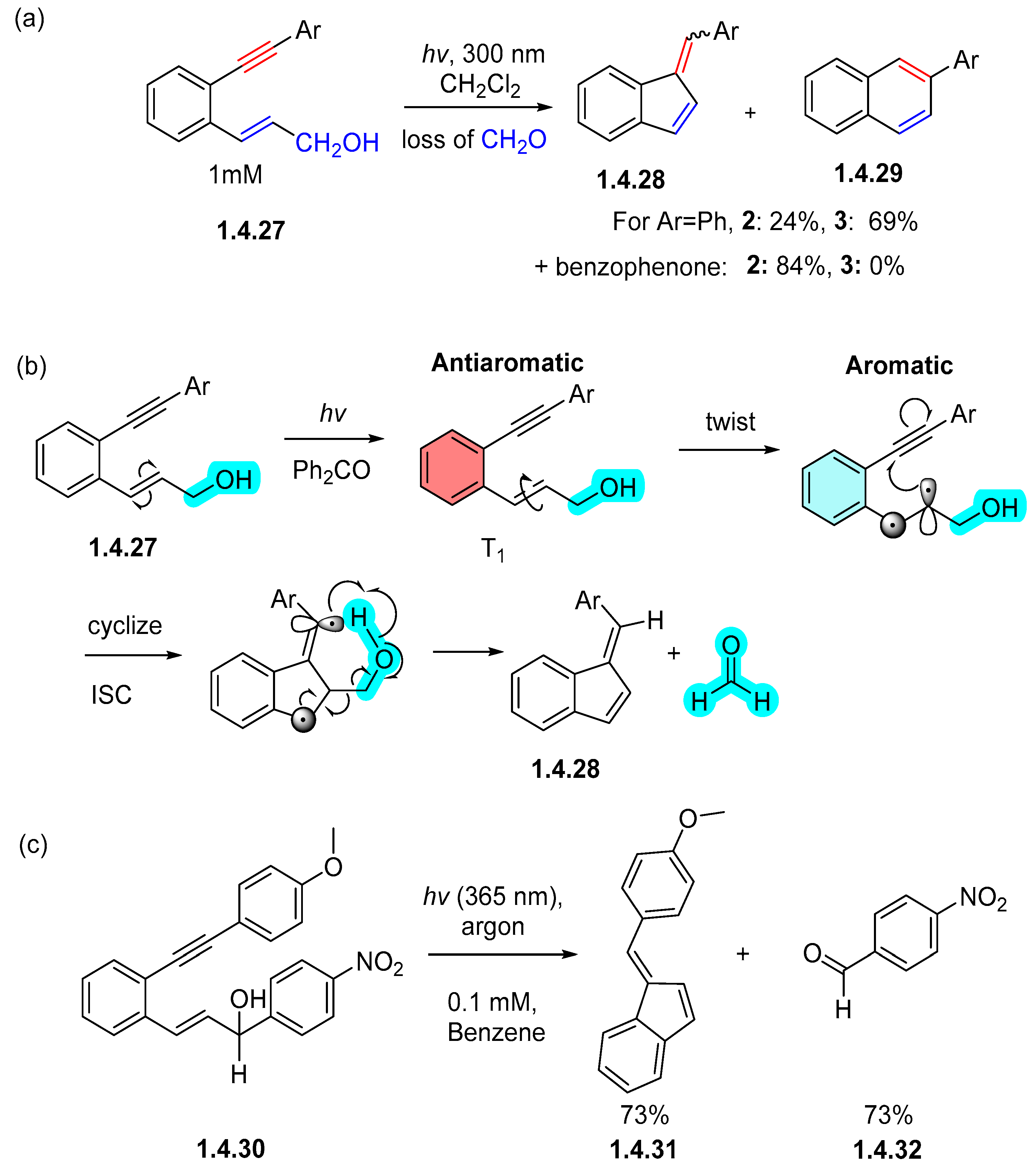
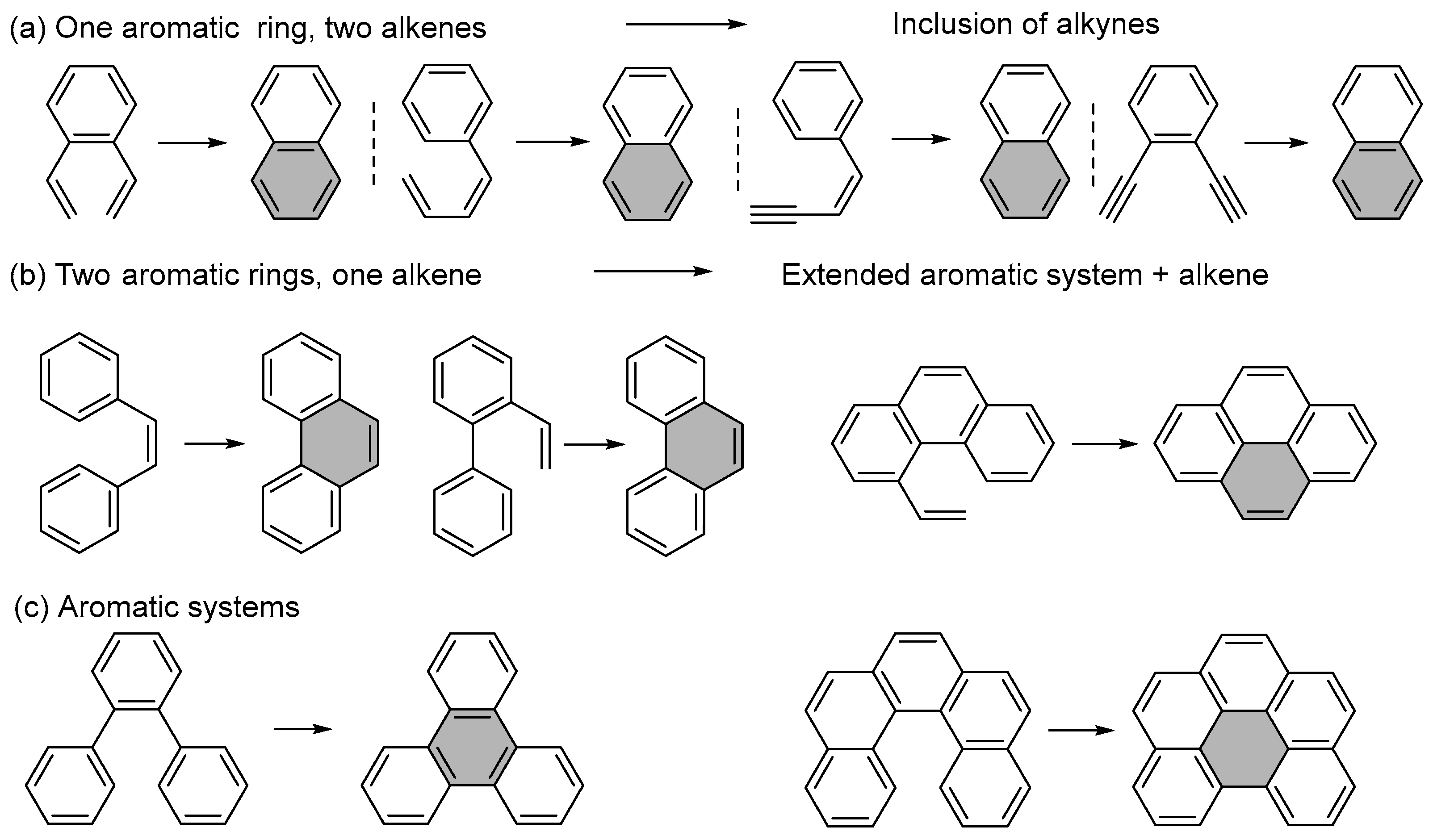
1.1. Photochemical Cyclizations Involving Tethered Alkenes and Arenes
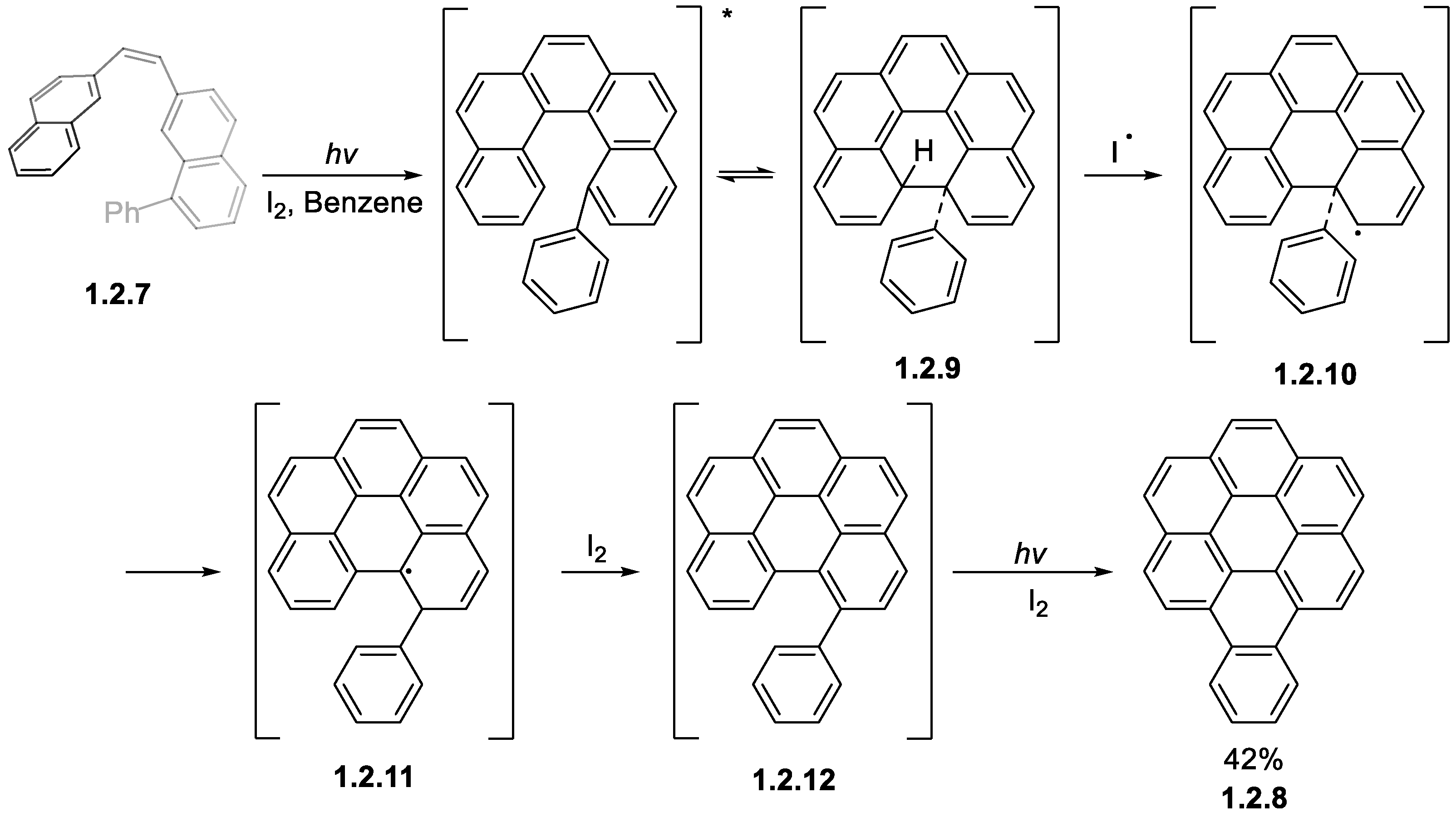
1.2. Photocyclizations Involving Extended Polyaromatics
1.3. Photochemical Ring Closures of Fully Aromatic Precursors
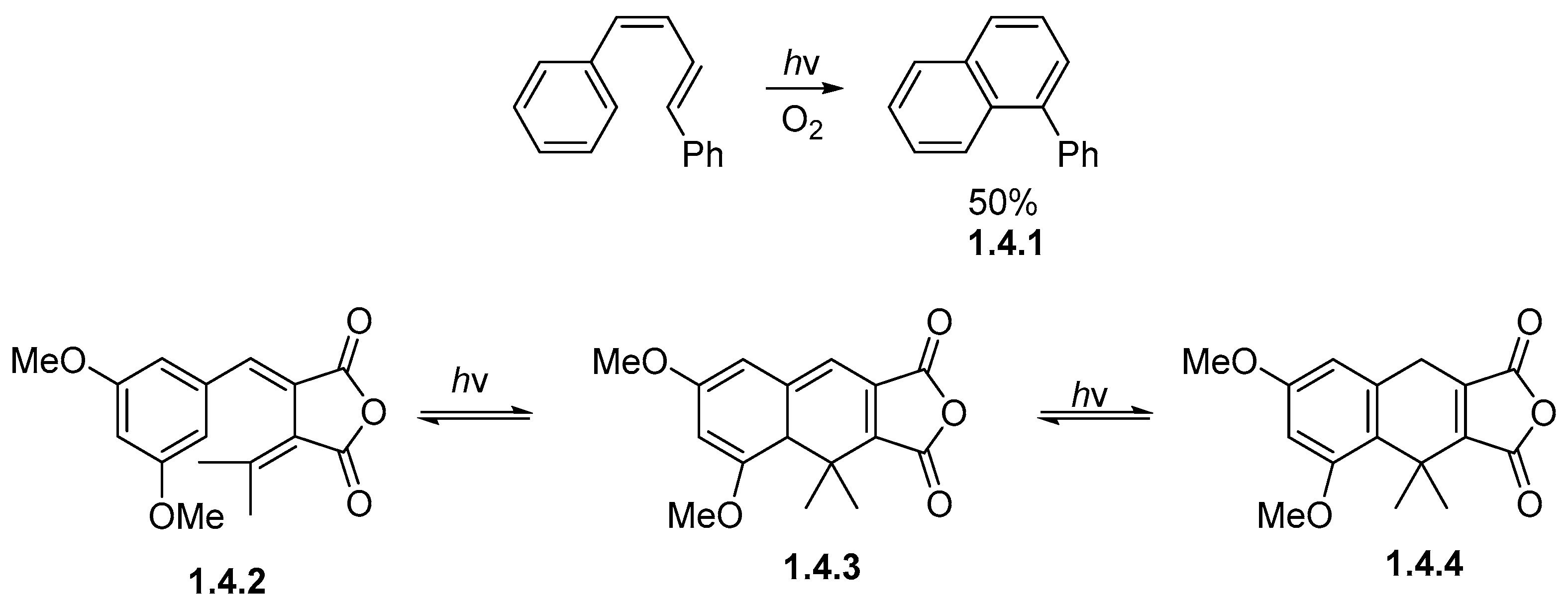
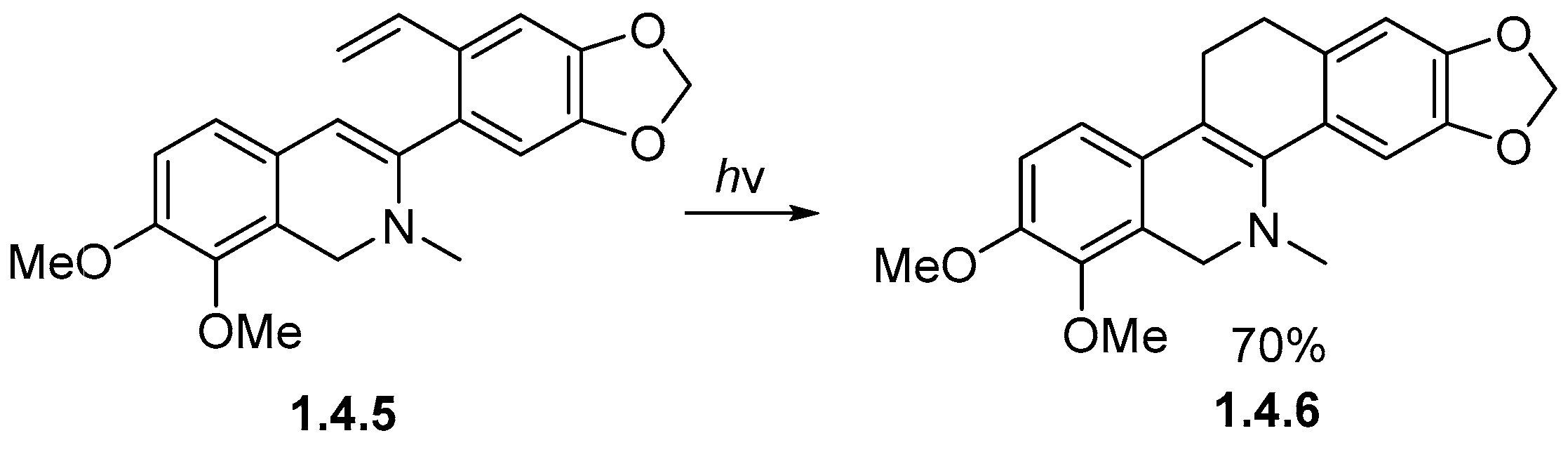
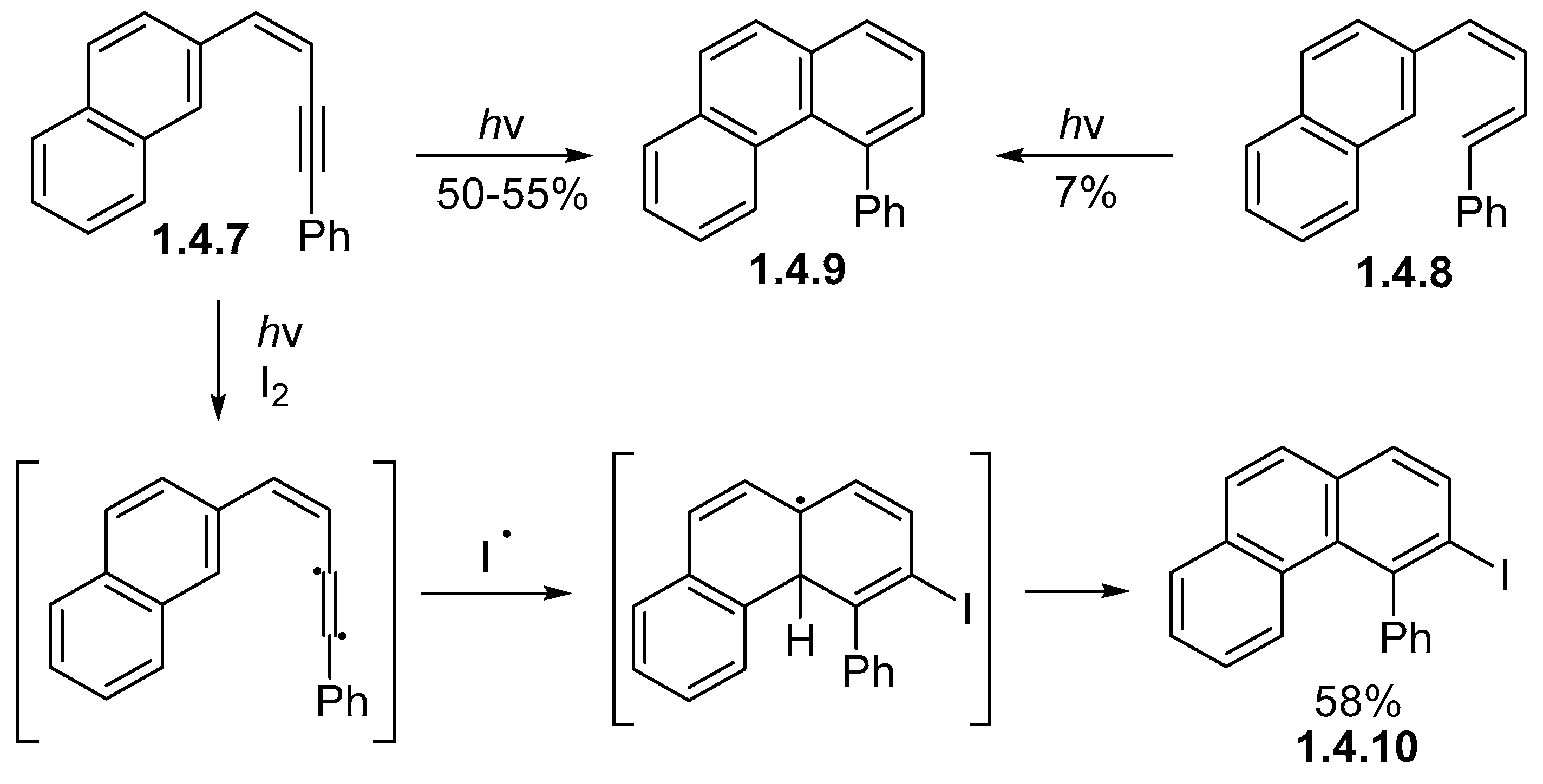
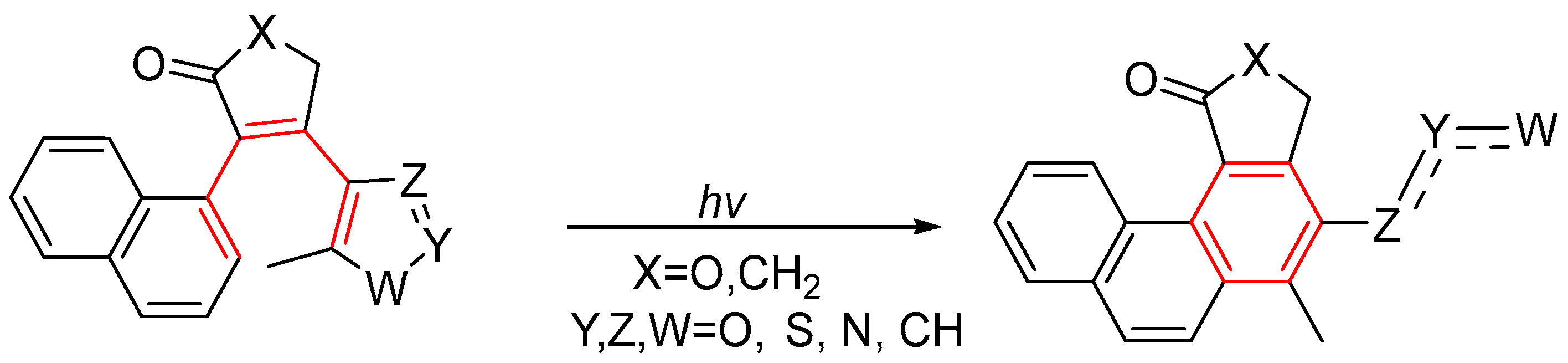
1.4. Single Aromatic Ring and Two Alkenes
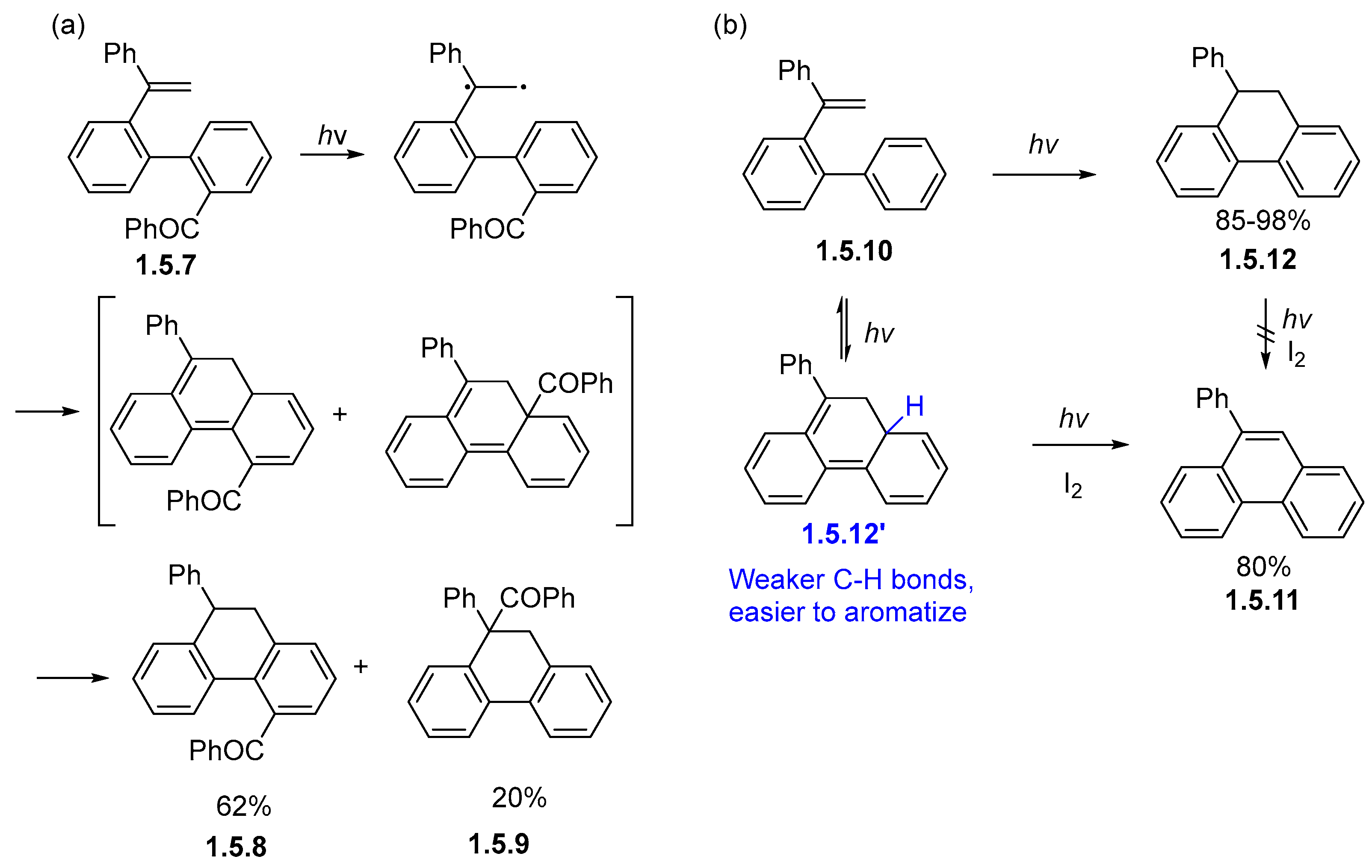
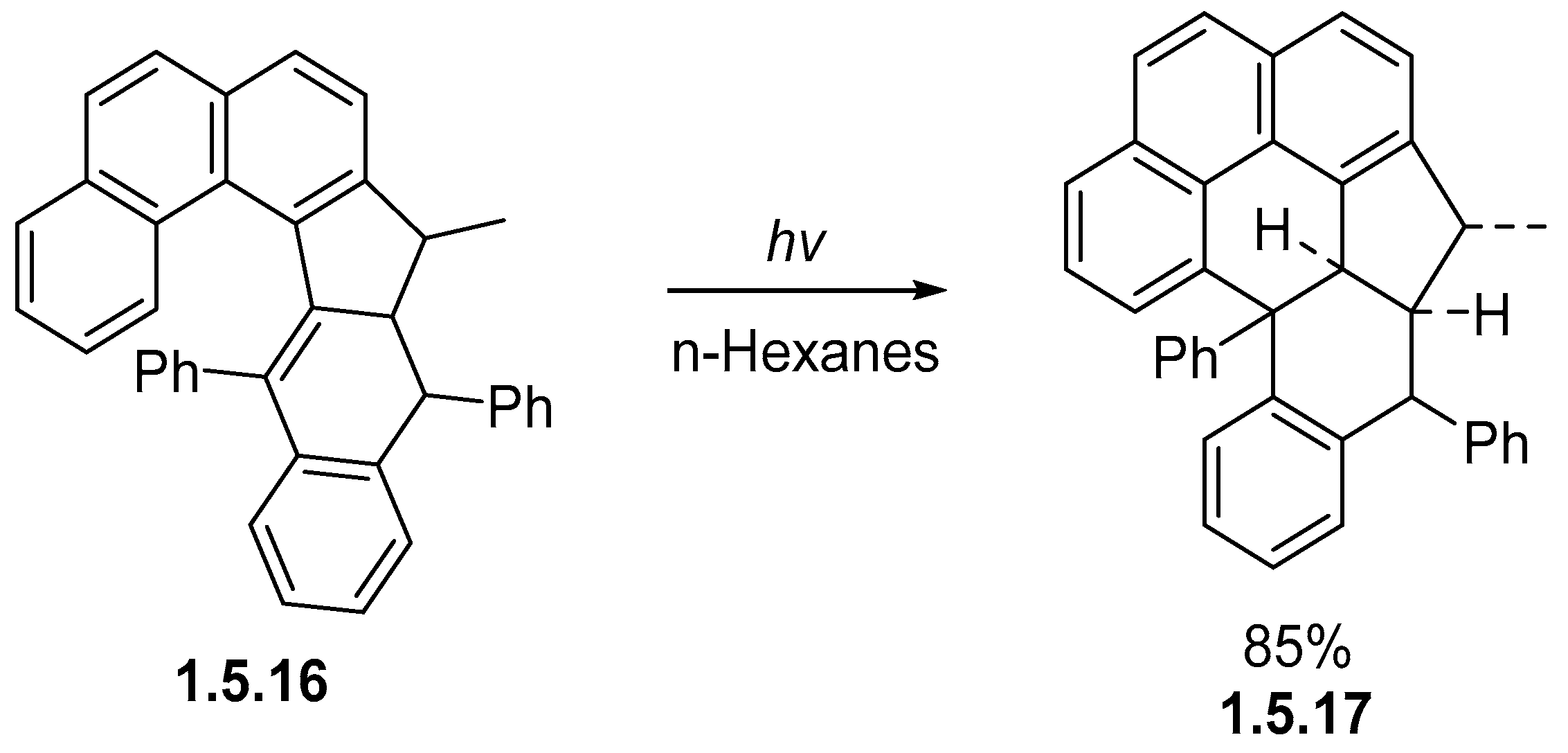
1.5. Alkene Photocyclizations Involving Biphenyl and Phenanthrene Systems
1.6. Heteroaromatic Stilbene Analogs
1.7. Photocyclizations Involving Pyrroles, Indoles, and Other Five-Membered Rings
1.8. Photocyclizations Involving Non-Alkene Linkages
1.9. Photocyclizations Involving Single Heteroatom Linkages
1.10. Additional Photocyclizations Forming Five-Membered Rings
2. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| HMO | Hückel molecular orbital |
| LUMO | Lowest Unoccupied molecular orbital |
| HOMO | Highest occupied molecular orbital |
| PAH | Polyaromatic hydrocarbon |
| DHP | Dihydrophenanthrene |
| UV | Ultra-violet |
| OLED | Organic light emitting diode |
References
- Hu, C.; Mena, J.; Alabugin, I.V. Design Principles of the Use of Alkynes in Radical Cascades. Nat. Rev. Chem. 2023, 7, 405–423. [Google Scholar] [CrossRef]
- Alabugin, I.V.; Gonzalez-Rodriguez, E.; Kawade, R.K.; Stepanov, A.A.; Vasilevsky, S.F. Alkynes as Synthetic Equivalents of Ketones and Aldehydes: A Hidden Entry into Carbonyl Chemistry. Molecules 2019, 24, 1036. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.; Matus, M.H.; Lester William, A.; Dixon, D.A. Heats of Formation of Triplet Ethylene, Ethylidene, and Acetylene. J. Phys. Chem. A 2008, 112, 2082–2087. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, H.E.; Alabugin, I.V. Excited State Energy Distribution and Redistribution and Chemical Reactivity; Mechanistic and Exploratory Organic Photochemistry1,2. J. Am. Chem. Soc. 2000, 122, 952–953. [Google Scholar] [CrossRef]
- Modern Molecular Photochemistry of Organic Molecules. University Science Books. Available online: https://uscibooks.aip.org/books/modern-molecular-photochemistry-of-organic-molecules/ (accessed on 24 January 2025).
- Bonačić-Koutecký, V.; Bruckmann, P.; Hiberty, P.; Koutecký, J.; Leforestier, C.; Salem, L. Sudden Polarization in the Zwitterionic Z1 Excited States of Organic Intermediates. Photochemical Implications. Angew. Chem. Int. Ed. Engl. 1975, 14, 575–576. [Google Scholar] [CrossRef]
- Schuddeboom, W.; Jonker, S.A.; Warman, J.M.; de Haas, M.P.; Vermeulen, M.J.W.; Jager, W.F.; de Lange, B.; Feringa, B.L.; Fessenden, R.W. Sudden Polarization in the Twisted, Phantom State of Tetraphenylethylene Detected by Time-Resolved Microwave Conductivity. J. Am. Chem. Soc. 1993, 115, 3286–3290. [Google Scholar] [CrossRef]
- Baird, N.C. Quantum Organic Photochemistry. II. Resonance and Aromaticity in the Lowest 3.Pi..Pi.* State of Cyclic Hydrocarbons. J. Am. Chem. Soc. 1972, 94, 4941–4948. [Google Scholar] [CrossRef]
- Karas, L.J.; Wu, J.I. Baird’s Rules at the Tipping Point. Nat. Chem. 2022, 14, 723–725. [Google Scholar] [CrossRef]
- Slanina, T.; Ayub, R.; Toldo, J.; Sundell, J.; Rabten, W.; Nicaso, M.; Alabugin, I.; Fdez Galván, I.; Gupta, A.K.; Lindh, R.; et al. Impact of Excited-State Antiaromaticity Relief in a Fundamental Benzene Photoreaction Leading to Substituted Bicyclo [3.1.0]Hexenes. J. Am. Chem. Soc. 2020, 142, 10942–10954. [Google Scholar] [CrossRef]
- Romero, N.A.; Nicewicz, D.A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef]
- Shaw, M.H.; Twilton, J.; MacMillan, D.W.C. Photoredox Catalysis in Organic Chemistry. J. Org. Chem. 2016, 81, 6898–6926. [Google Scholar] [CrossRef] [PubMed]
- Alabugin, I.V.; Eckhardt, P.; Christopher, K.M.; Opatz, T. The Photoredox Paradox: Electron and Hole Upconversion as the Hidden Secrets of Photoredox Catalysis. J. Am. Chem. Soc. 2024, 146, 27233–27254. [Google Scholar] [CrossRef]
- Parthiban, J.; Garg, D.; Sivaguru, J. Photoinduced Transformations with Diverse Maleimide Scaffolds. Molecules 2024, 29, 4895. [Google Scholar] [CrossRef] [PubMed]
- Ottosson, H. Exciting Excited-State Aromaticity. Nat. Chem. 2012, 4, 969–971. [Google Scholar] [CrossRef]
- Karadakov, P.B. Ground- and Excited-State Aromaticity and Antiaromaticity in Benzene and Cyclobutadiene. J. Phys. Chem. A 2008, 112, 7303–7309. [Google Scholar] [CrossRef]
- Karadakov, P.B. Aromaticity and Antiaromaticity in the Low-Lying Electronic States of Cyclooctatetraene. J. Phys. Chem. A 2008, 112, 12707–12713. [Google Scholar] [CrossRef] [PubMed]
- Karadakov, P.B.; Hearnshaw, P.; Horner, K.E. Magnetic Shielding, Aromaticity, Antiaromaticity, and Bonding in the Low-Lying Electronic States of Benzene and Cyclobutadiene. J. Org. Chem. 2016, 81, 11346–11352. [Google Scholar] [CrossRef]
- Karadakov, P.B.; Al-Yassiri, M.A.H.; Cooper, D.L. Magnetic Shielding, Aromaticity, Antiaromaticity and Bonding in the Low-Lying Electronic States of S2N2. Chem. A Eur. J. 2018, 24, 16791–16803. [Google Scholar] [CrossRef]
- Feixas, F.; Vandenbussche, J.; Bultinck, P.; Matito, E.; Solà, M. Electron Delocalization and Aromaticity in Low-Lying Excited States of Archetypal Organic Compounds. Phys. Chem. Chem. Phys. 2011, 13, 20690–20703. [Google Scholar] [CrossRef]
- Rosenberg, M.; Dahlstrand, C.; Kilså, K.; Ottosson, H. Excited State Aromaticity and Antiaromaticity: Opportunities for Photophysical and Photochemical Rationalizations. Chem. Rev. 2014, 114, 5379–5425. [Google Scholar] [CrossRef]
- Oh, J.; Sung, Y.M.; Hong, Y.; Kim, D. Spectroscopic Diagnosis of Excited-State Aromaticity: Capturing Electronic Structures and Conformations upon Aromaticity Reversal. Acc. Chem. Res. 2018, 51, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Brink, M.; Möllerstedt, H.; Ottosson, C.-H. Characteristics of the Electronic Structures of Diabatically and Adiabatically Z/E-Isomerizing Olefins in the T1 State. J. Phys. Chem. A 2001, 105, 4071–4083. [Google Scholar] [CrossRef]
- Mohamed, R.K.; Mondal, S.; Jorner, K.; Delgado, T.F.; Lobodin, V.V.; Ottosson, H.; Alabugin, I.V. The Missing C1–C5 Cycloaromatization Reaction: Triplet State Antiaromaticity Relief and Self-Terminating Photorelease of Formaldehyde for Synthesis of Fulvenes from Enynes. J. Am. Chem. Soc. 2015, 137, 15441–15450. [Google Scholar] [CrossRef]
- Poplata, S.; Tröster, A.; Zou, Y.-Q.; Bach, T. Recent Advances in the Synthesis of Cyclobutanes by Olefin [2 + 2] Photocycloaddition Reactions. Chem. Rev. 2016, 116, 9748–9815. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Brown, M.K. Photosensitized [2 + 2]-Cycloadditions of Dioxaborole: Reactivity Enabled by Boron Ring Constraint Strategy. J. Am. Chem. Soc. 2023, 145, 25061–25067. [Google Scholar] [CrossRef]
- Evenzahav, A.; Turro, N.J. Photochemical Rearrangement of Enediynes: Is a “Photo-Bergman” Cyclization a Possibility? J. Am. Chem. Soc. 1998, 120, 1835–1841. [Google Scholar] [CrossRef]
- Lewis, K.D.; Matzger, A.J. Bergman Cyclization of Sterically Hindered Substrates and Observation of Phenyl-Shifted Products. J. Am. Chem. Soc. 2005, 127, 9968–9969. [Google Scholar] [CrossRef]
- Bhattacharya, P.; Basak, A.; Campbell, A.; Alabugin, I.V. Photochemical Activation of Enediyne Warheads: A Potential Tool for Targeted Antitumor Therapy. Mol. Pharm. 2018, 15, 768–797. [Google Scholar] [CrossRef]
- Jones, G.B.; Wright, J.M.; Plourde, G.; Purohit, A.D.; Wyatt, J.K.; Hynd, G.; Fouad, F. Synthesis and Photochemical Activity of Designed Enediynes. J. Am. Chem. Soc. 2000, 122, 9872–9873. [Google Scholar] [CrossRef]
- Meier, H. The Photochemistry of Stilbenoid Compounds and Their Role in Materials Technology. Angew. Chem. Int. Ed. Engl. 1992, 31, 1399–1420. [Google Scholar] [CrossRef]
- Smakula, A. The Photochemical Transformation of Trans-Stilbene. Z. Phys. Chem. 1934, B 25, 90–98. [Google Scholar] [CrossRef]
- Seylar, J.; Stasiouk, D.; Simone, D.L.; Varshney, V.; Heckler, J.E.; McKenzie, R. Breaking the Bottleneck: Stilbene as a Model Compound for Optimizing 6π e − Photocyclization Efficiency. RSC Adv. 2021, 11, 6504–6508. [Google Scholar] [CrossRef] [PubMed]
- Sudhakar, A.; Katz, T.J. Directive Effect of Bromine on Stilbene Photocyclizations. an Improved Synthesis of [7]Helicene. Tetrahedron Lett. 1986, 27, 2231–2234. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Cheprakov, A.V. The Heck Reaction as a Sharpening Stone of Palladium Catalysis. Chem. Rev. 2000, 100, 3009–3066. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, K.B. Photochemical Oxidative Cyclisation of Stilbenes and Stilbenoids—The Mallory-Reaction. Molecules 2010, 15, 4334–4358. [Google Scholar] [CrossRef]
- Mallory, F.B.; Wood, C.S.; Gordon, J.T. Photochemistry of Stilbenes. III. Some Aspects of the Mechanism of Photocyclization to Phenanthrenes. J. Am. Chem. Soc. 1964, 86, 3094–3102. [Google Scholar] [CrossRef]
- Wood, C.S.; Mallory, F.B. Photochemistry of Stilbenes. IV. The Preparation of Substituted Phenanthrenes1a–c. J. Org. Chem. 1964, 29, 3373–3377. [Google Scholar] [CrossRef]
- Hulley, E.B.; Clennan, E.L. Dihydrophenanthrene Open-Shell Singlet Diradicals and Their Roles in the Mallory Photocyclization Reaction. J. Am. Chem. Soc. 2024, 146, 1122–1131. [Google Scholar] [CrossRef]
- Courtney, S.H.; Fleming, G.R. Photoisomerization of Stilbene in Low Viscosity Solvents: Comparison of Isolated and Solvated Molecules. J. Chem. Phys. 1985, 83, 215–222. [Google Scholar] [CrossRef]
- Saltiel, J.; Gupta, S. Photochemistry of the Stilbenes in Methanol. Trapping the Common Phantom Singlet State. J. Phys. Chem. A 2018, 122, 6089–6099. [Google Scholar] [CrossRef]
- Minezawa, N.; Gordon, M.S. Photoisomerization of Stilbene: A Spin-Flip Density Functional Theory Approach. J. Phys. Chem. A 2011, 115, 7901–7911. [Google Scholar] [CrossRef] [PubMed]
- Sauer, P.; Allen, R.E. Dynamics of the Photoinduced Ring-Opening of Stilbene, a Prototypical Diarylethene. Chem. Phys. Lett. 2007, 434, 260–264. [Google Scholar] [CrossRef]
- Weber, J.; Clennan, E.L. Origin of the Preferential Formation of Helicenes in Mallory Photocyclizations. Temperature as a Tool to Influence Reaction Regiochemistry. J. Org. Chem. 2019, 84, 817–830. [Google Scholar] [CrossRef]
- Scholz, M.; Mühlstädt, M.; Dietz, F. Chemie Angeregter Zustände. I. Mitt. Die Richtung Der Photocyclisierung Naphthalinsubstituierter Äthylene. Tetrahedron Lett. 1967, 8, 665–668. [Google Scholar] [CrossRef]
- Mayer, I. Improved Definition of Bond Orders for Correlated Wave Functions. Chem. Phys. Lett. 2012, 544, 83–86. [Google Scholar] [CrossRef]
- Cooper, D.L.; Ponec, R.; Karadakov, P.B. Investigating István Mayer’s “Improved” Definitions of Bond Orders and Free Valence for Correlated Singlet-State Wave Functions. Int. J. Quantum Chem. 2022, 122, e26612. [Google Scholar] [CrossRef]
- Laarhoven, W.H. Photochemical Cyclizations and Intramolecular Cycloadditions of Conjugated Arylolefins. Part I: Photocyclization with Dehydrogenation. Recl. Trav. Chim. Pays-Bas 1983, 102, 185–204. [Google Scholar] [CrossRef]
- Krongauz, V.A. Reactivity of Organic Compounds in an Excited Electronic State. Russ. Chem. Rev. 1972, 41, 439. [Google Scholar] [CrossRef]
- Bromberg, A.; Muszkat, K.A.; Fischer, E. Photocyclisation and Photocyclodehydrogenation of Stilbene and Related Compounds. Isr. J. Chem. 1972, 10, 765–773. [Google Scholar] [CrossRef]
- Lantos, I. A New Photochemically Based Synthesis of Phenanthrene-9,10-Quinones. Tetrahedron Lett. 1978, 19, 2761–2762. [Google Scholar] [CrossRef]
- Mallory, F.B.; Mallory, C.W. Photocyclization of Stilbenes and Related Molecules. In Organic Reactions; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2005; pp. 1–456. [Google Scholar] [CrossRef]
- Rio, G.; Hardy, J.C. 3,4-Triphenyl, (5 H), Furan-2-One, Cis-3-Benzoyl-2,3-Diphenyl Acrylic Acid and Related Compounds—Photochemical Cyclisation into Phenanthrene and 9,10-Dihydrophenanthrene Derivatives—Photochromy. J. Bull. Soc. Chim. Fr. 1970, 10, 3578. [Google Scholar]
- Horspool, W.M. Photolysis of Some Cyclopentadioxinones. J. Chem. Soc. C 1971, 400–404. [Google Scholar] [CrossRef]
- Horspool, W.M.; Khalaf, A.I. Substituent and Wavelength Effects in the Photochemistry of 5,6,7,8-Tetrachloro-3a,9a-Dihydro-2,3,9a-Triarylfuro(2,3-b)(1,4)Benzodioxin Derivatives. Tetrahedron Lett. 1983, 24, 3745–3748. [Google Scholar] [CrossRef]
- Martin, K.; Melan, C.; Cauchy, T.; Avarvari, N. Reactivity and Mechanistic Issues in the Photocyclization of Dihalostyryl-Naphthalenes towards Halo-[4]Helicenes: A Transposition on a Mallory Theme. ChemPhotoChem 2022, 6, e202100215. [Google Scholar] [CrossRef]
- Cava, M.P.; Stern, P.; Wakisaka, K. An Improved Photochemical Aporphine Synthesis: New Syntheses of Dicentrine and Cassameridine. Tetrahedron 1973, 29, 2245–2249. [Google Scholar] [CrossRef]
- Daigle, M.; Picard-Lafond, A.; Soligo, E.; Morin, J.-F. Regioselective Synthesis of Nanographenes by Photochemical Cyclodehydrochlorination. Angew. Chem. Int. Ed. 2016, 55, 2042–2047. [Google Scholar] [CrossRef]
- Kupchan, S.M.; Moniot, J.L.; Kanojia, R.M.; O’Brien, J.B. Photochemical Synthesis of Aporphines. J. Org. Chem. 1971, 36, 2413–2418. [Google Scholar] [CrossRef]
- Kawade, R.K.; Hu, C.; Dos Santos, N.R.; Watson, N.; Lin, X.; Hanson, K.; Alabugin, I.V. Phenalenannulations: Three-Point Double Annulation Reactions That Convert Benzenes into Pyrenes. Angew. Chem. Int. Ed. 2020, 59, 14352–14357. [Google Scholar] [CrossRef]
- Kupchan, S.M.; Wormser, H.C. Tumor Inhibitors. X.1 Photochemical Synthesis of Phenanthrenes. Synthesis of Aristolochic Acid and Related Compounds2-4. J. Org. Chem. 1965, 30, 3792–3800. [Google Scholar] [CrossRef]
- Laarhoven, W.H.; Cuppen, T.J.H.M. Photodehydrocyclisations of Stilbene-like Compounds XVI: Photoreactions of α-(9-Phenanthryl)Stilbene and 1-(9-Phenanthryl)-1-Phenylethylene. Recl. Trav. Chim. Pays-Bas 1976, 95, 165–168. [Google Scholar] [CrossRef]
- Blum, J.; Grauer, F.; Bergmann, E.D. The Synthesis of 12-Fluoro-7-Methyl- and 7-Fluoro-Benz[a]Anthracene. Tetrahedron 1969, 25, 3501–3507. [Google Scholar] [CrossRef]
- Thulin, B.; Wennerström, O. Propellicene or Bi-2,13-pentahelicenylene. Acta Chem. Scand. B 1976, 30, 688–690. [Google Scholar] [CrossRef]
- Marx, G.S.; Bergmann, E.D. Synthesis of 5- and 6-Fluorobenzo[c]Phenanthrene by Photocyclization. J. Org. Chem. 1972, 37, 1807–1810. [Google Scholar] [CrossRef]
- Sargent, M.V.; Timmons, C.J. 1063. Studies in Photochemistry. Part I. The Stilbenes. J. Chem. Soc. 1964, 5544–5552. [Google Scholar] [CrossRef]
- Wismontski-Knittel, T.; Kaganowitch, M.; Seger, G.; Fischer, E. The Reversible Photochemistry of 1,2-Di-(2-Naphthyl)Cyclopentene. Recl. Trav. Chim. Pays-Bas 1979, 98, 114–117. [Google Scholar] [CrossRef]
- Op het Veld, P.H.G.; Laarhoven, W.H. The Mechanism of the Photoconversion of .Alpha.-Phenylcinnamic Esters into 9,10-Dihydrophenanthrenes. J. Am. Chem. Soc. 1977, 99, 7221–7224. [Google Scholar] [CrossRef]
- Mallory, F.B.; Mallory, C.W. Photochemistry of Stilbenes. VI. Steric Effects on the Photocyclizations of Some m-Substituted Stilbenes. J. Am. Chem. Soc. 1972, 94, 6041–6048. [Google Scholar] [CrossRef]
- Yang, N.C.; Lenz, G.R.; Shani, A. Photochemical Synthesis of Dehydroaporphanes. Tetrahedron Lett. 1966, 7, 2941–2946. [Google Scholar] [CrossRef]
- Cava, M.P.; Mitchell, M.J.; Havlicek, S.C.; Lindert, A.; Spangler, R.J. Photochemical Routes to Aporphines. New Syntheses of Nuciferine and Glaucine. J. Org. Chem. 1970, 35, 175–179. [Google Scholar] [CrossRef]
- Kende, A.S.; Curran, D.P. Regiochemical Control in Dihydrophenanthrene Synthesis. A Photochemical Total Synthesis of Juncusol. J. Am. Chem. Soc. 1979, 101, 1857–1864. [Google Scholar] [CrossRef]
- Dey, A.S.; Neumeyer, J.L. Synthesis and Antimalarial Evaluation of 9,10-Dihydrophenanthrene Amino Alcohols. J. Med. Chem. 1974, 17, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Hakimelahi, G.; Boyce, C.; Kasmai, H. Photolytic Closure of 4,5-Diphenyl-Oxazol-2-Ones to Phenanthro [9,10-d]Oxazol-2-Ones and an Improved Synthesis of Benzoins. Helv. Chim. Acta 1977, 60, 342. [Google Scholar] [CrossRef]
- Buquet, A.; Couture, A.; Lablache-Combier, A. Photocyclization Reactions in Primary Amines. Convenient Synthesis of 1,4-Dihydrophenanthrene. J. Org. Chem. 1979, 44, 2300–2303. [Google Scholar] [CrossRef]
- Laarhoven, W.H.; Cuppen, T.J.H.M.; Nivard, R.J.F. Photodehydrocyclizations in Stilbene-like Compounds—II: Photochemistry of Distyrylbenzenes. Tetrahedron 1970, 26, 1069–1083. [Google Scholar] [CrossRef]
- Moradpour, A.; Kagan, H.; Baes, M.; Morren, G.; Martin, R.H. Photochemistry with Circularly Polarized Light—III: Synthesis of Helicenes Using Bis(Arylvinyl) Arenes as Precursors. Tetrahedron 1975, 31, 2139–2143. [Google Scholar] [CrossRef]
- Kagan, H.; Moradpour, A.; Nicoud, J.F.; Balavoine, G.; Martin, R.H.; Cosyn, J.P. Photochemistry with Circularly Polarised Light. II) Asymmetric Synthesis of Octa and Nonahelicene. Tetrahedron Lett. 1971, 12, 2479–2482. [Google Scholar] [CrossRef]
- Tinnemans, A.H.A.; Laarhoven, W.H. Photodehydrocyclizations in Stilbene-like Compounds. IX. 1,2-Phenyl Shifts in the Cyclization of 1-Phenylpentahelicenes. J. Am. Chem. Soc. 1974, 96, 4611–4616. [Google Scholar] [CrossRef]
- Korenstein, R.; Muszkat, K.A.; Fischer, E. The Irreversible Photochemistry of Bianthrone. Helv. Chim. Acta 1976, 59, 1826–1831. [Google Scholar] [CrossRef]
- Hayward, R.J.; Leznoff, C.C. Photocyclization Reactions of Aryl Polyenes—III: The Photocyclization of 1,8-Diphenyl-1,3,5,7-Octatetraene and 1,10-Diphenyl-1,3,5,7,9-Decapentaene. Tetrahedron 1971, 27, 5115–5120. [Google Scholar] [CrossRef]
- Darcy, P.J.; Hart, R.J.; Heller, H.G. Overcrowded Molecules. Part 14. Photochromic Systems Involving (Z)-1-Methylpropylidene(Diphenylmethylene)Succinic and (E)-3,5-Dimethoxybenzylidene(Alkyl-Substituted Methylene)Succinic Anhydrides. J. Chem. Soc. Perkin Trans. 1 1978, 571–576. [Google Scholar] [CrossRef]
- Onda, M.; Yonezawa, K.; Abe, K. Utilization of Protopine and Related Alkaloids. IV. Transformation of Protopine and Protoberberine Alkaloid to Benzo [c] Phenan-Thridine Alkaloid. Chem. Pharm. Bull. 1971, 19, 31–36. [Google Scholar] [CrossRef]
- Tinnemans, A.H.A.; Laarhoven, W.H. Photocyclisations of 1,4-Diarylbut-1-En-3-Ynes. Part III. Scope and Limitations of the Reaction. J. Chem. Soc. Perkin Trans. 2 1976, 1115–1120. [Google Scholar] [CrossRef]
- Zakharov, A.V.; Yadykov, A.V.; Lvov, A.G.; Mitina, E.A.; Shirinian, V.Z. Photochemical Rearrangement of Diarylethenes: Synthesis of Functionalized Phenanthrenes. Org. Biomol. Chem. 2020, 18, 3098–3103. [Google Scholar] [CrossRef] [PubMed]
- Zakharov, A.V.; Timofeeva, S.M.; Yadykov, A.V.; Krayushkin, M.M.; Shirinian, V.Z. Skeletal Photoinduced Rearrangement of Diarylethenes: Ethene Bridge Effects. Org. Biomol. Chem. 2023, 21, 2015–2023. [Google Scholar] [CrossRef] [PubMed]
- Yadykov, A.V.; Scherbakov, A.M.; Trofimova, V.V.; Lvov, A.G.; Markosyan, A.I.; Zavarzin, I.V.; Shirinian, V.Z. Photoswitching off the Antiproliferative Activity of Combretastatin A-4 Analogues. Org. Lett. 2019, 21, 9608–9612. [Google Scholar] [CrossRef]
- op den Brouw, P.M.; de Zeeuw, P.; Laarhoven, W.H. Photochemistry of 2-Vinylstilbenes: Importance of the Ground State Conformation for the Type of Photoreaction. J. Photochem. 1984, 27, 327–341. [Google Scholar] [CrossRef]
- Sajimon, M.C.; Lewis, F.D. Photocyclization of 2-Vinyldiphenylacetylenes and Behavior of the Isonaphthalene Intermediates. Photochem. Photobiol. Sci. 2005, 4, 629–636. [Google Scholar] [CrossRef]
- Fukazawa, A.; Karasawa, T.; Zhang, H.; Minemura, K.; Camacho, C.; Wang, J.; Irle, S.; Yamaguchi, S. Photochemical Double 5-Exo Cyclization of Alkenyl-Substituted Dithienylacetylenes: Efficient Synthesis of Diarylated Dithienofulvalenes. Angew. Chem. Int. Ed. 2013, 52, 10519–10523. [Google Scholar] [CrossRef]
- op den Brouw, P.M.; Laarhoven, W.H. The Photochemistry of 2-Vinyldiphenylacetylene and Related Compounds. J. Chem. Soc. Perkin Trans. 2 1982, 795–799. [Google Scholar] [CrossRef]
- Campbell, A.; Dos Santos, N.R.; Alabugin, I. Photochemical Uncaging of Aldehydes and Ketones via Photocyclization/Fragmentation Cascades of Enyne Alcohols: An Unusual Application for a Cycloaromatization Process. Molecules 2023, 28, 5704. [Google Scholar] [CrossRef]
- Alabugin, A. Near-IR Photochemistry for Biology: Exploiting the Optical Window of Tissue. Photochem. Photobiol. 2019, 95, 722–732. [Google Scholar] [CrossRef] [PubMed]
- van Arendonk, R.J.F.M.; Laarhoven, W.H. The Formation of a Cyclopropapyrone in the Photocyclization of 1-(9-Phenanthryl)-4-Phenylbut-1-En-3-Yne. Tetrahedron Lett. 1977, 18, 2629–2630. [Google Scholar] [CrossRef]
- Laarhoven, W.H.; Cuppen, T.J.H.M. Photodehydrocyclizations in Stilbene-like Compounds. Part V. Photochemistry of 2,2′-Distyrylbiphenyl. J. Chem. Soc. Perkin Trans. 1 1972, 2074–2079. [Google Scholar] [CrossRef]
- Padwa, A.; Doubleday, C.; Mazzu, A. Photocyclization Reactions of Substituted 2,2’-Divinylbiphenyl Derivatives. J. Org. Chem. 1977, 42, 3271–3279. [Google Scholar] [CrossRef]
- Koussini, R.; Lapouyade, R.; Fornier de Violet, P. Intramolecular Photocyclization of 2-Vinylbiphenyl-like Compounds. 1. A Quantitative Study of the Cyclization under Steady-State Illumination. J. Am. Chem. Soc. 1978, 100, 6679–6683. [Google Scholar] [CrossRef]
- Lapouyade, R.; Koussini, R.; Nourmamode, A.; Courseille, C. Divergent Stereochemistry of Photocyclization from Singlet and Triplet States of 2-Vinylbiphenyls. X-Ray Crystal Structure of Cis-9-Phenyl-10-Methyl-9,10-Dihydrophenanthrene. J. Chem. Soc. Chem. Commun. 1980, 740–742. [Google Scholar] [CrossRef]
- Fornier de Violet, P.; Lapouyade, R.; Rayez, J.-C. The Effect of Changing the Electronic Configurations of the Excited States on the Rate Constant of Photocyclization of Aryl Ethylenes. J. Photochem. 1982, 19, 253–266. [Google Scholar] [CrossRef]
- Mitchell, R.H.; Boekelheide, V. Transformation of Sulfide Linkages to Carbon-Carbon Double Bond. Syntheses of Cis- and Trans-15,16-Dimethyldihydropyrene and Trans-15,16-Dihydropyrene. J. Am. Chem. Soc. 1974, 96, 1547–1557. [Google Scholar] [CrossRef]
- Ayub, K.; Mitchell, R.H. Syntheses of Dihydropyrene–Cyclophanediene Negative Photochromes Containing Internal Alkenyl and Alkynyl Groups and Comparison of Their Photochemical and Thermochemical Properties. J. Org. Chem. 2014, 79, 664–678. [Google Scholar] [CrossRef]
- Heller, H.G.; Salisbury, K. Overcrowded Molecules. Part VI. Photocyclisation of Cis-9a,10-Dihydro-9-Methyl-10,15-Diphenyl-9H-Benzo [5,6]Indeno [2,1-c] Phenanthrene. J. Chem. Soc. C 1970, 1997–2000. [Google Scholar] [CrossRef]
- Vögtle, F.; Neumann, P. Stereochemistry of [2.2]Metacyclophanes. Angew. Chem. Int. Ed. Engl. 1972, 11, 73–83. [Google Scholar] [CrossRef]
- Kawano, S.; Yang, C.; Ribas, M.; Baluschev, S.; Baumgarten, M.; Müllen, K. Blue-Emitting Poly(2,7-Pyrenylene)s: Synthesis and Optical Properties. Macromolecules 2008, 41, 7933–7937. [Google Scholar] [CrossRef]
- Allinger, N.L.; Gorden, B.J.; Hu, S.-E.; Ford, R.A. Some Chemistry of Compounds Related to [2.2]Metacyclophane. J. Org. Chem. 1967, 32, 2272–2278. [Google Scholar] [CrossRef]
- Umemoto, T.; Kawashima, T.; Sakata, Y.; Misumi, S. Layered Compounds. XXV. Peropyrene and Teropyrene -a New Synthetic Route of Pyrene-like Polynuclear Aromatic Hydrocarbons. Tetrahedron Lett. 1975, 16, 1005–1006. [Google Scholar] [CrossRef]
- Yao, T.; Yu, H.; Vermeij, R.J.; Bodwell, G.J. Nonplanar aromatic compounds. Part 10: A strategy for the synthesis of aromatic belts-all wrapped up or down the tubes? Pure Appl. Chem. 2008, 80, 533–546. [Google Scholar] [CrossRef]
- Laarhoven, W.H.; Cuppen, T.J.H.M. Chiral Solvent-Induced Asymmetric Synthesis. Part 2. Photosynthesis of Optically Enriched Hexahelicenes. J. Chem. Soc. Perkin Trans. 2 1978, 315–318. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Zhang, C. Intramolecular Cyclization of 2,2’-Dibenzoylbiphenyl Units as a New Route to Increase the Rigidity and Solvent Resistance in Poly(Arylene Ethers). Macromolecules 1992, 25, 5851–5854. [Google Scholar] [CrossRef]
- Walker, D.B.; Howgego, J.; Davis, A.P. Synthesis of Regioselectively Functionalized Pyrenes via Transition-Metal-Catalyzed Electrocyclization. Synthesis 2010, 2010, 3686–3692. [Google Scholar] [CrossRef]
- Gonzalez-Rodriguez, E.; Abdo, M.A.; dos Passos Gomes, G.; Ayad, S.; White, F.D.; Tsvetkov, N.P.; Hanson, K.; Alabugin, I.V. Twofold π-Extension of Polyarenes via Double and Triple Radical Alkyne Peri-Annulations: Radical Cascades Converging on the Same Aromatic Core. J. Am. Chem. Soc. 2020, 142, 8352–8366. [Google Scholar] [CrossRef]
- Wu, X.; Shi, G. Synthesis of a Carboxyl-Containing Conducting Oligomer and Non-Covalent Sidewall Functionalization of Single-Walled Carbon Nanotubes. J. Mater. Chem. 2005, 15, 1833. [Google Scholar] [CrossRef]
- Morgan, D.D.; Horgan, S.W.; Orchin, M. Photocyclization of Stilbene Analogs I. The Oxidative Photocyclization of 1,3-Distyrylbenzene. Tetrahedron Lett. 1970, 11, 4347–4350. [Google Scholar] [CrossRef]
- Dos Santos, N.R.; Schober, J.V.; Laconsay, C.J.; Palazzo, A.M.; Kuhn, L.; Chu, A.; Hanks, B.; Hanson, K.; Wu, J.; Alabugin, I.V. Assembly of Pyrenes through a Quadruple Photochemical Cascade: Blocking Groups Allow Diversion from the Double Mallory Path to Photocyclization at the Bay Region. J. Am. Chem. Soc. 2025, 147, 1074–1091. [Google Scholar] [CrossRef]
- Kumler, P.L.; Dybas, R.A. Photochemical Cyclizations. II. Effect of Structural Features on the Photocyclization of 2-Stilbazole Derivatives. J. Org. Chem. 1970, 35, 3825–3831. [Google Scholar] [CrossRef]
- Loader, C.E.; Timmons, C.J. Studies in Photochemistry. Part II. The Photocyclisation of Stilbazoles to Azaphenanthrenes. J. Chem. Soc. C 1966, 1078–1081. [Google Scholar] [CrossRef]
- Galiazzo, G.; Bortolus, P.; Cauzzo, G. Two-Ways Photocyclization of 3-Styrylpyridine. Tetrahedron Lett. 1966, 7, 3717–3721. [Google Scholar] [CrossRef]
- Bortolus, P.; Cauzzo, G.; Mazzucato, U.; Galiazzo, G. Cis-Trans Photoisomerization of Styrylpyridines. Z. Phys. Chem. Neue Folge 1966, 51, 264–273. [Google Scholar] [CrossRef]
- Muzkat, K.A.; Sharafi-Ozeri, S. Electronic Overlap Population as Measure of Reactivity in Electrocyclic Reactions, Stilbenes and Analogs. Chem. Phys. Lett. 1973, 20, 397–400. [Google Scholar] [CrossRef]
- Doolittle, R.E.; Bradsher, C.K. Photochemical Methods in the Synthesis of Heterocyclic Compounds. I. Phenanthridizinium Perchlorates1. J. Org. Chem. 1966, 31, 2616–2618. [Google Scholar] [CrossRef]
- Mariano, P.S.; Krochmal, E., Jr.; Leone, A. Stilbenelike Photocyclizations of 1-Phenylvinyl-2-Pyridinones. Preparation of 4H-Benzo[a]Quinolizin-4-Ones. J. Org. Chem. 1977, 42, 1122–1125. [Google Scholar] [CrossRef]
- Perkampus, H.-H.; Bluhm, T. Zur Photochemie Der Styryldiazine: Darstellung Der Edukte Und Produkte. Tetrahedron 1972, 28, 2099–2110. [Google Scholar] [CrossRef]
- Perkampus, H.-H.; Bluhm, T.; Knop, J.V. Die Elektronenanregungsspektren Der Diazaphenanthrene. Spectrochim. Acta Part A Mol. Spectrosc. 1972, 28, 2179–2187. [Google Scholar] [CrossRef]
- Mudry, C.A.; Frasca, A.R. Photochemical Reactions of 2,3-Diphenylindoles. Tetrahedron 1974, 30, 2983–2991. [Google Scholar] [CrossRef]
- Cooper, J.L.; Wasserman, H.H. Photolysis of 4,5-Diphenyl-Oxazoles, -Thiazoles, and -Imidazoles. Formation of Phenanthro [9,10-d]-Heterocyclic Systems. J. Chem. Soc. D 1969, 200–201. [Google Scholar] [CrossRef]
- Grimshaw, J.; Mannus, D. Photocyclisation and Photoisomerisation of 1,3,4- and 1,4,5-Triphenylpyrazole. J. Chem. Soc. Perkin Trans. 1 1977, 2096–2099. [Google Scholar] [CrossRef]
- Jerchel, D.; Fischer, H. 2.3-Azadiphenylen-tetrazoliumsalze und Triazaphenanthrene. Chem. Berichte 1956, 89, 563–570. [Google Scholar] [CrossRef]
- Neugebauer, F.A. Substituted 5-T-Butyl Tetrazolinyl and Phototetrazolinyl Radicals. Tetrahedron 1970, 26, 4843–4851. [Google Scholar] [CrossRef]
- Bolotova, I.A.; Lvov, A.G. Oxidative Photocyclization of 4,5-Diphenylimidazoles: A Commentary to the Paper “Invisible Photochromism and on–off Switch Based on Thiophene-Imidazole Fused Derivatives: Synthesis, Properties and Applications”. J. Photochem. Photobiol. A Chem. 2024, 456, 115855. [Google Scholar] [CrossRef]
- Silva, O.D.; Snieckus, V. Photochemical Synthesis of Benzo[c]Carbazole and Pyridocarbazoles. Synthesis 2002, 1971, 254–255. [Google Scholar] [CrossRef]
- Silva, S.O.d.; Snieckus, V. The Wittig Synthesis and Photocyclodehydrogenation of 3-(2-Aryl)Vinylindole Derivatives. Can. J. Chem. 1974, 52, 1294–1306. [Google Scholar] [CrossRef]
- Fedorova, O.A.; Fedorov, Y.V.; Andryukhina, E.N.; Gromov, S.P.; Alfimov, M.V.; Lapouyade, R. Photochemical Electrocyclization of the Indolinylphenylethenes Involving a C−N Bond Formation. Org. Lett. 2003, 5, 4533–4535. [Google Scholar] [CrossRef]
- Loader, C.E.; Timmons, C.J. Studies in Photochemistry. Part V. The Photocyclodehydrogenation of 2-Furyl- and 2-Thienyl-Ethylenes: The Mass Spectra of the Products. J. Chem. Soc. C 1967, 1677–1681. [Google Scholar] [CrossRef]
- Padwa, A.; Hartman, R. Ground-State and Photochemical Reactions in the Epoxypyrone Series1–3. J. Am. Chem. Soc. 1966, 88, 3759–3765. [Google Scholar] [CrossRef]
- Couture, A.; Lablache-Combier, A.; Ofenberg, H. Reductive Photocyclization of Benzo[b]Furans and Their Analogs. Tetrahedron 1975, 31, 2023–2028. [Google Scholar] [CrossRef]
- Couture, A.; Delevallee, A.; Lablache-Combier, A.; Párkányl, C. Etude Comparative des Photoréactions de Thiophènes et de Furannes Avec la Propylamine. Tetrahedron 1975, 31, 785–796. [Google Scholar] [CrossRef]
- Wynberg, H. Some Observations on the Chemical, Photochemical, and Spectral Properties of Thiophenes. Acc. Chem. Res. 1971, 4, 65–73. [Google Scholar] [CrossRef]
- Lehman, P.G.; Wynberg, H. The Synthesis of a Series of Regularly Annelated 2-Methylheterohelicenes. Aust. J. Chem. 1974, 27, 315–322. [Google Scholar] [CrossRef]
- Lucas, L.N.; Esch, J.v.; Kellogg, R.M.; Feringa, B.L. A New Class of Photochromic 1,2-Diarylethenes; Synthesis and Switching Properties of Bis(3-Thienyl)Cyclopentenes. Chem. Commun. 1998, 2313–2314. [Google Scholar] [CrossRef]
- Lucas, L.N.; Esch, J.v.; Kellogg, R.M.; Feringa, B.L. Photocontrolled Self-Assembly of Molecular Switches. Chem. Commun. 2001, 759–760. [Google Scholar] [CrossRef]
- Norsten, T.B.; Branda, N.R. Photoregulation of Fluorescence in a Porphyrinic Dithienylethene Photochrome. J. Am. Chem. Soc. 2001, 123, 1784–1785. [Google Scholar] [CrossRef]
- Irie, M. Diarylethenes for Memories and Switches. Chem. Rev. 2000, 100, 1685–1716. [Google Scholar] [CrossRef]
- Cava, M.P.; Schlessinger, R.H. The Synthesis of a Phenanthridine from an Aromatic Schiff Base by a Photooxidative Ring Closure. Tetrahedron Lett. 1964, 5, 2109–2111. [Google Scholar] [CrossRef]
- Ahmed, M.; Kricka, L.J.; Vernon, J.M. Autoxidation of Polysubstituted Isoindoles. Part II. Products from 1,3-Diphenyl- and 1,2,3-Triphenyl-Isoindoles. J. Chem. Soc. Perkin Trans. 1 1975, 71–75. [Google Scholar] [CrossRef]
- Prabhakar, S.; Lobo, A.M.; Tavares, M.R. Boron Complexes as Control Synthons in Photocyclisations: An Improved Phenanthridine Synthesis. J. Chem. Soc. Chem. Commun. 1978, 884–885. [Google Scholar] [CrossRef]
- Kos, M.; Žádný, J.; Storch, J.; Církva, V.; Cuřínová, P.; Sýkora, J.; Císařová, I.; Kuriakose, F.; Alabugin, I.V. Oxidative Photocyclization of Aromatic Schiff Bases in Synthesis of Phenanthridines and Other Aza-PAHs. Int. J. Mol. Sci. 2020, 21, 5868. [Google Scholar] [CrossRef] [PubMed]
- Glogowski, M.E.; Grisdale, P.J.; Williams, J.L.R.; Regan, T.H. Boron Photochemistry: X. Anilinodiaryl Boranes: Their Synthesis and Photochemistry. J. Organomet. Chem. 1973, 54, 51–60. [Google Scholar] [CrossRef]
- Glogowski, M.E.; Grisdale, P.J.; Williams, J.L.R.; Costa, L. Boron Photochemistry: XI. A Study of the Unusual Luminescence and Spectral Properties of Anilinodimesitylboranes. J. Organomet. Chem. 1974, 74, 175–183. [Google Scholar] [CrossRef]
- Cava, M.P.; Havlicek, S.C. A Novel Photochemical Cyclization Reaction. Tetrahedron Lett. 1967, 8, 2625–2627. [Google Scholar] [CrossRef]
- Tada, M.; Saiki, H.; Miura, K.; Shinozaki, H. The Photoreaction of Benzyl 1-Cycloalkenyl Ketone in Acidic or Protic Media. Bull. Chem. Soc. Jpn. 1976, 49, 1666–1670. [Google Scholar] [CrossRef]
- Yang, N.C.; Lin, L.C.; Shani, A.; Yang, S.S. Photoacylation. II. Intramolecular Photoacylation of Enolates. J. Org. Chem. 1969, 34, 1845–1848. [Google Scholar] [CrossRef]
- Kanaoka, Y.; Itoh, K. Photocyclization of Benzanilides to Phenanthridones with Elimination of the Ortho-Methoxy-Group. J. Chem. Soc. Chem. Commun. 1973, 647–648. [Google Scholar] [CrossRef]
- Kanaoka, Y.; San-nohe, K. Photocyclization of Benzofurancarboxanilides. Photorearrangement of the Benzodihydrofuran System to 3-o-Hydroxyphenylquinolones. Tetrahedron Lett. 1980, 21, 3893–3896. [Google Scholar] [CrossRef]
- Ninomiya, I.; Naito, T.; Kiguchi, T. Photocyclisation of Enamides. Part III. Photocyclisation of N-Benzoylenamines of Cyclohexanone and 2-Substituted Cyclohexanones. J. Chem. Soc. Perkin Trans. 1 1973, 2257–2261. [Google Scholar] [CrossRef]
- Ninomiya, I.; Kiguchi, T.; Yamamoto, O.; Naito, T. Photocyclisation of Enamides. Part 13. Substituent Effects in the Photocyclisation of N-Benzoylenamines. J. Chem. Soc. Perkin Trans. 1 1979, 1723–1728. [Google Scholar] [CrossRef]
- Ninomiya, I.; Naito, T. Chapter 4 Application of Enamide Cyclizations in Alkaloid Synthesis. In The Alkaloids: Chemistry and Pharmacology; Brossi, A., Ed.; Academic Press: Cambridge, MA, USA, 1984; Volume 22, pp. 189–279. [Google Scholar] [CrossRef]
- Ninomiya, I.; Kiguchi, T.; Yamauchi, S.; Naito, T. Photocyclisation of Enamides. Part 14. Substituent Effects in the Photocyclisation of N-α,β-Unsaturated Acylanilides. J. Chem. Soc. Perkin Trans. 1 1980, 197–202. [Google Scholar] [CrossRef]
- Lenz, G.R. Lead Tetra-Acetate-Mediated Oxidative Cyclizations of Isoquinoline Alkyl Substituted Methylene Urethanes (Enamides) to Isoquinoline Hydroxyoxazolidinones. J. Chem. Soc. Perkin Trans. 1 1990, 33–38. [Google Scholar] [CrossRef]
- Kametani, T.; Fukumoto, K. Photochemical Synthesis of Isoquinoline Alkaloids. Acc. Chem. Res. 1972, 5, 212–219. [Google Scholar] [CrossRef]
- Kametani, T.; Nemoto, H.; Fukumoto, K. A New Method for Selective Epoxidation and a Biogenetic-Type Synthesis of Linalyloxides. Bioorg. Chem. 1978, 7, 215–220. [Google Scholar] [CrossRef]
- Swenton, J.S.; Ikeler, T.J.; Smyser, G.L. Effect of Biphenyl Geometry and Substituents on the Multiplicity and Efficiency of the Photocyclization Reactions of 2-Substituted Biphenyls. J. Org. Chem. 1973, 38, 1157–1166. [Google Scholar] [CrossRef]
- Schultz, A.G.; Lucci, R.D.; Fu, W.Y.; Berger, M.H.; Erhardt, J.; Hagmann, W.K. Heteroatom Directed Photoarylation. Synthetic Potential of the Heteroatom Oxygen. J. Am. Chem. Soc. 1978, 100, 2150–2162. [Google Scholar] [CrossRef]
- Schultz, A.G.; DeTar, M.B. Thiocarbonyl Ylides. Photogeneration, Rearrangement, and Cycloaddition Reactions. J. Am. Chem. Soc. 1976, 98, 3564–3572. [Google Scholar] [CrossRef]
- Romero, I.E.; Postigo, A.; Bonesi, S.M. Solvent Effects on the Photoinduced [6π]-Electrocyclization Reactions of Mono-, Di-, and Trisubstituted Arylamines: Photophysical, Preparative Photochemistry, and Mechanistic Investigations. J. Org. Chem. 2023, 88, 4405–4421. [Google Scholar] [CrossRef] [PubMed]
- Schultz, A.G.; Chiu, I.-C. Heteroatom Directed Photoarylation; an Approach to the Synthesis of Aspidosperma Alkaloids. J. Chem. Soc. Chem. Commun. 1978, 29. [Google Scholar] [CrossRef]
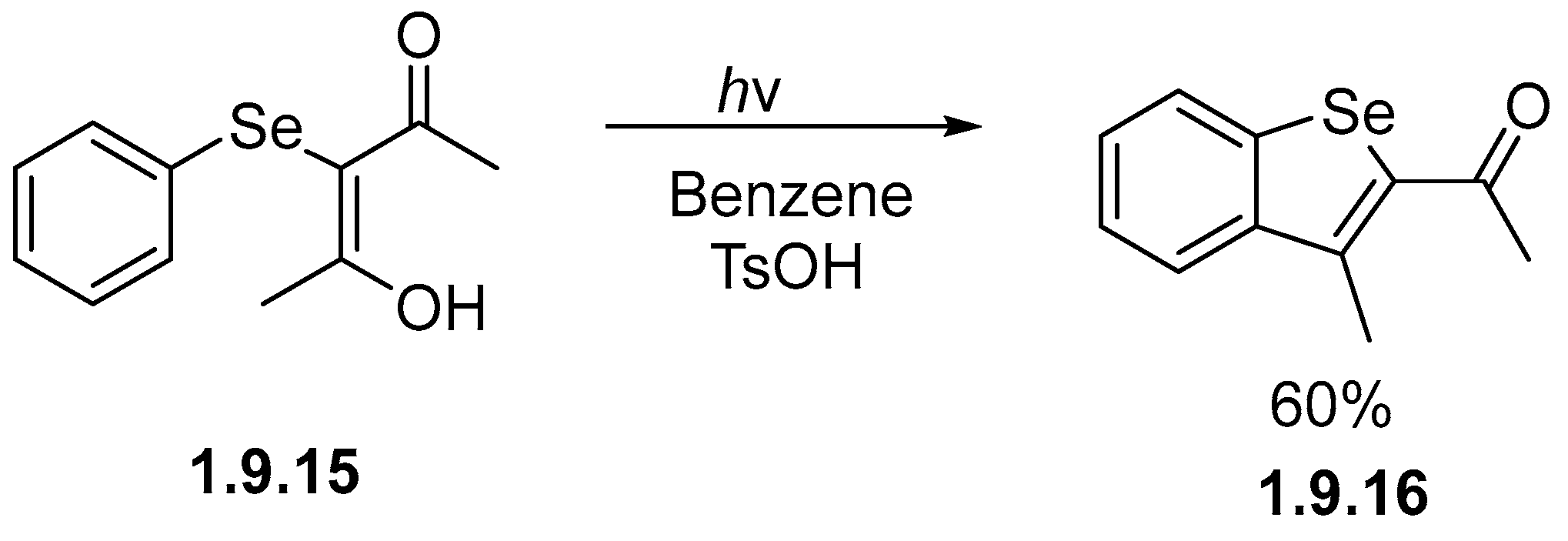
- Schultz, A.G. Heteroatom Directed Photoarylation. Photochemistry of an Organoselenide. J. Org. Chem. 1975, 40, 3466–3467. [Google Scholar] [CrossRef]
- Schultz, A.G.; Fu, W.Y.; Lucci, R.D.; Kurr, B.G.; Lo, K.M.; Boxer, M. Heteroatom Directed Photoarylation. Synthetic Potential of the Heteroatom Sulfur. J. Am. Chem. Soc. 1978, 100, 2140–2149. [Google Scholar] [CrossRef]
- Lapouyade, R.; Koussini, R.; Bouas-Laurent, H. Primary and Tertiary Amines as Catalysts of Hydrogen Transfer in the Photocyclization of 1,1-Diarylethylenes. J. Am. Chem. Soc. 1977, 99, 7374–7376. [Google Scholar] [CrossRef]
- Grovenstein, E., Jr.; Campbell, T.C.; Shibata, T. Photochemical Reactions of Dimethyl Acetylenedicarboxylate with Benzene and Naphthalene. J. Org. Chem. 1969, 34, 2418–2428. [Google Scholar] [CrossRef]
- Cox, A.; Kemp, D.R.; Lapouyade, R.; Mayo, P.d.; Joussot-Dubien, J.; Bonneau, R. Thione Photochemistry: The Peri Cyclization of Some Polycyclic Aromatic Thiones. Can. J. Chem. 1975, 53, 2386–2393. [Google Scholar] [CrossRef]


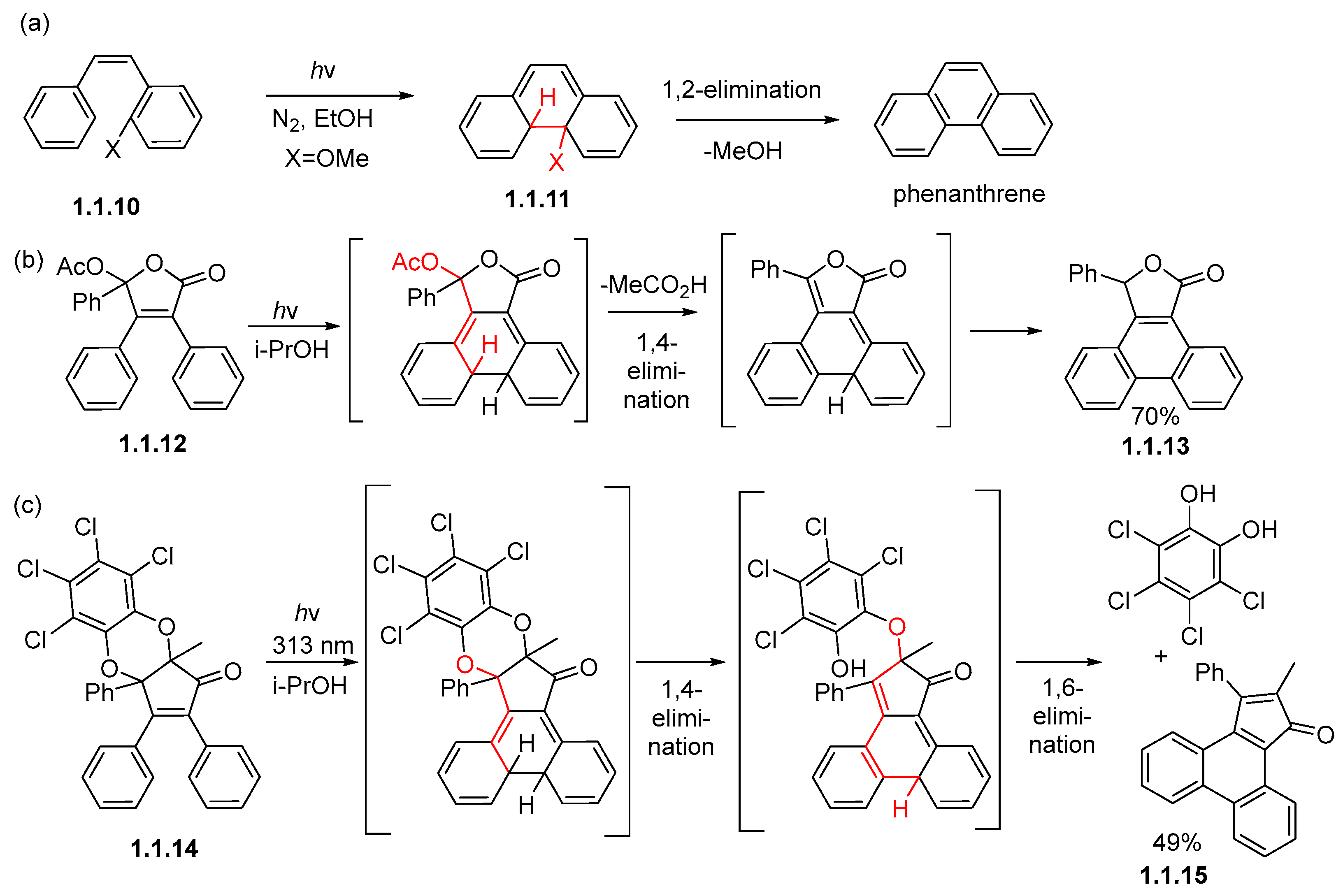






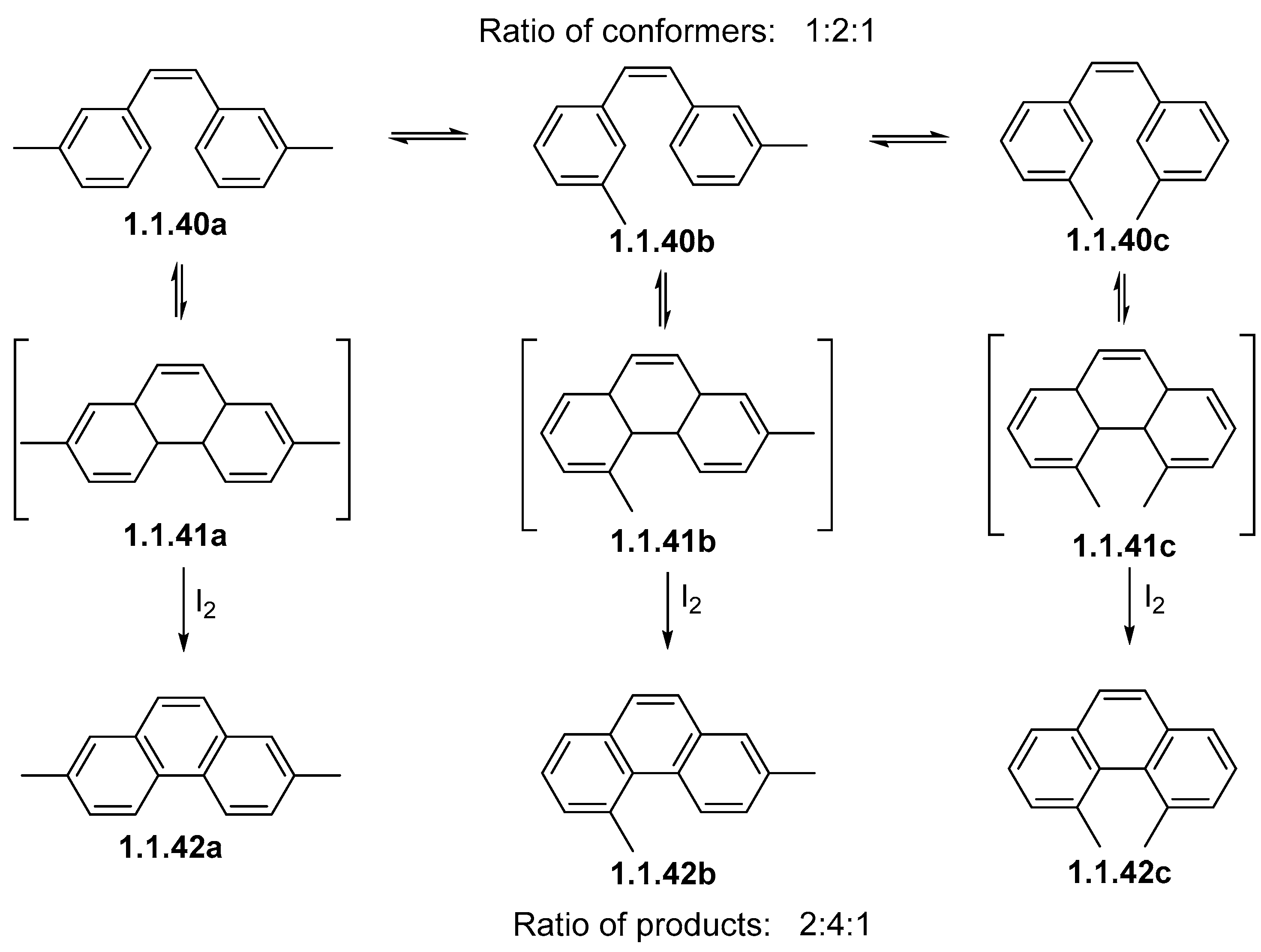
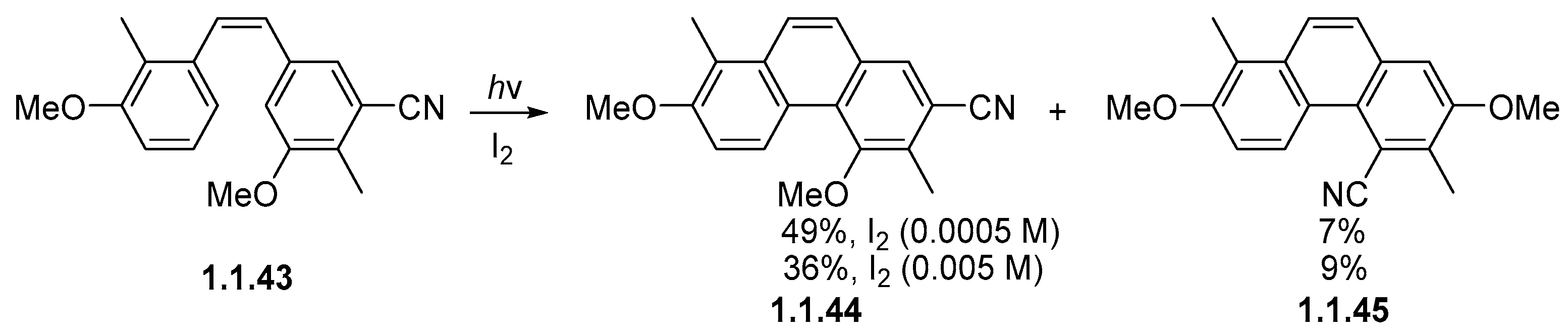

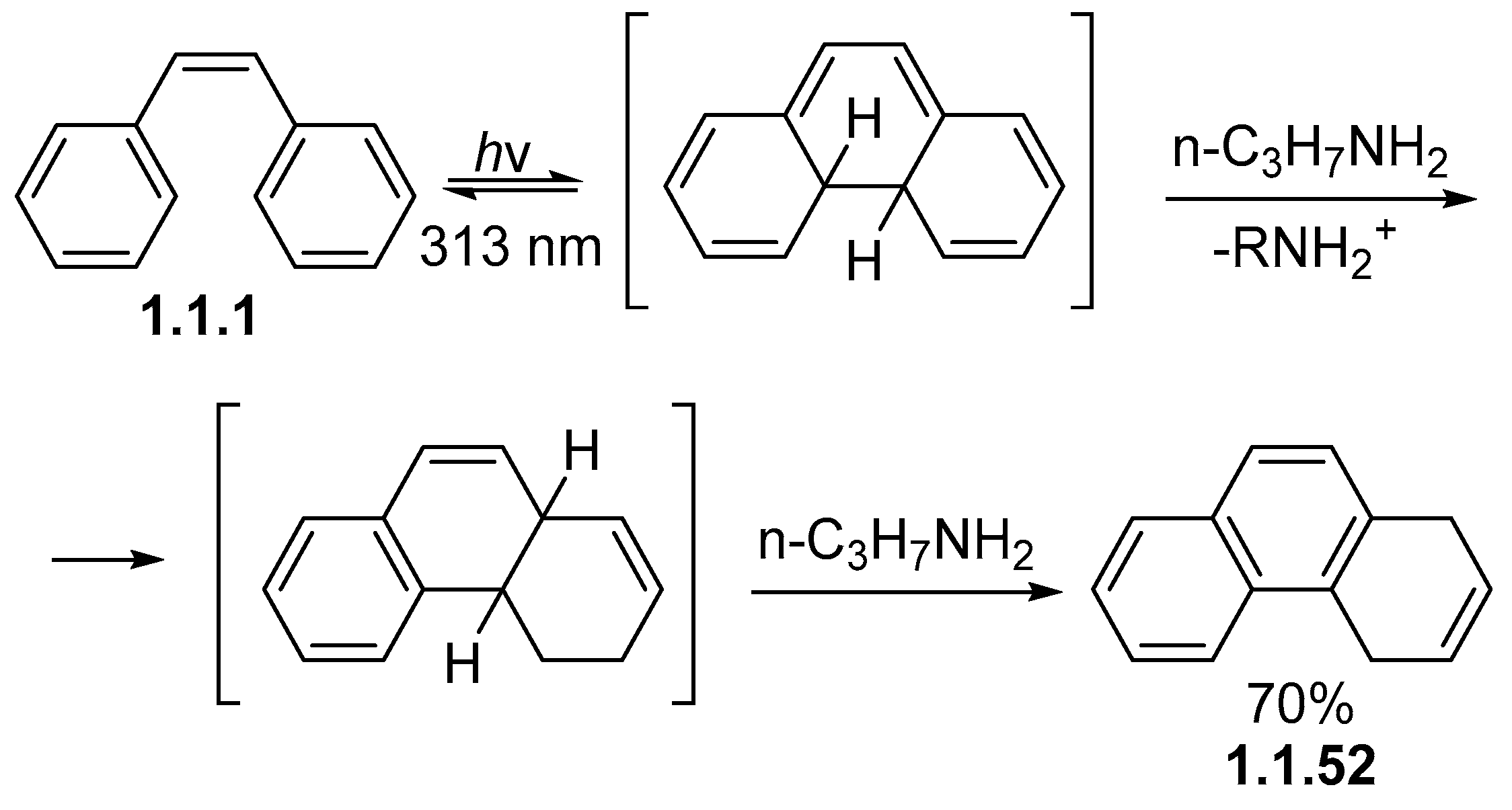










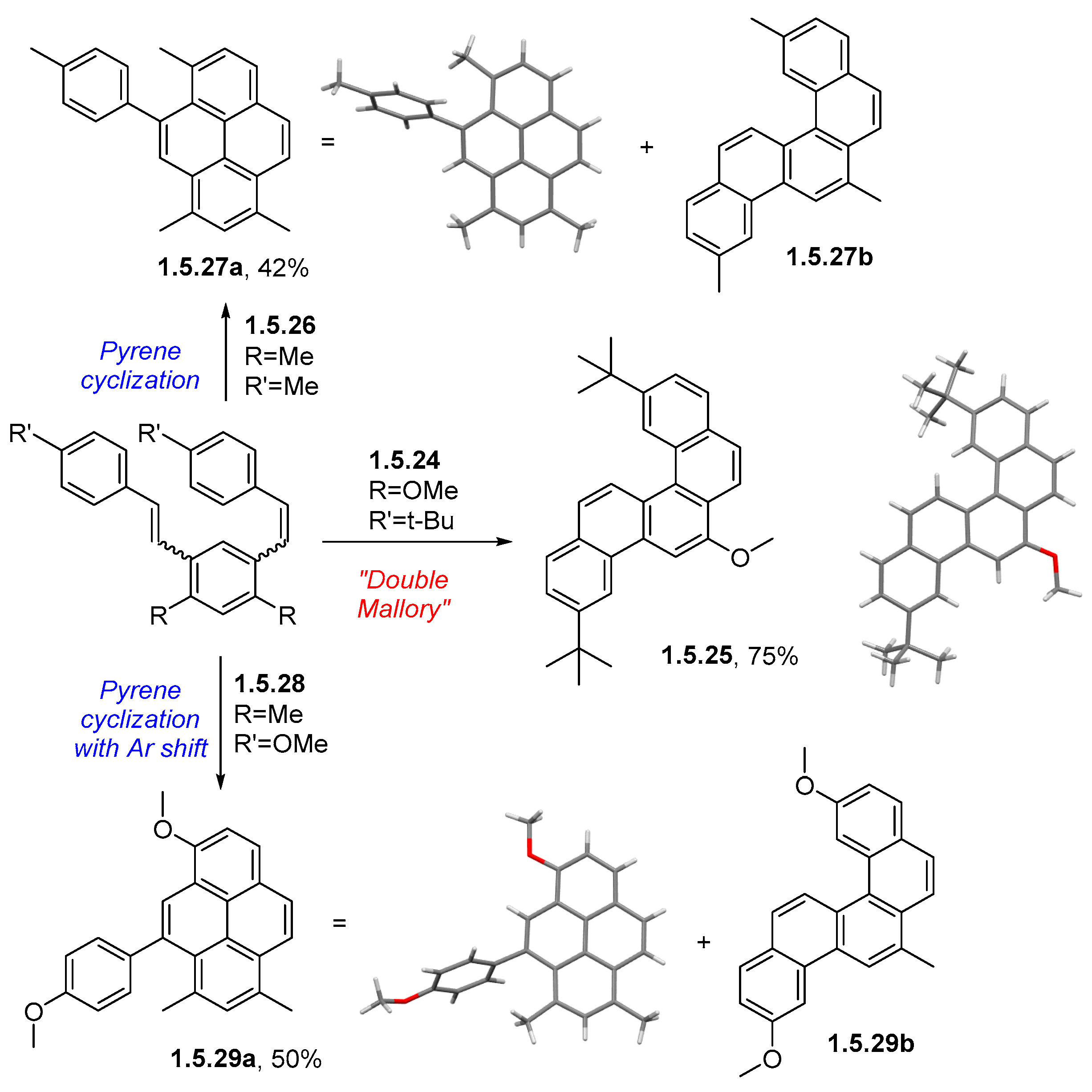


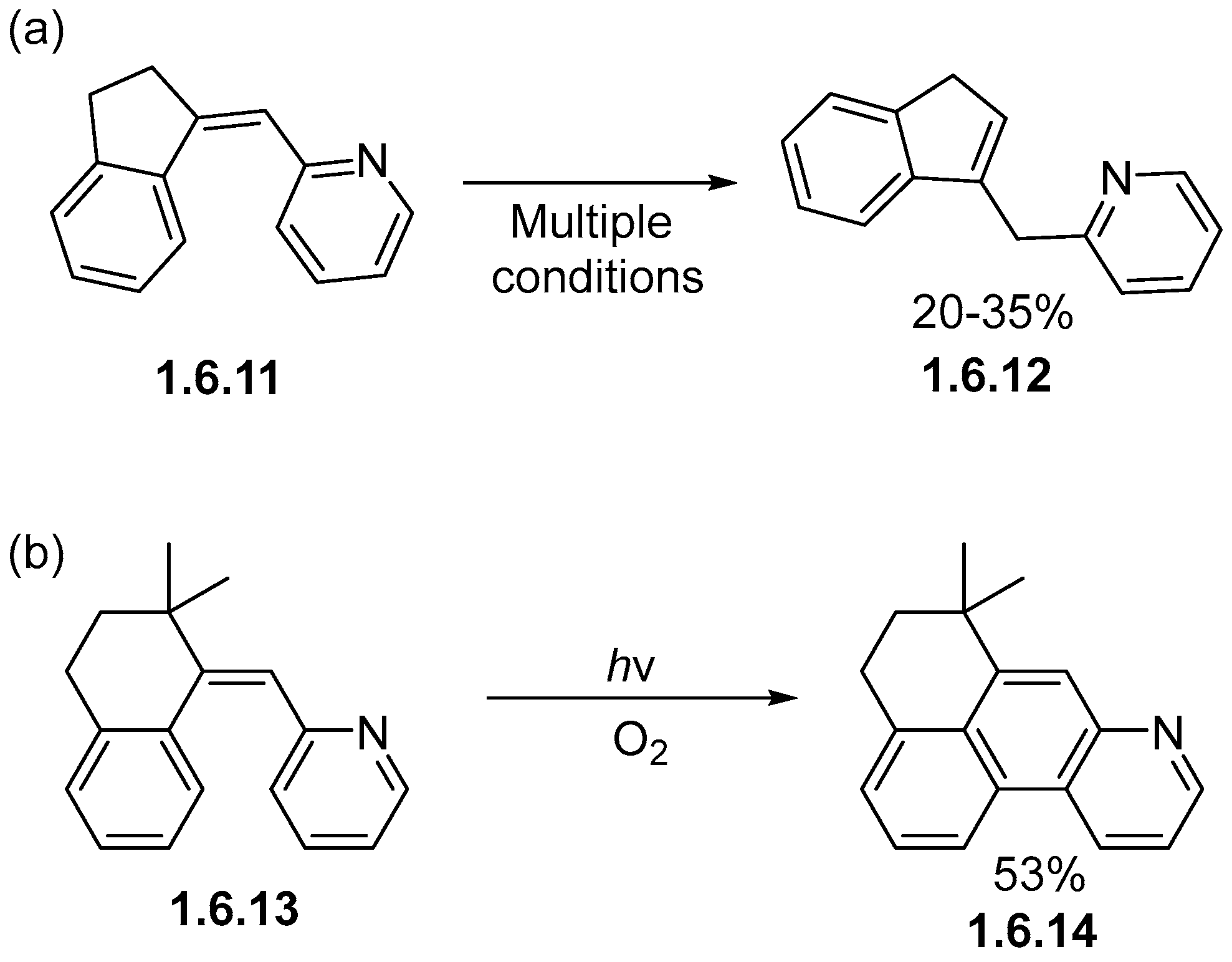
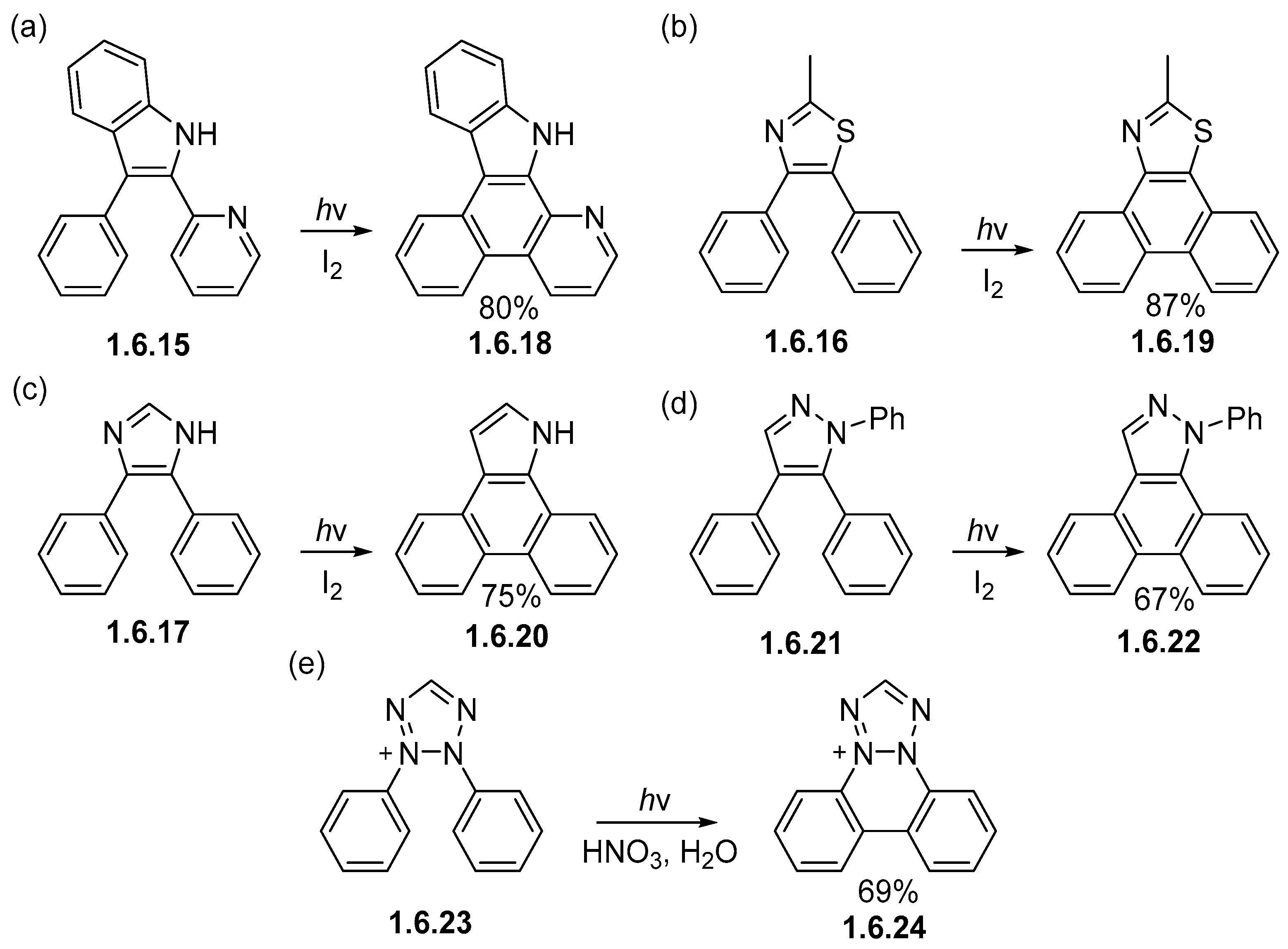
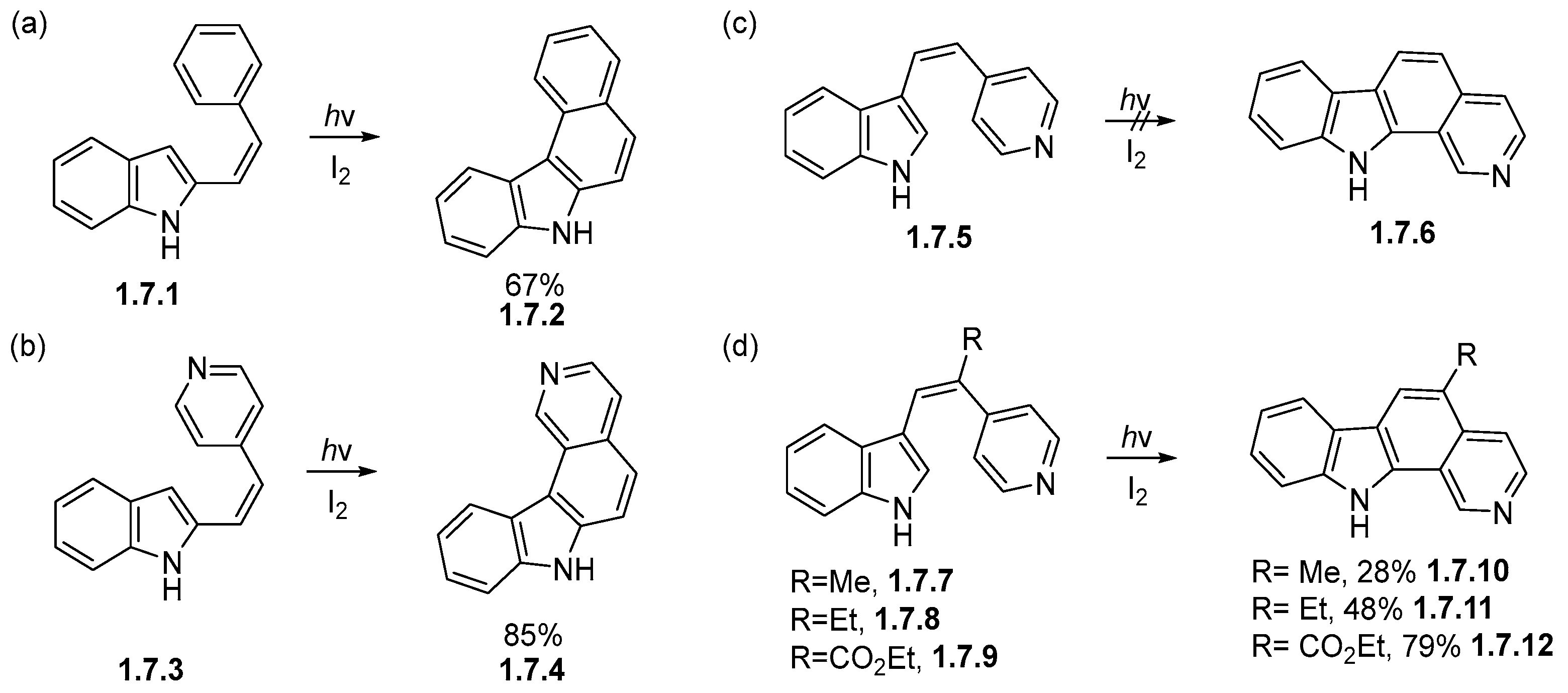

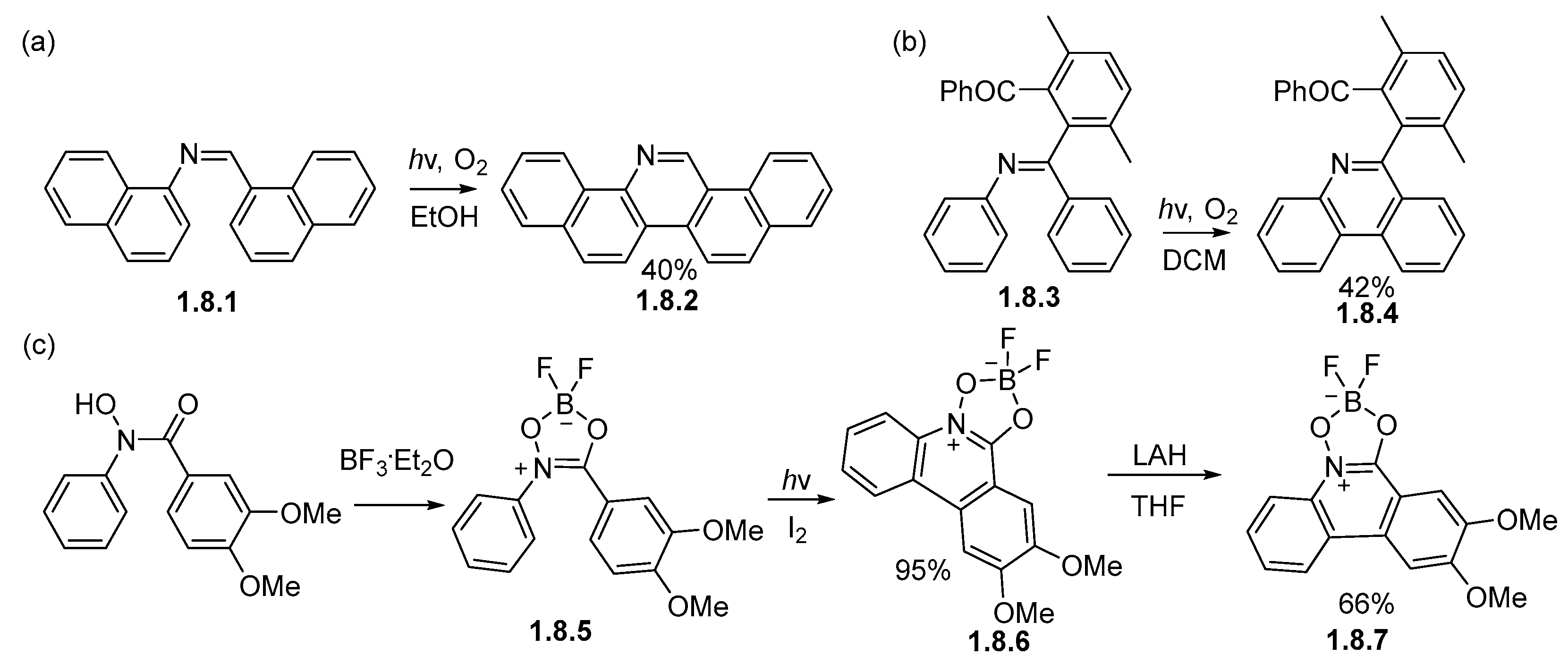
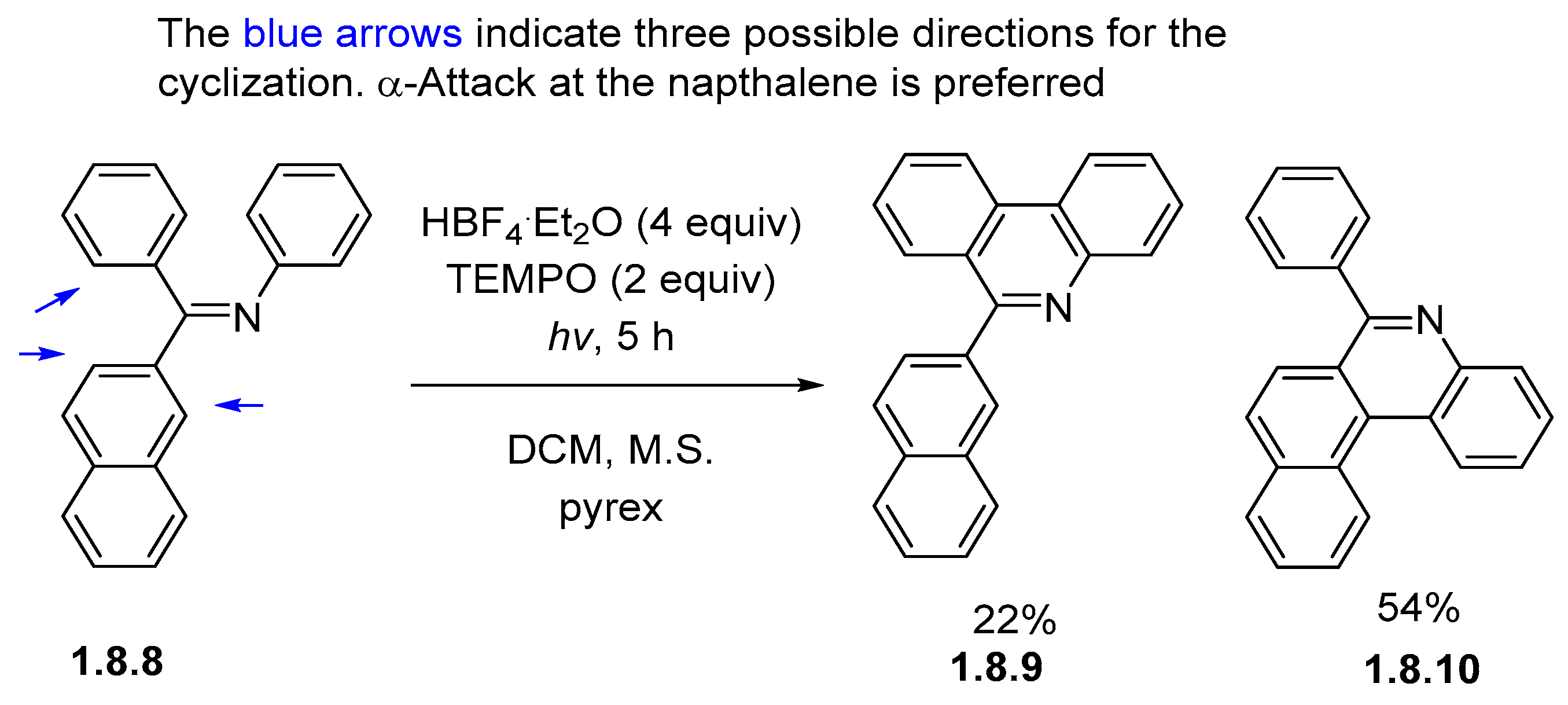

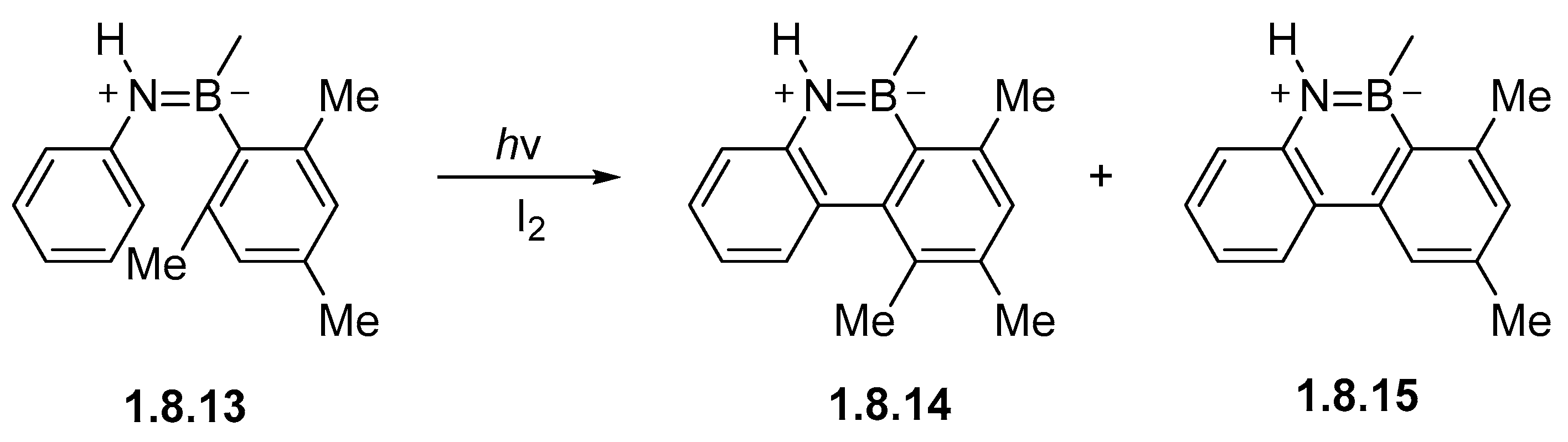

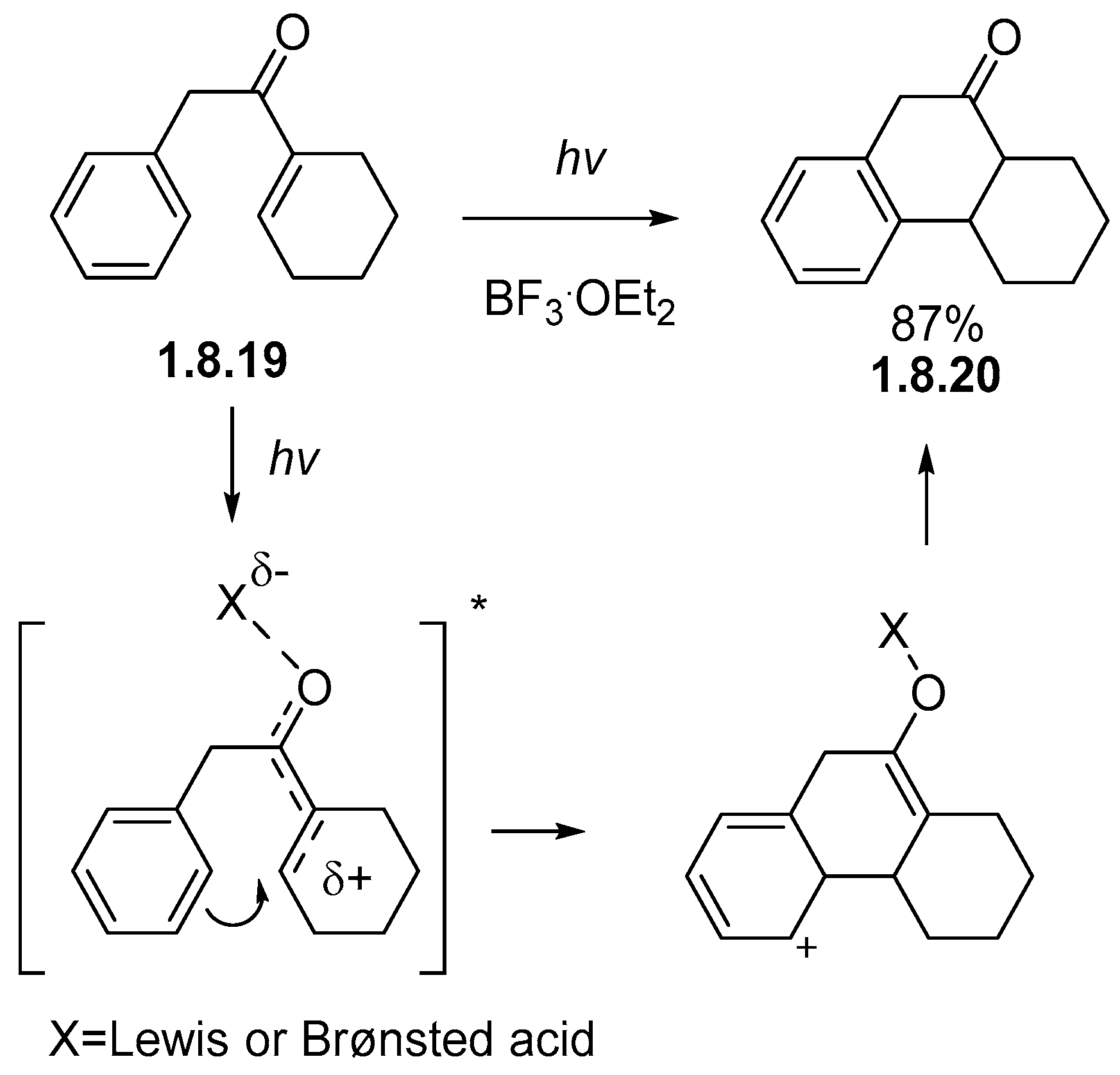






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dos Santos, N.R.; Wu, J.I.; Alabugin, I.V. Photocyclization of Alkenes and Arenes: Penetrating Through Aromatic Armor with the Help of Excited State Antiaromaticity. Chemistry 2025, 7, 79. https://doi.org/10.3390/chemistry7030079
Dos Santos NR, Wu JI, Alabugin IV. Photocyclization of Alkenes and Arenes: Penetrating Through Aromatic Armor with the Help of Excited State Antiaromaticity. Chemistry. 2025; 7(3):79. https://doi.org/10.3390/chemistry7030079
Chicago/Turabian StyleDos Santos, Nikolas R., Judy I. Wu, and Igor V. Alabugin. 2025. "Photocyclization of Alkenes and Arenes: Penetrating Through Aromatic Armor with the Help of Excited State Antiaromaticity" Chemistry 7, no. 3: 79. https://doi.org/10.3390/chemistry7030079
APA StyleDos Santos, N. R., Wu, J. I., & Alabugin, I. V. (2025). Photocyclization of Alkenes and Arenes: Penetrating Through Aromatic Armor with the Help of Excited State Antiaromaticity. Chemistry, 7(3), 79. https://doi.org/10.3390/chemistry7030079