
Green Synthesis, Characterization, and Biological Activity of 4-Aminoquinoline Derivatives: Exploring Antibacterial Efficacy, MRSA Inhibition, and PBP2a Docking Insights

 ,
,  ,
,  , and
, and
Abstract

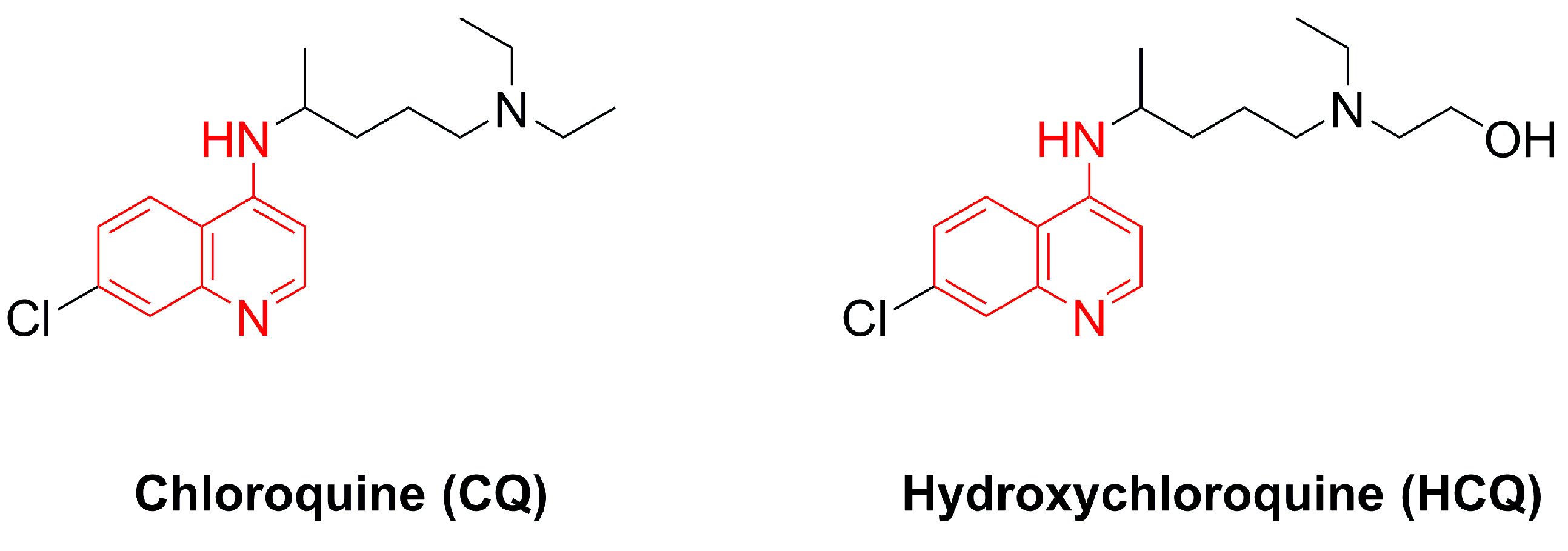
1. Introduction
2. Materials and Methods
2.1. Chemicals and Materials
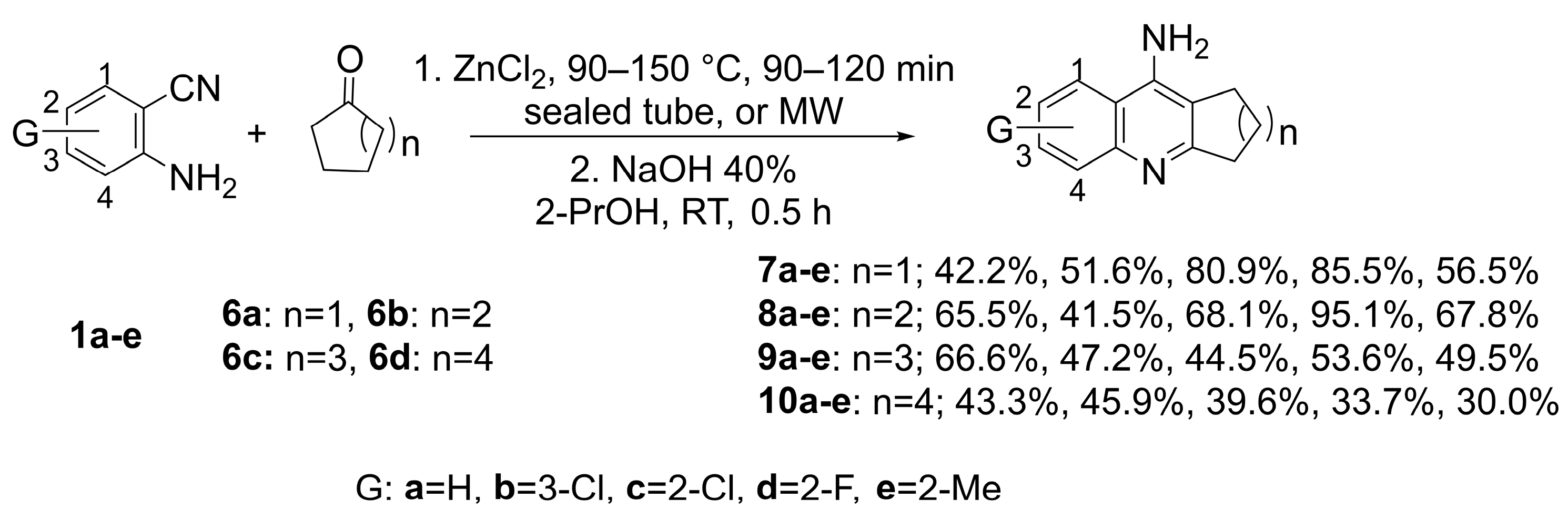
2.2. Synthesis
2.2.1. General Procedure (GP)
2.2.2. Synthesis and Spectral Analysis
- 2-Methylquinolin-4-amine (4a)
- 7-Chloro-2-methylquinolin-4-amine (4b)
- 6-Chloro-2-methylquinolin-4-amine (4c)
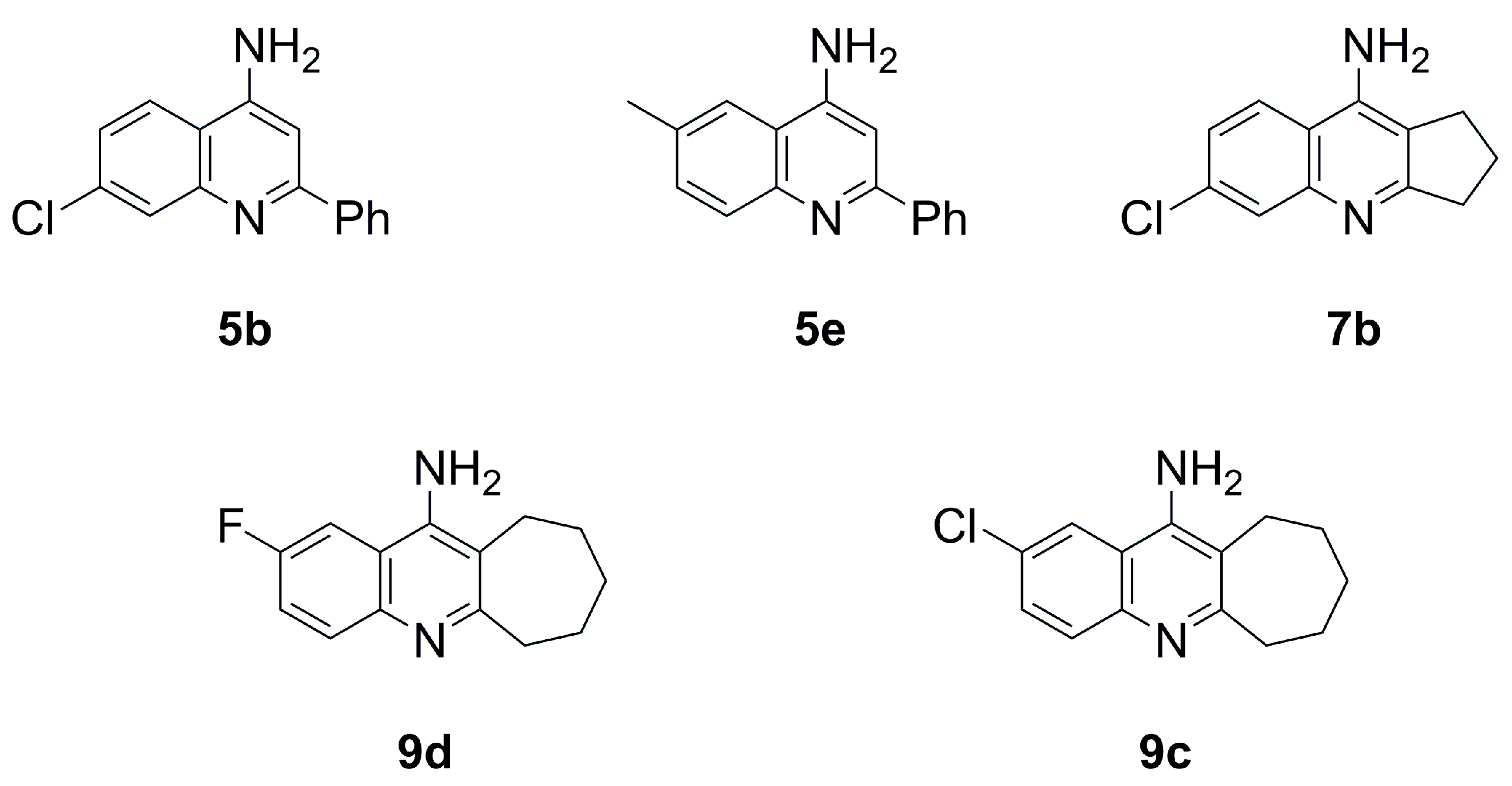
- 6-Fluoro-2-methylquinolin-4-amine (4d)
- 2,6-Dimethylquinolin-4-amine (4e)
- 2-Phenylquinolin-4-amine (5a)
- 7-Chloro-2-phenylquinolin-4-amine (5b)
- 6-Chloro-2-phenylquinolin-4-amine (5c)
- 6-Fluoro-2-phenylquinolin-4-amine (5d)
- 6-Methyl-2-phenylquinolin-4-amine (5e)
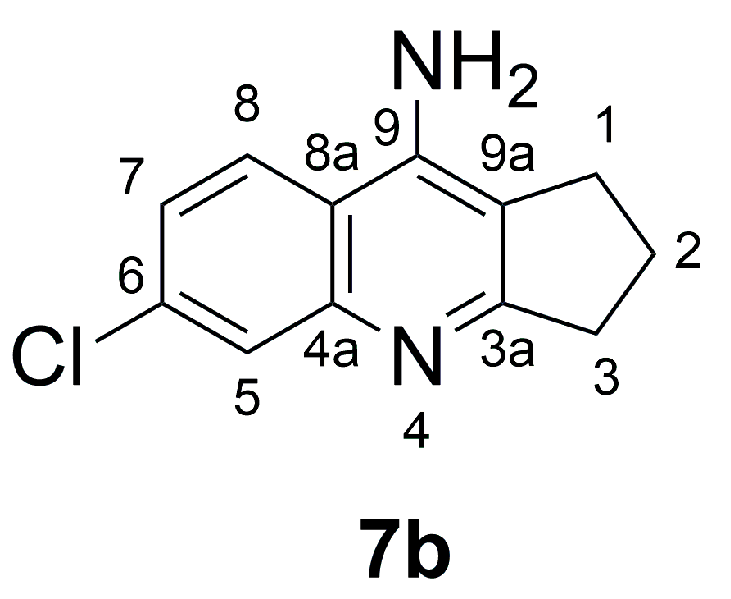
- 6-Chloro-2,3-dihydro-1H-cyclopenta[b]quinolin-9-amine (7b)
- 3-Chloro-7,8,9,10-tetrahydro-6H-cyclohepta[b]quinolin-11-amine (9b)
- 3-Chloro-6,7,8,9,10,11-hexahydrocycloocta[b]quinolin-12-amine (10b)
- 2-Chloro-6,7,8,9,10,11-hexahydrocycloocta[b]quinolin-12-amine (10c)
- 2-Fluoro-6,7,8,9,10,11-hexahydrocycloocta[b]quinolin-12-amine (10d)
- 2-Methyl-6,7,8,9,10,11-hexahydrocycloocta[b]quinolin-12-amine (10e)
2.3. Antibacterial and Antifungal Activities
2.3.1. Microorganisms
2.3.2. Well Diffusion Method
2.3.3. Minimum Inhibitory Concentration (MIC)
2.3.4. Minimum Bactericidal Concentration (MBC)
2.4. Molecular Docking Methodology
2.4.1. Software Packages
We Used the Following Software
- CS ChemDraw® Ultra Cambridge Soft Corp. (www.cambridgesoft.com, accessed in 15 October 2024) as a 2D chemical drawing tool.
2.4.2. Ligand Preparation and Molecular Docking
Protein Preparation
Ligand Preparation
Binding Site Definition
- PBP2a Binding Site:
- 3NKH Integrase Enzyme Binding Site:
2.5. Docking Procedure
2.5.1. Scoring and Evaluation
2.5.2. Ligand Pose Analysis
3. Results
3.1. Synthesis and Characterization
3.2. Antibacterial and Antifungal Activity
3.3. Results of the Molecular Docking Experiment
3.3.1. Docking of Anti-MRSA Active Compounds into the Binding Pocket of MRSA PBP2a
3.3.2. Docking of the Anti-MRSA Active Compounds into the Binding Pocket of MRSA Integrase
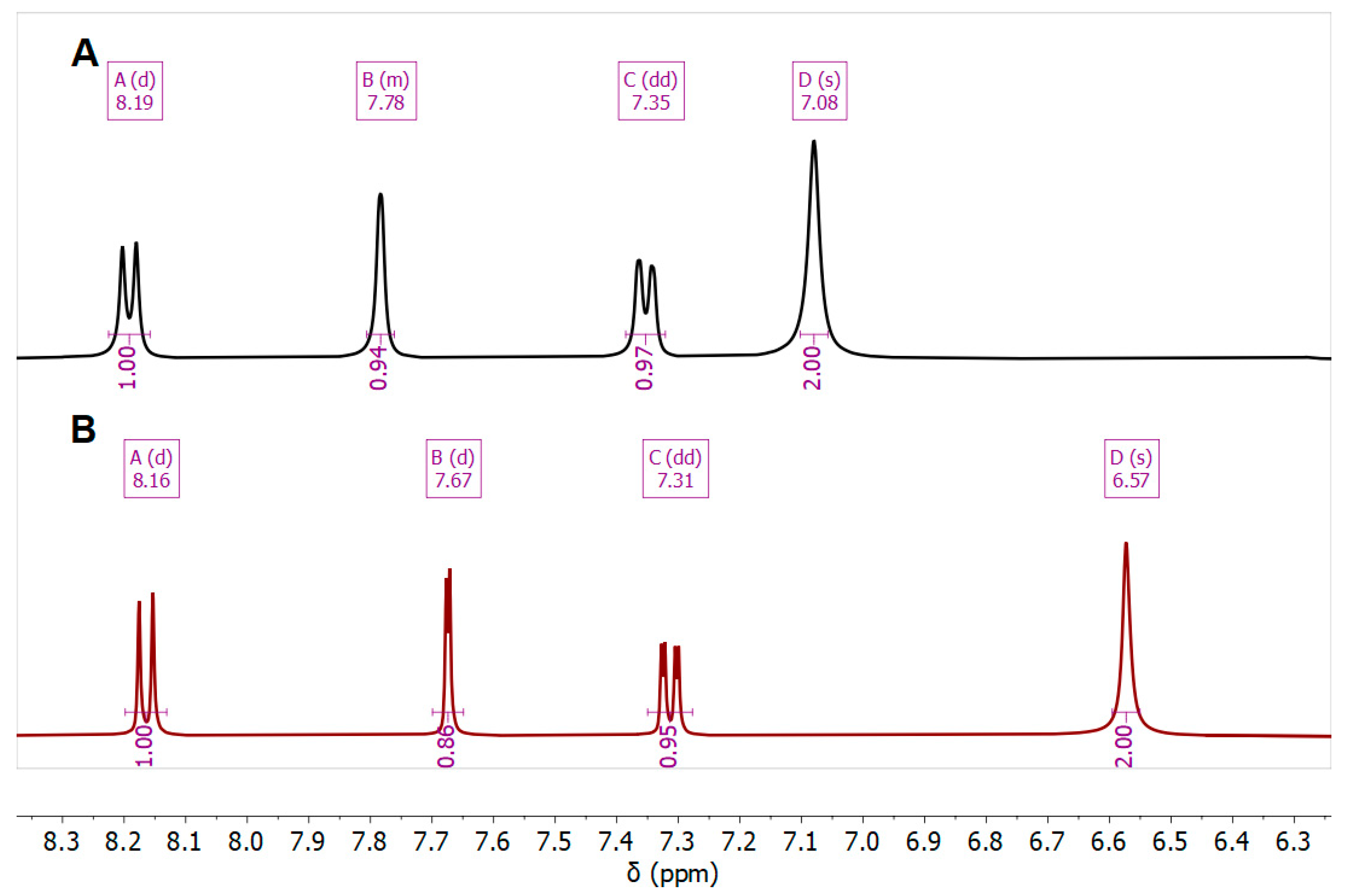
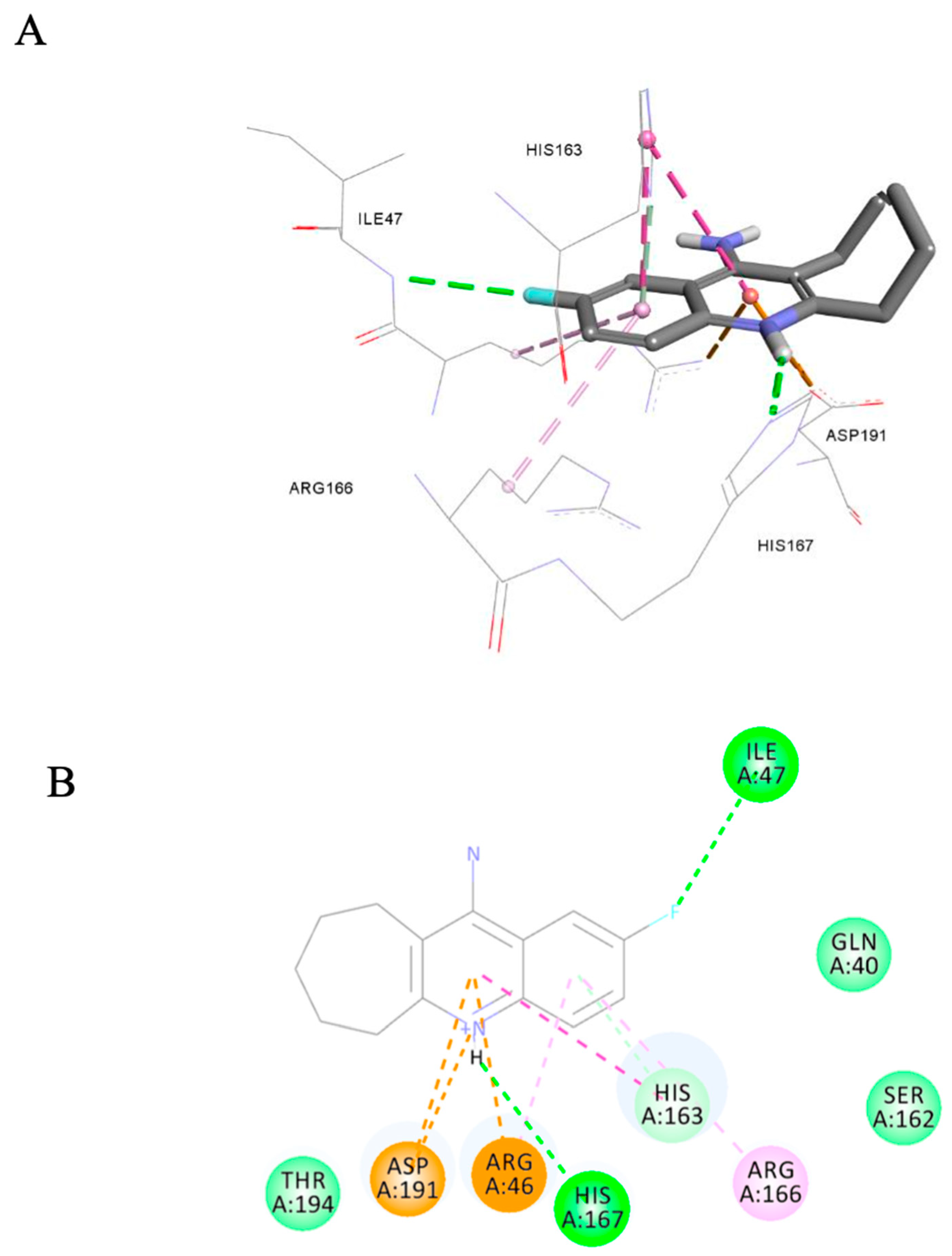
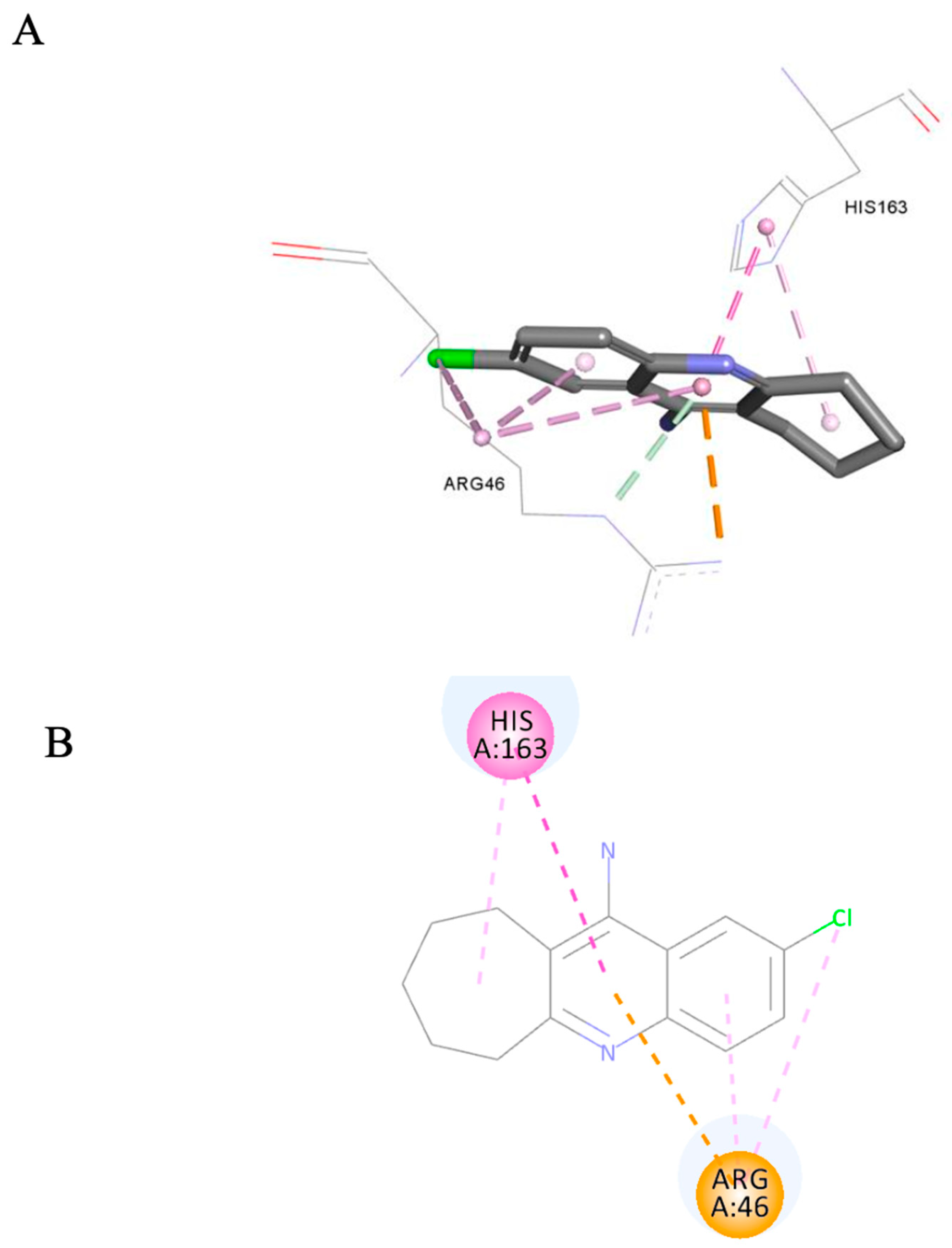
3.3.3. Ligand Pose Analysis Inside the Binding Pocket of MRSA–Penicillin Binding Protein 2a (PBP2a)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and Practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Bassyouni, F.A.; Abu-Bakr, S.M.; Rehim, M.A. Evolution of microwave irradiation and its application in green chemistry and biosciences. Res. Chem. Intermed. 2012, 38, 283–322. [Google Scholar] [CrossRef]
- Van Praet, S.; Preegel, G.; Rammal, F.; Sels, B.F. One-Pot Consecutive Reductive Amination Synthesis of Pharmaceuticals: From Biobased Glycolaldehyde to Hydroxychloroquine. ACS Sustain. Chem. Eng. 2022, 10, 6503–6508. [Google Scholar] [CrossRef]
- Ray, R.L. Alkaloids—The World’s Pain Killers. J. Chem. Educ. 1960, 37, 451–454. [Google Scholar] [CrossRef]
- Nainwal, L.M.; Tasneem, S.; Akhtar, W.; Verma, G.; Khan, M.F.; Parvez, S.; Shaquiquzzaman, M.; Akhter, M.; Alam, M.M. Green Recipes to Quinoline: A Review. Eur. J. Med. Chem. 2019, 164, 121–170. [Google Scholar] [CrossRef] [PubMed]
- Matada, B.S.; Pattanashettar, R.; Yernale, N.G. A Comprehensive Review on the Biological Interest of Quinoline and Its Derivatives. Bioorg. Med. Chem. 2021, 32, 115973. [Google Scholar] [CrossRef] [PubMed]
- Kaur, K.; Jain, M.; Reddy, R.P.; Jain, R. Quinolines and Structurally Related Heterocycles as Antimalarials. Eur. J. Med. Chem. 2010, 45, 3245–3264. [Google Scholar] [CrossRef]
- Browning, D.J. Pharmacology of Chloroquine and Hydroxychloroquine. In Hydroxychloroquine and Chloroquine Retinopathy; Browning, D.J., Ed.; Springer: New York, NY, USA, 2014; pp. 35–63. ISBN 978-1-4939-0597-3. [Google Scholar] [CrossRef]
- Pandey, A.V.; Bisht, H.; Babbarwal, V.K.; Srivastava, J.; Pandey, K.C.; Chauhan, V.S. Mechanism of Malarial Haem Detoxification Inhibition by Chloroquine. Biochem. J. 2001, 355, 333–338. [Google Scholar] [CrossRef]
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Isaka, Y. Chloroquine in Cancer Therapy: A Double-Edged Sword of Autophagy. Cancer Res. 2013, 73, 3–7. [Google Scholar] [CrossRef]
- Li, X.; Wang, Y.; Agostinis, P.; Agostinis, P.; Rabson, A.; Melino, G.; Carafoli, E.; Shi, Y.; Sun, E. Is hydroxychloroquine beneficial for COVID-19 patients? Cell Death Dis. 2020, 11, 512. [Google Scholar] [CrossRef]
- Tzekov, R. Ocular Toxicity Due to Chloroquine and Hydroxychloroquine: Electrophysiological and Visual Function Correlates. Doc. Ophthalmol. 2005, 110, 111–120. [Google Scholar] [CrossRef]
- Verbaanderd, C.; Maes, H.; Schaaf, M.B.; Sukhatme, V.P.; Pantziarka, P.; Sukhatme, V.; Agostinis, P.; Bouche, G. Repurposing Drugs in Oncology (ReDO)—Chloroquine and Hydroxychloroquine as Anti-Cancer Agents. Ecancermedicalscience 2017, 11, 781. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Wang, H.; Yang, Y.; Chen, Z.-S.; Zou, C.; Zhang, J. Chloroquine against Malaria, Cancers and Viral Diseases. Drug Discov. Today 2020, 25, 2012–2022. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.-N.; Wu, Z.-X.; Dong, S.; Yang, D.-H.; Zhang, L.; Ke, Z.; Zou, C.; Chen, Z.-S. Chloroquine and Hydroxychloroquine in the Treatment of Malaria and Repurposing in Treating COVID-19. Pharmacol. Ther. 2020, 216, 107672. [Google Scholar] [CrossRef] [PubMed]
- Nardi, N.; Baumgarten, L.G.; Dreyer, J.P.; Santana, E.R.; Winiarski, J.P.; Vieira, I.C. Nanocomposite Based on Green Synthesis of Gold Nanoparticles Decorated with Functionalized Multi-Walled Carbon Nanotubes for the Electrochemical Determination of Hydroxychloroquine. J. Pharm. Biomed. Anal. 2023, 236, 115681. [Google Scholar] [CrossRef]
- Bray, P.; Park, B.; Asadollaly, E.; Biagini, G.; Jeyadevan, J.; Berry, N.; Ward, S.; O’Neill, P. A Medicinal Chemistry Perspective on 4-Aminoquinoline Antimalarial Drugs. Curr. Top. Med. Chem. 2006, 6, 479–507. [Google Scholar] [CrossRef]
- De, D.; Krogstad, F.M.; Byers, L.D.; Krogstad, D.J. Structure−Activity Relationships for Antiplasmodial Activity among 7-Substituted 4-Aminoquinolines. J. Med. Chem. 1998, 41, 4918–4926. [Google Scholar] [CrossRef]
- O’Neill, P.M.; Willock, D.J.; Hawley, S.R.; Bray, P.G.; Storr, R.C.; Ward, S.A.; Park, B.K. Synthesis, Antimalarial Activity, and Molecular Modeling of Tebuquine Analogues. J. Med. Chem. 1997, 40, 437–448. [Google Scholar] [CrossRef]
- Zhao, Z.; Song, J.; Shang, Y.; Shan, M.; Tang, L.; Pang, T.; Ye, Q.; Cheng, G.; Jiang, G. Design, Synthesis, Biological Evaluation and Docking Study of a New Series of 9-Aminoacridine Derivatives as Anti-Inflammatory Agents. ChemistrySelect 2024, 9, e202302387. [Google Scholar] [CrossRef]
- Gorecki, L.; Misiachna, A.; Damborsky, J.; Dolezal, R.; Korabecny, J.; Cejkova, L.; Hakenova, K.; Chvojkova, M.; Karasova, J.Z.; Prchal, L.; et al. Structure-activity relationships of dually-acting acetylcholinesterase inhibitors derived from tacrine on N-methyl-d-Aspartate receptors. Eur. J. Med. Chem. 2021, 219, 113434. [Google Scholar] [CrossRef]
- Hasaninejad, A.; Zare, A.; Shekouhy, M.; Ameri-Rad, J. Sulfuric acid-modified PEG-6000 (PEG-OSO3H): An efficient, biodegradable and reusable polymeric catalyst for the solvent-free synthesis of poly-substituted quinolines under microwave irradiation. Green Chem. 2011, 13, 958–964. [Google Scholar] [CrossRef]
- Khalilzadeh, M.A.; Hosseini, A.; Tajbakhsh, M. Synthesis of tacrine derivatives under solventless conditions. J. Heterocycl. Chem. 2007, 44, 535–538. [Google Scholar] [CrossRef]
- Moore, J.A.; Kornreich, L.D. A direct synthesis of 4-aminoquinolines. Tetrahedron Lett. 1963, 4, 1277–1281. [Google Scholar] [CrossRef]
- Malhotra, R.C.; Ramachandran, P.K. Amination of hydroxyquinolines. Indian J. Chem. Sec. B Org. Chem. Incl. Med. Chem. 1978, 16B, 329. [Google Scholar]
- Royer, R. The preparation of 4-amino-2-styrylquinoline and some related compounds. J. Chem. Soc. 1949, 386, 1803–1806. [Google Scholar] [CrossRef]
- White, H.C.; Bergstrom, F.W. A Hofmann Type Rearrangement in Liquid Ammonia. J. Org. Chem. 1942, 6, 497–507. [Google Scholar] [CrossRef]
- Elderfield, R.C.; Gensler, W.J.; Bembry, T.H.; Kremer, C.B.; Head, J.D.; Brody, F.; Frohardt, R. Synthesis of 2-Phenyl-4-chloroquinolines. J. Am. Chem. Soc. 1946, 68, 1272–1276. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.W.; Kirby, W.M.M.; Sherris, J.C.; Turck, M. Antibiotic Susceptibility Testing by a Standardized Single Disk Method. Am. J. Clin. Pathol. 1966, 45, 493–496. [Google Scholar] [CrossRef]
- Gedam, V.V.; Punyapwar, S.V.; Raut, P.A.; Chahande, A.D. Rapid In Vitro Proliferation of Tinospora cordifolia Callus Biomass from Stem and Leaf with Phytochemical and Antimicrobial Assessment. In Waste Valorisation and Recycling; Springer: Singapore, 2019; Volume 2, pp. 421–431. ISBN 978-981-13-2783-4. Available online: https://link.springer.com/chapter/10.1007/978-981-13-2784-1_39 (accessed on 20 May 2024).
- Ibrahim, A.I.M.; Abul-Futouh, H.; Bourghli, L.M.S.; Abu-Sini, M.; Sunoqrot, S.; Ikhmais, B.; Jha, V.; Sarayrah, Q.; Abulebdah, D.H.; Ismail, W.H. Design and Synthesis of Thionated Levofloxacin: Insights into a New Generation of Quinolones with Potential Therapeutic and Analytical Applications. Curr. Issues Mol. Biol. 2022, 44, 4626–4638. [Google Scholar] [CrossRef]
- Magwaza, R.N.; Abubaker, M.; Hussain, B.; Haley, M.; Couper, K.; Freeman, S.; Nirmalan, N.J. Evaluation of 4-Aminoquinoline Hydrazone Analogues as Potential Leads for Drug-Resistant Malaria. Molecules 2023, 28, 6471. [Google Scholar] [CrossRef]
- Wu, G.; Robertson, D.H.; Brooks III, C.L.; Vieth, M. Detailed Analysis of Grid-Based Molecular Docking: A Case Study of CDOCKER—A CHARMm-Based MD Docking Algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef] [PubMed]
- Krammer, A.; Kirchhoff, P.D.; Jiang, X.; Venkatachalam, C.M.; Waldman, M. LigScore: A Novel Scoring Function for Predicting Binding Affinities. J. Mol. Graph. Model. 2005, 23, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Gehlhaarl, D.K.; Verkhivkerl, G.M.; Rejtol, P.A.; Sherman, C.J.; Fogel, D.B.; Fogel, L.J.; Freer’, S.T. Molecular Recognition of the Inhibitor AC-1343 by HIV-l Protease: Conformationally Flexible Docking by Evolutionary Programming. Chem. Biol. 1995, 2, 317–324. [Google Scholar] [CrossRef]
- Jain, A.N. Scoring Functions for Protein-Ligand Docking. Curr. Protein Pept. Sci. 2006, 7, 407–420. [Google Scholar] [CrossRef]
- Muegge, I.; Martin, Y.C. A General and Fast Scoring Function for Protein-Ligand Interactions: A Simplified Potential Approach. J. Med. Chem. 1999, 42, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Bæk, K.T.; Gründling, A.; Mogensen, R.G.; Thøgersen, L.; Petersen, A.; Paulander, W.; Frees, D. β-Lactam Resistance in Methicillin-Resistant Staphylococcus Aureus USA300 Is Increased by Inactivation of the ClpXP Protease. Antimicrob. Agents Chemother. 2014, 58, 4593–4603. [Google Scholar] [CrossRef]
- Lovering, A.L.; Gretes, M.C.; Safadi, S.S.; Danel, F.; De Castro, L.; Page, M.G.P.; Strynadka, N.C.J. Structural Insights into the Anti-Methicillin-Resistant Staphylococcus Aureus (MRSA) Activity of Ceftobiprole. J. Biol. Chem. 2012, 287, 32096–32102. [Google Scholar] [CrossRef]
- Purrello, S.M.; Daum, R.S.; Edwards, G.F.S.; Lina, G.; Lindsay, J.; Peters, G.; Stefani, S. Meticillin-Resistant Staphylococcus Aureus (MRSA) Update: New Insights into Bacterial Adaptation and Therapeutic Targets. J. Glob. Antimicrob. Resist. 2014, 2, 61–69. [Google Scholar] [CrossRef]
- Sigal, M.V., Jr.; Brent, B.I.; Marchini, P. 7,8,9,10-tetrahalo-6h-cyclohepta-(b)-quinolines U.S. Patent No. 3,232,945, 1 February.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
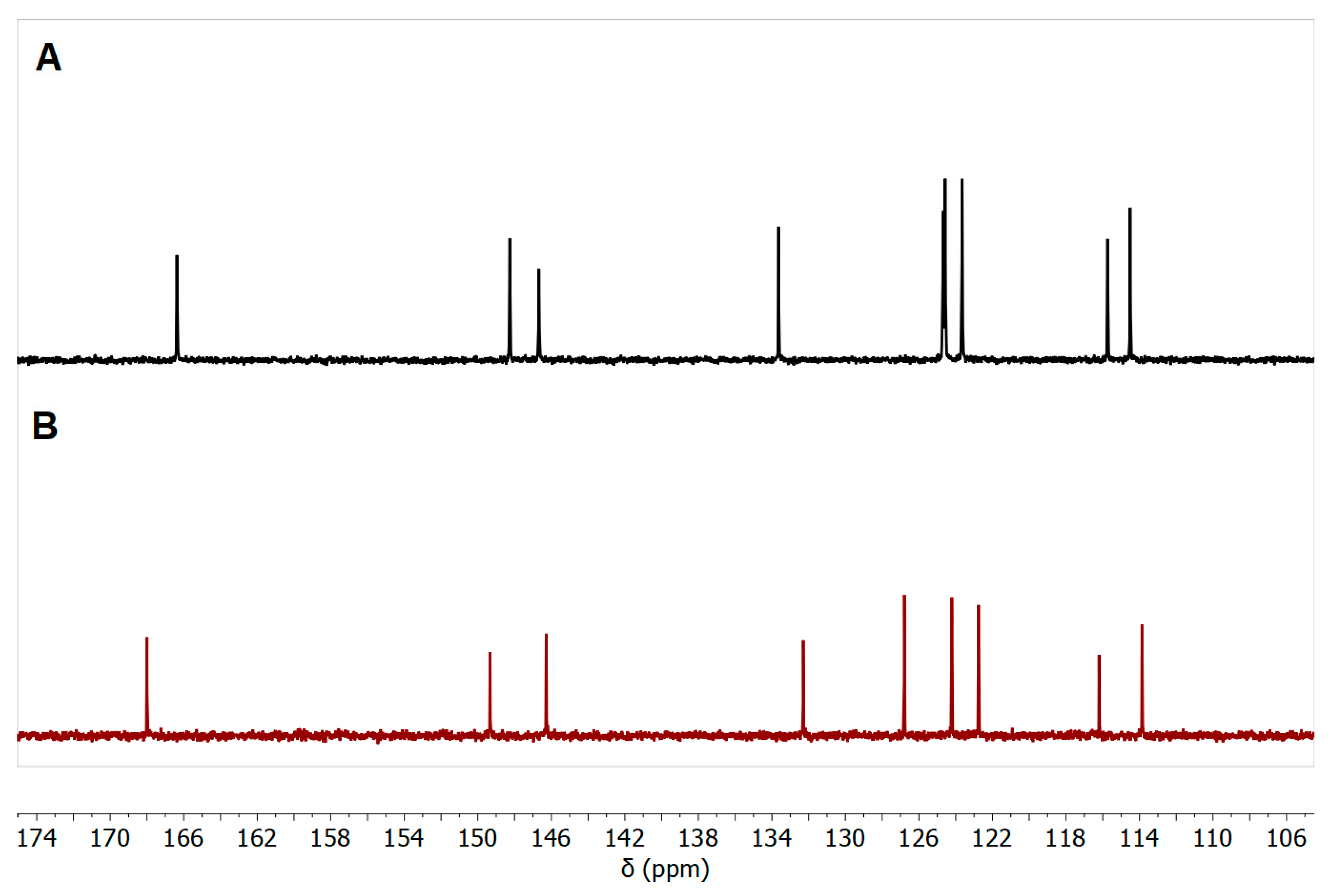
| Cmpd. | NH2 | H-5 | H-7 | H-8 |
|---|---|---|---|---|
| 7b Zn-bound | 7.08 | 7.78 | 7.35 | 8.18 |
| 7b Zn-free | 6.57 | 7.67 | 7.31 | 8.16 |
| Cmpd. | MRSA ATCC 33591 | MRSA Clinical | S. aureus ATCC 6538 | S. pyogenes ATCC 19615 |
|---|---|---|---|---|
| 5b | 12.5 | NZ | NZ | NZ |
| 5e | 8 | NZ | NZ | NZ |
| 7b | 20 | NZ | NZ | NZ |
| 9c | NZ | NZ | NZ | NZ |
| 9d | NZ | 14 | 17 | 22 |
| Imipenem | 26 | 28 | 34 | 29 |
| Cmpd. | MRSA ATCC 33591 | MRSA Clinical | S. aureus ATCC 6538 | S. pyogenes ATCC 19615 | ||||
|---|---|---|---|---|---|---|---|---|
| MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | |
| 7b | 0.125 | 0.5 | NT | NT | NT | NT | NT | NT |
| 9d | NT | NT | 1 | - | 0.5 | 1 | 0.25 | 0.5 |
| Cmpd. | Ligscore1 Dreiding | Ligscore2 Dreiding | PLP1 | PLP2 | Jain | PMF | Docking Score | Ligand Internal Energy |
|---|---|---|---|---|---|---|---|---|
| 7b | 1.5 | 4.03 | 59.15 | 60.01 | −0.3 | 9.93 | 34.317 | −1.916 |
| 9d | 0.69 | 2.05 | 51.16 | 65.23 | 2.15 | −16.25 | 8.764 | −1.148 |
| Cmpd. | Ligscore1 Dreiding | Ligscore2 Dreiding | PLP1 | PLP2 | Jain | PMF | Dock Score | Ligand Internal Energy |
|---|---|---|---|---|---|---|---|---|
| 5e | 0.78 | 2.4 | 16.85 | 20.76 | −1.18 | 56.81 | 13.094 | −0.294 |
| 9d | 2.42 | 3.09 | 24.22 | 42.31 | 2.44 | 78.12 | 20.617 | −1.148 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Momani, L.A.; Shawar, U.A.A.; Sarhan, A.H.A.; Shahin, R.; Koutentis, P.A.; Abu-Sini, M.K.; Mohammad, N.J. Green Synthesis, Characterization, and Biological Activity of 4-Aminoquinoline Derivatives: Exploring Antibacterial Efficacy, MRSA Inhibition, and PBP2a Docking Insights. Chemistry 2025, 7, 71. https://doi.org/10.3390/chemistry7030071
Al-Momani LA, Shawar UAA, Sarhan AHA, Shahin R, Koutentis PA, Abu-Sini MK, Mohammad NJ. Green Synthesis, Characterization, and Biological Activity of 4-Aminoquinoline Derivatives: Exploring Antibacterial Efficacy, MRSA Inhibition, and PBP2a Docking Insights. Chemistry. 2025; 7(3):71. https://doi.org/10.3390/chemistry7030071
Chicago/Turabian StyleAl-Momani, Lo’ay A., Ula A. Abu Shawar, Ayman H. Abu Sarhan, Rand Shahin, Panayiotis A. Koutentis, Mohammad K. Abu-Sini, and Nada J. Mohammad. 2025. "Green Synthesis, Characterization, and Biological Activity of 4-Aminoquinoline Derivatives: Exploring Antibacterial Efficacy, MRSA Inhibition, and PBP2a Docking Insights" Chemistry 7, no. 3: 71. https://doi.org/10.3390/chemistry7030071
APA StyleAl-Momani, L. A., Shawar, U. A. A., Sarhan, A. H. A., Shahin, R., Koutentis, P. A., Abu-Sini, M. K., & Mohammad, N. J. (2025). Green Synthesis, Characterization, and Biological Activity of 4-Aminoquinoline Derivatives: Exploring Antibacterial Efficacy, MRSA Inhibition, and PBP2a Docking Insights. Chemistry, 7(3), 71. https://doi.org/10.3390/chemistry7030071