
8-(Pyridin-2-yl)quinolin-7-ol and Beyond: Theoretical Design of Tautomeric Molecular Switches with Pyridine as a Proton Crane Unit



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
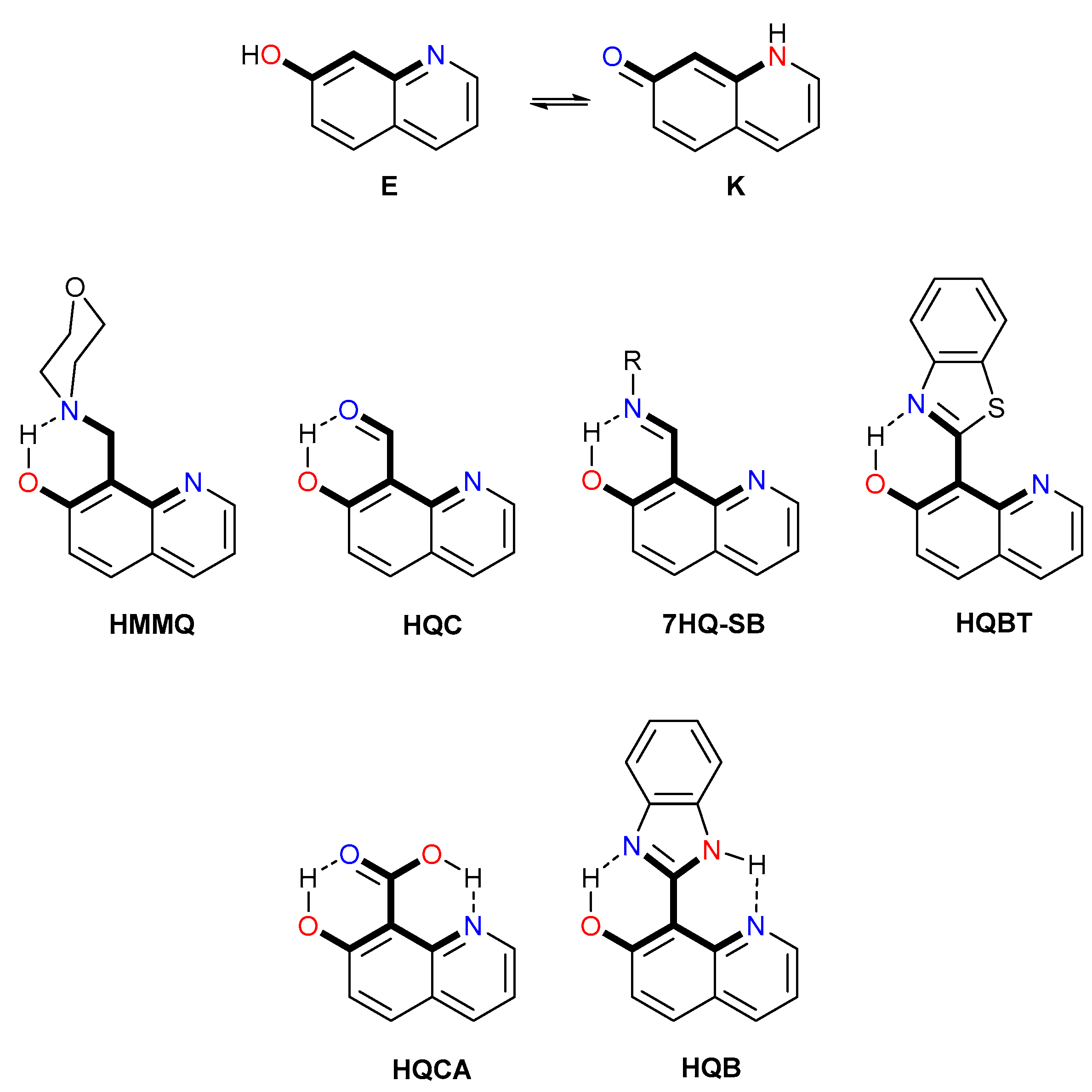

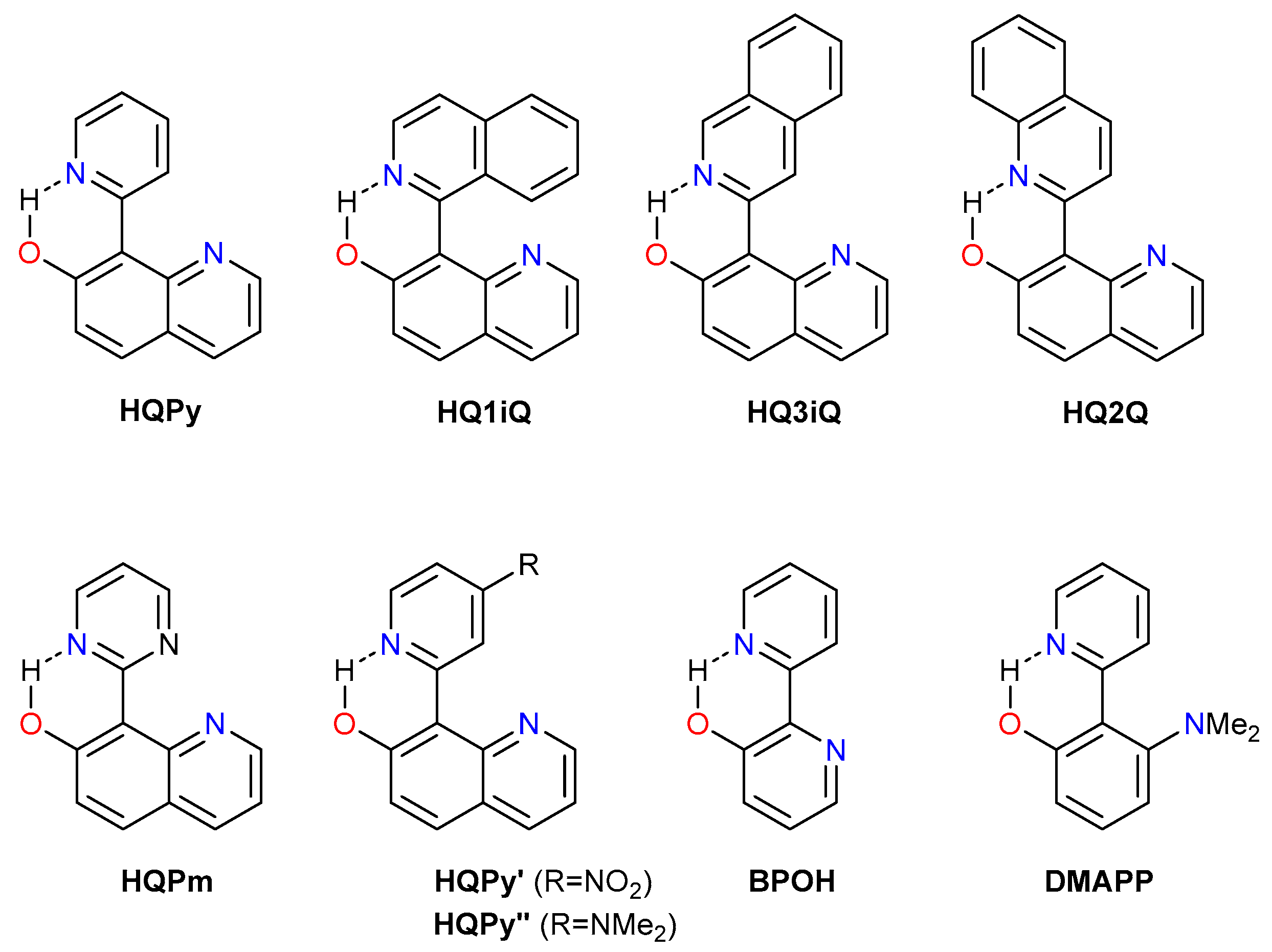
2. Theoretical Methodology
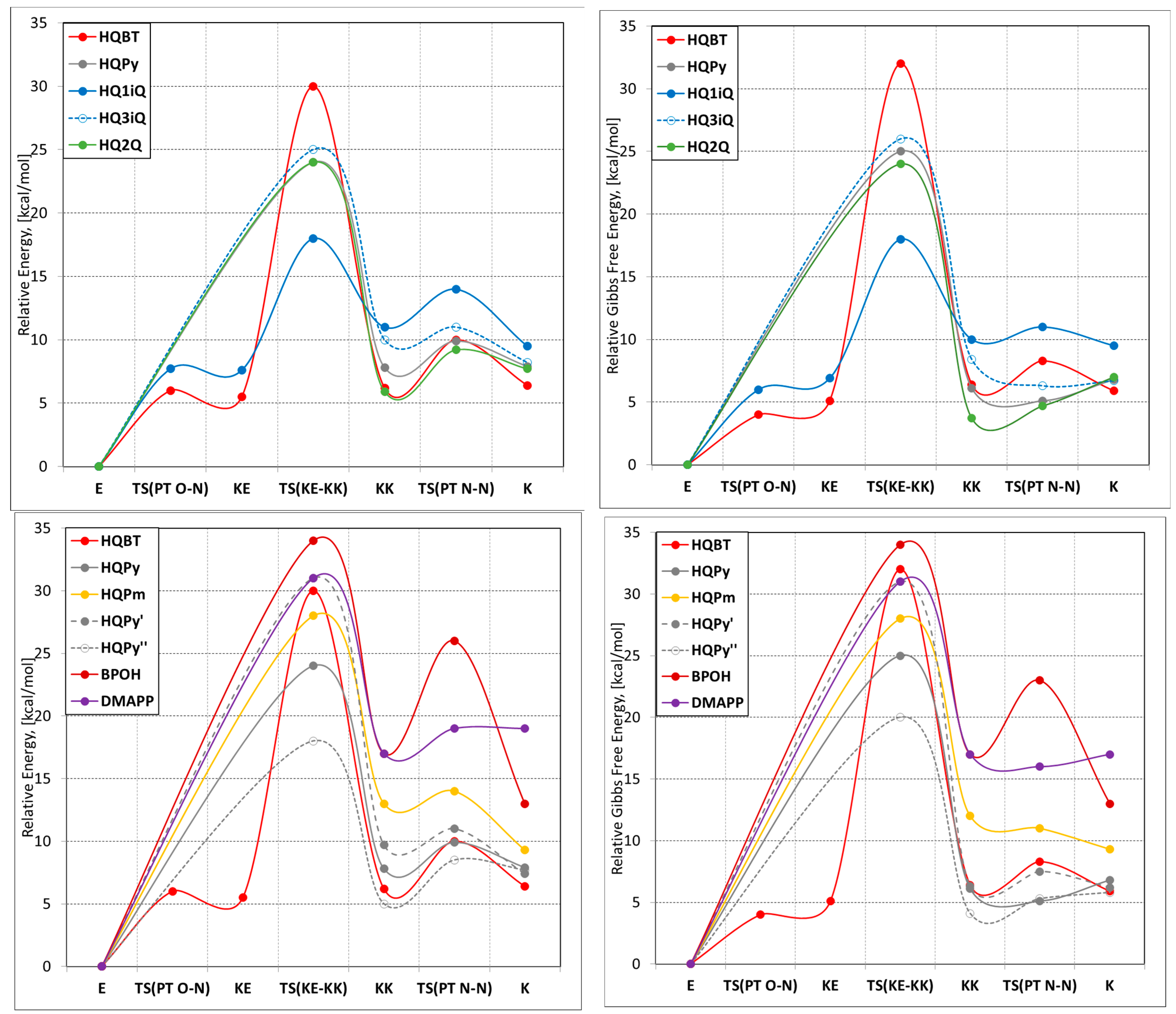
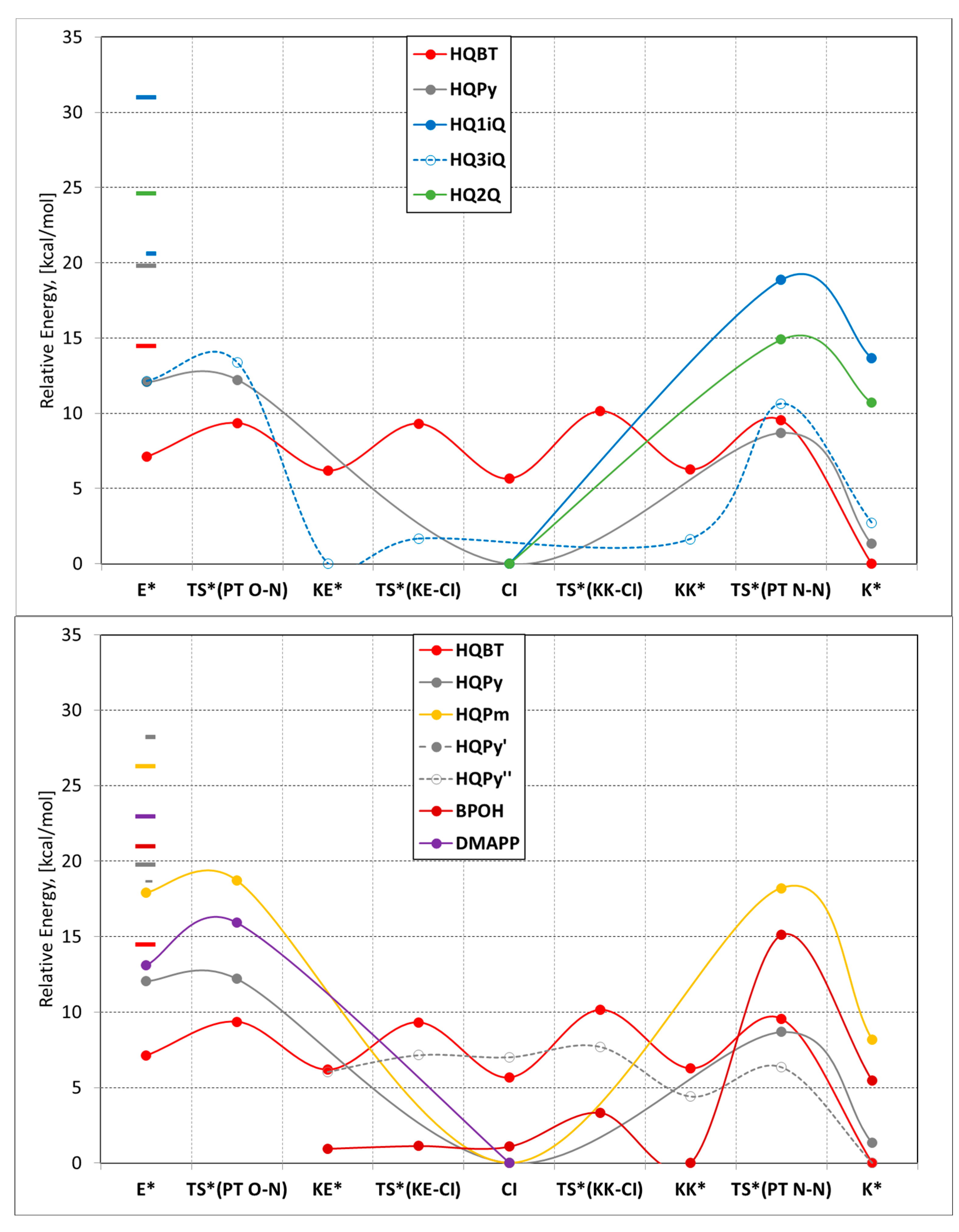
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Molecular Switches, 2nd ed.; Feringa, B.L., Browne, W.R., Eds.; Wiley-VCH: Weinheim, Germany, 2011; ISBN 978-3-527-63440-8. [Google Scholar]
- Molecular Photoswitches; Pianowski, Z.L., Ed.; Wiley-VCH: Weinheim, Germany, 2022; Volume 1, ISBN 978-3-527-35103-9. [Google Scholar]
- Kathan, M.; Hecht, S. Photoswitchable Molecules as Key Ingredients to Drive Systems Away from the Global Thermodynamic Minimum. Chem. Soc. Rev. 2017, 46, 5536–5550. [Google Scholar] [CrossRef] [PubMed]
- Crespi, S.; Simeth, N.A.; König, B. Heteroaryl Azo Dyes as Molecular Photoswitches. Nat. Rev. Chem. 2019, 3, 133–146. [Google Scholar] [CrossRef]
- Lameijer, L.N.; Budzak, S.; Simeth, N.A.; Hansen, M.J.; Feringa, B.L.; Jacquemin, D.; Szymanski, W. General Principles for the Design of Visible-Light-Responsive Photoswitches: Tetra-Ortho-Chloro-Azobenzenes. Angew. Chem. Int. Ed. 2020, 59, 21663–21670. [Google Scholar] [CrossRef] [PubMed]
- Feringa, B.L. The Art of Building Small: From Molecular Switches to Motors (Nobel Lecture). Angew. Chem. Int. Ed. 2017, 56, 11060–11078. [Google Scholar] [CrossRef] [PubMed]
- de Bekker, E.J.A.; Geerlings, J.D.; Varma, C.A.G.O. Mechanism of a Photoinduced Solvent-Assisted Transfer of a Proton to a Specified Remote Target. J. Phys. Chem. A 2000, 104, 5916–5927. [Google Scholar] [CrossRef]
- de Bekker, E.J.A.; Pugzlys, A.; Varma, C.A.G.O. Elementary Processes in Photoinduced Proton Transfers in 2-Hydroxy-1-(N-Morpholinomethyl)Naphthalene and 7-Hydroxy-8-(N-Morpholinomethyl)Quinoline in Liquid Solutions. J. Phys. Chem. A 2001, 105, 399–409. [Google Scholar] [CrossRef]
- van der Loop, T.H.; Ruesink, F.; Amirjalayer, S.; Sanders, H.J.; Buma, W.J.; Woutersen, S. Unraveling the Mechanism of a Reversible Photoactivated Molecular Proton Crane. J. Phys. Chem. B 2014, 118, 12965–12971. [Google Scholar] [CrossRef]
- Vetokhina, V.; Nowacki, J.; Pietrzak, M.; Rode, M.F.; Sobolewski, A.L.; Waluk, J.; Herbich, J. 7-Hydroxyquinoline-8-Carbaldehydes. 1. Ground- and Excited-State Long-Range Prototropic Tautomerization. J. Phys. Chem. A 2013, 117, 9127–9146. [Google Scholar] [CrossRef]
- Georgiev, A.; Yordanov, D.; Ivanova, N.; Deneva, V.; Vassilev, N.; Kamounah, F.S.; Pittelkow, M.; Crochet, A.; Fromm, K.M.; Antonov, L. 7-OH Quinoline Schiff Bases: Are They the Long Awaited Tautomeric Bistable Switches? Dye. Pigment. 2021, 195, 109739. [Google Scholar] [CrossRef]
- Nakashima, K.; Petek, A.; Hori, Y.; Georgiev, A.; Hirashima, S.; Matsushima, Y.; Yordanov, D.; Miura, T.; Antonov, L. Acylhydrazone Subunits as a Proton Cargo Delivery System in 7-Hydroxyquinoline. Chem. Eur. J. 2021, 27, 11559–11566. [Google Scholar] [CrossRef]
- Rehhagen, C.; Argüello Cordero, M.A.; Kamounah, F.S.; Deneva, V.; Angelov, I.; Krupp, M.; Svenningsen, S.W.; Pittelkow, M.; Lochbrunner, S.; Antonov, L. Reversible Switching Based on Truly Intramolecular Long-Range Proton Transfer─Turning the Theoretical Concept into Experimental Reality. J. Am. Chem. Soc. 2024, 146, 2043–2053. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.-C.; Chen, C.-L.; Chuang, H.-H.; Chen, J.-L.; Chen, Y.-J.; Lin, Y.-C.; Shen, J.-Y.; Hu, W.-P.; Chou, P.-T. A Genuine Intramolecular Proton Relay System Undergoing Excited-State Double Proton Transfer Reaction. J. Phys. Chem. Lett. 2011, 2, 3063–3068. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Jia, X.; Ma, Q.; He, Y.; Zhai, H.; Zhang, Y.; Liu, Y. Theoretical Study of the Excited State Intramolecular Double Proton Transfer and Spectral Behaviors of 7-Hydroxyquinoline-8-Carboxylic Acid. J. Mol. Liq. 2020, 302, 112552. [Google Scholar] [CrossRef]
- Kumar, G.; Paul, K.; Luxami, V. Deciphering the Excited State Intramolecular Charge-Coupled Double Proton Transfer in an Asymmetric Quinoline–Benzimidazole System. New J. Chem. 2020, 44, 12866–12874. [Google Scholar] [CrossRef]
- Chou, P.-T. The Host/Guest Type of Excited-State Proton Transfer; a General Review. J. Chin. Chem. Soc. 2001, 48, 651–682. [Google Scholar] [CrossRef]
- Lim, S.-J.; Seo, J.; Park, S.Y. Photochromic Switching of Excited-State Intramolecular Proton-Transfer (ESIPT) Fluorescence: A Unique Route to High-Contrast Memory Switching and Nondestructive Readout. J. Am. Chem. Soc. 2006, 128, 14542–14547. [Google Scholar] [CrossRef]
- Demchenko, A.P.; Tang, K.-C.; Chou, P.-T. Excited-State Proton Coupled Charge Transfer Modulated by Molecular Structure and Media Polarization. Chem. Soc. Rev. 2013, 42, 1379–1408. [Google Scholar] [CrossRef]
- Sedgwick, A.C.; Wu, L.; Han, H.-H.; Bull, S.D.; He, X.-P.; James, T.D.; Sessler, J.L.; Tang, B.Z.; Tian, H.; Yoon, J. Excited-State Intramolecular Proton-Transfer (ESIPT) Based Fluorescence Sensors and Imaging Agents. Chem. Soc. Rev. 2018, 47, 8842–8880. [Google Scholar] [CrossRef]
- Jankowska, J.; Sobolewski, A.L. Modern Theoretical Approaches to Modeling the Excited-State Intramolecular Proton Transfer: An Overview. Molecules 2021, 26, 5140. [Google Scholar] [CrossRef]
- Joshi, H.C.; Antonov, L. Excited-State Intramolecular Proton Transfer: A Short Introductory Review. Molecules 2021, 26, 1475. [Google Scholar] [CrossRef]
- Böhnke, H.; Bahrenburg, J.; Ma, X.; Röttger, K.; Näther, C.; Rode, M.F.; Sobolewski, A.L.; Temps, F. Ultrafast Dynamics of the ESIPT Photoswitch N-(3-Pyridinyl)-2-Pyridinecarboxamide. Phys. Chem. Chem. Phys. 2018, 20, 2646–2655. [Google Scholar] [CrossRef] [PubMed]
- Slavova, S.; Antonov, L. Azaindolizine Proton Cranes Attached to 7-Hydroxyquinoline and 3-Hydroxypyridine: A Comparative Theoretical Study. Phys. Chem. Chem. Phys. 2024, 26, 7177–7189. [Google Scholar] [CrossRef] [PubMed]
- Sobolewski, A.L. Reversible Molecular Switch Driven by Excited-State Hydrogen Transfer. Phys. Chem. Chem. Phys. 2008, 10, 1243–1247. [Google Scholar] [CrossRef] [PubMed]
- Rode, M.F.; Nedeltcheva-Antonova, D.; Antonov, L. Long-Range Proton Transfer in 7-Hydroxy-Quinoline-Based Azomethine Dyes: A Hidden Reason for the Low Efficiency. Molecules 2022, 27, 8225. [Google Scholar] [CrossRef] [PubMed]
- Bulska, H.; Grabowska, A.; Grabowski, Z.R. Single and Double Proton Transfer in Excited Hydroxy Derivatives of Bipyridyl. J. Lumin. 1986, 35, 189–197. [Google Scholar] [CrossRef]
- Lipkowski, J.; Grabowska, A.; Waluk, J.; Calestani, G.; Hess, B.A. Ground State Structures of Molecules ?Prepared? For Phototautomerization: 2,2?-Bipyridyl-3,3?-Diol and 2,2?-Bipyridyl-3-Ol. J. Crystallogr. Spectrosc. Res. 1992, 22, 563–572. [Google Scholar] [CrossRef]
- Kaczmarek, Ł.; Balicki, R.; Lipkowski, J.; Borowicz, P.; Grabowska, A. Structure and Photophysics of Deazabipyridyls. Excited Internally Hydrogen-Bonded Systems with One Proton Transfer Reaction Site. J. Chem. Soc. Perkin Trans. 2 1994, 2, 1603–1610. [Google Scholar] [CrossRef]
- Borowicz, P.; Grabowska, A.; Leś, A.; Kaczmarek, Ł.; Zagrodzki, B. New Phototautomerizing Systems: Non-Symmetric Derivatives of [2,2′-Bipyridyl]-3,3′-Diol. Chem. Phys. Lett. 1998, 291, 351–359. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Peng, C.; Ayala, P.Y.; Schlegel, H.B.; Frisch, M.J. Using Redundant Internal Coordinates to Optimize Equilibrium Geometries and Transition States. J. Comput. Chem. 1996, 17, 49–56. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Density Functionals with Broad Applicability in Chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, S.; Antonov, L. Description of the Tautomerism in Some Azonaphthols. J. Phys. Org. Chem. 2013, 26, 643–652. [Google Scholar] [CrossRef]
- Rayne, S.; Forest, K. A Comparative Examination of Density Functional Performance against the ISOL24/11 Isomerization Energy Benchmark. Comput. Theor. Chem. 2016, 1090, 147–152. [Google Scholar] [CrossRef]
- Antonov, L. Tautomerism in Azo and Azomethyne Dyes: When and If Theory Meets Experiment. Molecules 2019, 24, 2252. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of Electronic Excitations within the Adiabatic Approximation of Time Dependent Density Functional Theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Improta, R. UV-Visible Absorption and Emission Energies in Condensed Phase by PCM/TD-DFT Methods. In Computational Strategies for Spectroscopy; Barone, V., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 37–75. ISBN 978-1-118-00872-0. [Google Scholar]
- Liu, J.; Liang, W. Analytical Approach for the Excited-State Hessian in Time-Dependent Density Functional Theory: Formalism, Implementation, and Performance. J. Chem. Phys. 2011, 135, 184111. [Google Scholar] [CrossRef]
- Guo, Y.; Riplinger, C.; Becker, U.; Liakos, D.G.; Minenkov, Y.; Cavallo, L.; Neese, F. Communication: An Improved Linear Scaling Perturbative Triples Correction for the Domain Based Local Pair-Natural Orbital Based Singles and Doubles Coupled Cluster Method [DLPNO-CCSD(T)]. J. Chem. Phys. 2018, 148, 011101. [Google Scholar] [CrossRef]
- Berraud-Pache, R.; Neese, F.; Bistoni, G.; Izsák, R. Unveiling the Photophysical Properties of Boron-Dipyrromethene Dyes Using a New Accurate Excited State Coupled Cluster Method. J. Chem. Theory Comput. 2020, 16, 564–575. [Google Scholar] [CrossRef]
- Krygowski, T.M.; Cyrański, M.K. Structural Aspects of Aromaticity. Chem. Rev. 2001, 101, 1385–1420. [Google Scholar] [CrossRef] [PubMed]
- Krygowski, T.M.; Szatylowicz, H. Aromaticity: What Does It Mean? ChemTexts 2015, 1, 12. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Chen, S.; Chen, D.; Lin, L.; Xiao, K.; Zhao, L.; Solà, M.; Zhu, J. The Application of Aromaticity and Antiaromaticity to Reaction Mechanisms. Fundam. Res. 2023, 3, 926–938. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Slanina, T.; Bergman, J.; Ottosson, H. Photochemistry Driven by Excited-State Aromaticity Gain or Antiaromaticity Relief. Chem. A Eur. J. 2023, 29, e202203748. [Google Scholar] [CrossRef] [PubMed]
- Lu, T. A Comprehensive Electron Wavefunction Analysis Toolbox for Chemists, Multiwfn. J. Chem. Phys. 2024, 161, 082503. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Antonov, L.; Kawauchi, S.; Okuno, Y. Prediction of the Color of Dyes by Using Time-Dependent Density Functional Theory. Bulg. Chem. Commun. 2014, 46, 228–237. [Google Scholar]
- Becke, A.D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Nedeltcheva-Antonova, D.; Antonov, L. Ground-State Tautomerism and Excited-State Proton Transfer in 7-Hydroxy-4-Methyl-8-((Phenylimino)Methyl)-2H-Chromen-2-One as a Potential Proton Crane. Physchem 2024, 4, 91–105. [Google Scholar] [CrossRef]
- Marx, D. Proton Transfer 200 Years after von Grotthuss: Insights from Ab Initio Simulations. ChemPhysChem 2007, 8, 209–210. [Google Scholar] [CrossRef]
- Ekimova, M.; Hoffmann, F.; Bekçioğlu-Neff, G.; Rafferty, A.; Kornilov, O.; Nibbering, E.T.J.; Sebastiani, D. Ultrafast Proton Transport between a Hydroxy Acid and a Nitrogen Base along Solvent Bridges Governed by the Hydroxide/Methoxide Transfer Mechanism. J. Am. Chem. Soc. 2019, 141, 14581–14592. [Google Scholar] [CrossRef] [PubMed]
- Meuwly, M.; Bach, A.; Leutwyler, S. Grotthus-Type and Diffusive Proton Transfer in 7-Hydroxyquinoline·(NH3)n Clusters. J. Am. Chem. Soc. 2001, 123, 11446–11453. [Google Scholar] [CrossRef] [PubMed]
- Codescu, M.-A.; Weiß, M.; Brehm, M.; Kornilov, O.; Sebastiani, D.; Nibbering, E.T.J. Switching between Proton Vacancy and Excess Proton Transfer Pathways in the Reaction between 7-Hydroxyquinoline and Formate. J. Phys. Chem. A 2021, 125, 1845–1859. [Google Scholar] [CrossRef] [PubMed]
- Codescu, M.-A.; Kunze, T.; Weiß, M.; Brehm, M.; Kornilov, O.; Sebastiani, D.; Nibbering, E.T.J. Ultrafast Proton Transfer Pathways Mediated by Amphoteric Imidazole. J. Phys. Chem. Lett. 2023, 14, 4775–4785. [Google Scholar] [CrossRef]
- Shekhovtsov, N.A.; Bushuev, M.B. Enol or Keto? Interplay between Solvents and Substituents as a Factor Controlling ESIPT. J. Mol. Liq. 2022, 361, 119611. [Google Scholar] [CrossRef]
- Shekhovtsov, N.A.; Nikolaenkova, E.B.; Ryadun, A.A.; Samsonenko, D.G.; Tikhonov, A.Y.; Bushuev, M.B. ESIPT-Capable 4-(2-Hydroxyphenyl)-2-(Pyridin-2-Yl)-1H-Imidazoles with Single and Double Proton Transfer: Synthesis, Selective Reduction of the Imidazolic OH Group and Luminescence. Molecules 2023, 28, 1793. [Google Scholar] [CrossRef]
- Shekhovtsov, N.A.; Nikolaenkova, E.B.; Vorobyeva, S.; Ryadun, A.A.; Bushuev, M.B. Coordination-Driven Molecular Switch on the Base of an ESIPT-Capable Pyrimidine Ligand: Synthesis, Fine-Tuning of Emission by the Halide Anion and Theoretical Studies. J. Photochem. Photobiol. A Chem. 2025, 459, 116091. [Google Scholar] [CrossRef]
- Kaczmarek, Ł.; Zagrodzki, B.; Kamieński, B.; Pietrzak, M.; Schilf, W.; Leś, A. Synthesis and NMR Study of New Derivatives of [2,2′-Bipyridyl]-3,3′-Diol and [2,2′-Bipyridyl]-3-Ol. J. Mol. Struct. 2000, 553, 61–72. [Google Scholar] [CrossRef]
- Taylor, P.J.; van der Zwan, G.; Antonov, L. Tautomerism: Introduction, History, and Recent Developments in Experimental and Theoretical Methods. In Tautomerism: Methods and Theories; Antonov, L., Ed.; Wiley: Weinheim, Germany, 2013; pp. 1–24. ISBN 978-3-527-65882-4. [Google Scholar]
- Gordon, P.F.; Gregory, P. Azo Dyes. In Organic Chemistry in Colour; Springer: Berlin/Heidelberg, Germany, 1987; ISBN 978-3-642-82959-8. [Google Scholar]
- Fabian, J.; Hartmann, H. Light Absorption of Organic Colorants: Theoretical Treatment and Empirical Rules; Springer: Berlin/Heidelberg, Germany, 1980; ISBN 978-3-642-67587-4. [Google Scholar]
- Antonov, L.; Lyčka, A. A Correction of the Neglected Tautomerism in the Spectra Prediction of O-Methoxyaniline Terminated Monoazonaphthols: DFT and 15N NMR Study. J. Mol. Liq. 2024, 406, 125134. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Kosińska, W.; Ośmiałowski, B.; Gawinecki, R. Tautomeric Equilibria in Relation to Pi-Electron Delocalization. Chem. Rev. 2005, 105, 3561–3612. [Google Scholar] [CrossRef]
- Elguero, J. Tautomerism: A Historical Perspective. In Tautomerism: Concepts and Applications in Science and Technology; Antonov, L., Ed.; Wiley-VCH: Weinheim, Germany, 2016; pp. 1–10. ISBN 978-3-527-69571-3. [Google Scholar]
- Gorelik, M.V. Current State of the Problem of Aromaticity. Russ. Chem. Rev. 1990, 59, 116–133. [Google Scholar] [CrossRef]
- Elguero, J.; Katritzky, A.R.; Denisko, O.V. Prototropic Tautomerism of Heterocycles: Heteroaromatic Tautomerism—General Overview and Methodology. In Advances in Heterocyclic Chemistry; Elsevier: Amsterdam, The Netherlands, 2000; Volume 76, pp. 1–84. ISBN 978-0-12-020776-3. [Google Scholar]
- Filarowski, A.; Kochel, A.; Cieslik, K.; Koll, A. Steric and Aromatic Impact on Intramolecular Hydrogen Bonds in o-hydroxyaryl Ketones and Ketimines. J. Phys. Org. Chem. 2005, 18, 986–993. [Google Scholar] [CrossRef]
- Filarowski, A.; Kochel, A.; Kluba, M.; Kamounah, F.S. Structural and Aromatic Aspects of Tautomeric Equilibrium in Hydroxy Aryl Schiff Bases. J. Phys. Org. Chem. 2008, 21, 939–944. [Google Scholar] [CrossRef]
- Martyniak, A.; Majerz, I.; Filarowski, A. Peculiarities of Quasi-Aromatic Hydrogen Bonding. RSC Adv. 2012, 2, 8135. [Google Scholar] [CrossRef]
- Kalapos, P.P.; Kunfi, A.; Bogner, M.M.; Holczbauer, T.; Kochman, M.A.; Durbeej, B.; London, G. Salicylideneaniline/Dithienylethene Hybrid Molecular Switches: Design, Synthesis, and Photochromism. J. Org. Chem. 2024, 89, 16–26. [Google Scholar] [CrossRef]
- Albert, A.; Goldacre, R.; Phillips, J. 455. The Strength of Heterocyclic Bases. J. Chem. Soc. 1948, 2240–2249. [Google Scholar] [CrossRef]
- Perrin, D.D. Dissociation Constants of Organic Bases in Aqueous Solution—Supplement 1972; Pure and Applied Chemistry Supplement; Butterworths: London, UK, 1972; ISBN 978-0-408-70408-3. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaharieva, L.; Nedeltcheva-Antonova, D.; Antonov, L. 8-(Pyridin-2-yl)quinolin-7-ol and Beyond: Theoretical Design of Tautomeric Molecular Switches with Pyridine as a Proton Crane Unit. Chemistry 2024, 6, 1608-1621. https://doi.org/10.3390/chemistry6060097
Zaharieva L, Nedeltcheva-Antonova D, Antonov L. 8-(Pyridin-2-yl)quinolin-7-ol and Beyond: Theoretical Design of Tautomeric Molecular Switches with Pyridine as a Proton Crane Unit. Chemistry. 2024; 6(6):1608-1621. https://doi.org/10.3390/chemistry6060097
Chicago/Turabian StyleZaharieva, Lidia, Daniela Nedeltcheva-Antonova, and Liudmil Antonov. 2024. "8-(Pyridin-2-yl)quinolin-7-ol and Beyond: Theoretical Design of Tautomeric Molecular Switches with Pyridine as a Proton Crane Unit" Chemistry 6, no. 6: 1608-1621. https://doi.org/10.3390/chemistry6060097
APA StyleZaharieva, L., Nedeltcheva-Antonova, D., & Antonov, L. (2024). 8-(Pyridin-2-yl)quinolin-7-ol and Beyond: Theoretical Design of Tautomeric Molecular Switches with Pyridine as a Proton Crane Unit. Chemistry, 6(6), 1608-1621. https://doi.org/10.3390/chemistry6060097