
DFT Investigation of the Mechanism of Methoxycarbonylation of Styrene by Palladium Chloride


Abstract

1. Introduction
2. Computational Details
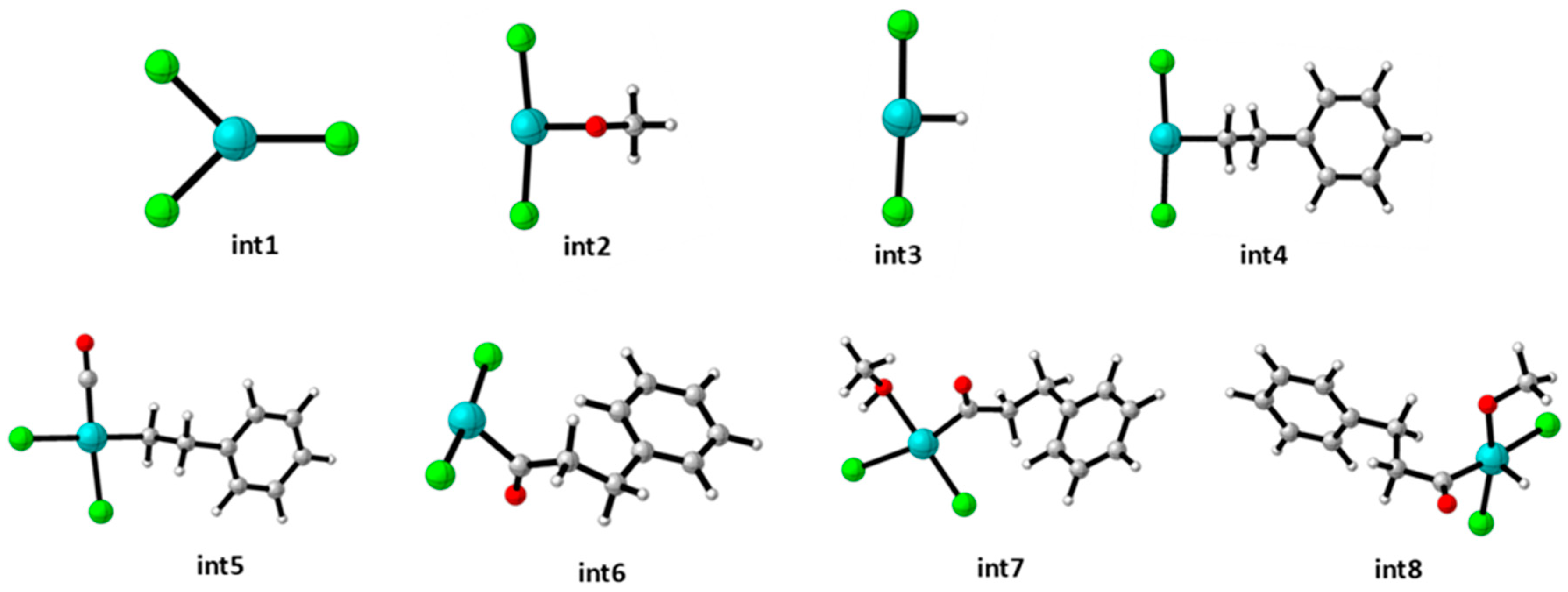
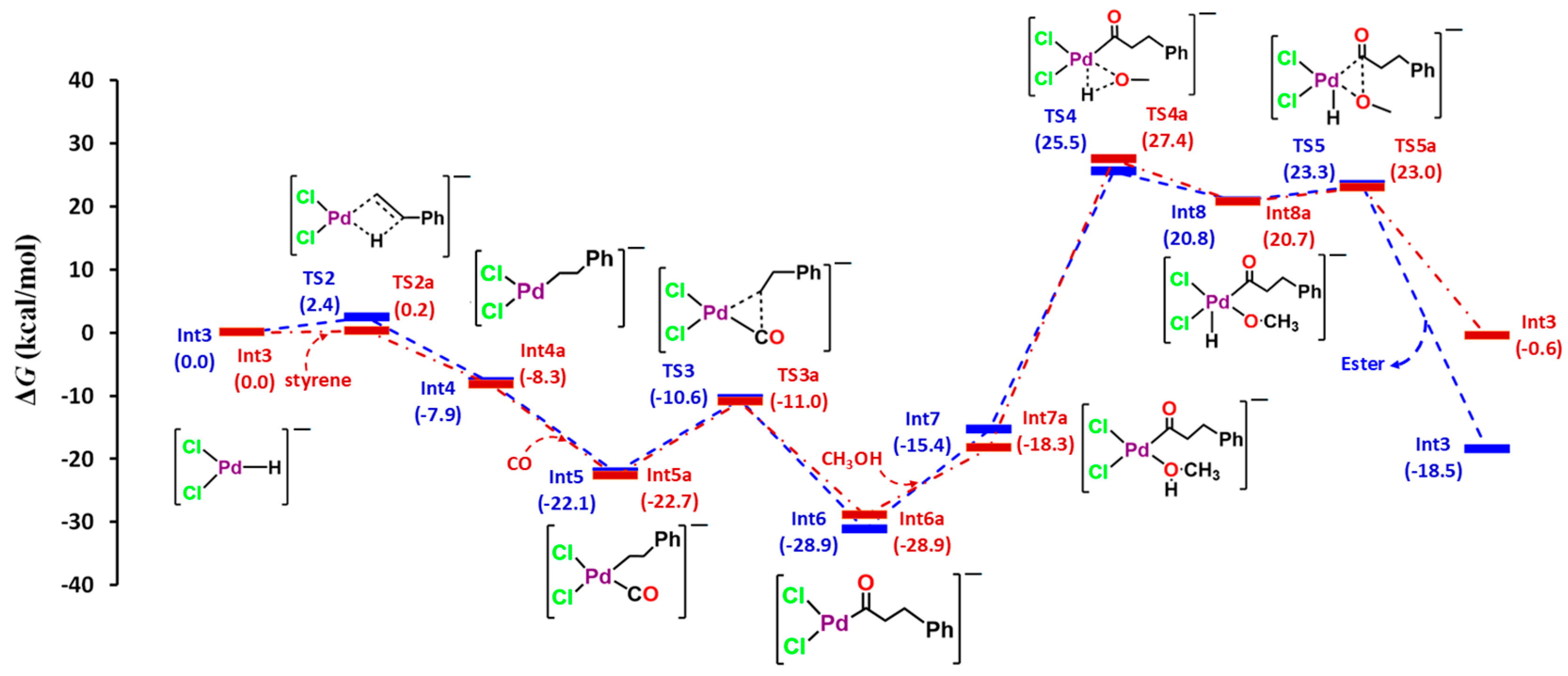
3. Results and Discussions
3.1. Energy Decomposition Analysis (EDA) and Natural Orbital for Chemical Valence (NOCV) Analysis
3.2. Global Electron Density Transfer (GEDT) Analysis
3.3. Quantum Theory of Atom in Molecule (QTAIM) Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peng, J.-B.; Geng, H.-Q.; Wu, X.-F. The Chemistry of CO: Carbonylation. Chem 2019, 5, 526–552. [Google Scholar] [CrossRef]
- Jurado, L.; Posada-Pérez, S.; Axet, M.R. Carbonylation Reactions Using Single-Atom Catalysts. ChemCatChem 2024, e202400543. [Google Scholar] [CrossRef]
- Ma, K.; Martin, B.S.; Yin, X.; Dai, M. Natural product syntheses via carbonylative cyclizations. Nat. Prod. Rep. 2019, 36, 174–219. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-F.; Fang, X.; Wu, L.; Jackstell, R.; Neumann, H.; Beller, M. Transition-Metal-Catalyzed Carbonylation Reactions of Olefins and Alkynes: A Personal Account. Acc. Chem. Res. 2014, 47, 1041–1053. [Google Scholar] [CrossRef] [PubMed]
- Beller, M.; Wu, X.-F. Transition Metal Catalyzed Carbonylation Reactions; Springer: Berlin/Heidelberg, Germany, 2013; ISBN 978-3-642-39015-9. [Google Scholar]
- Wei, W.-M.; Dong, F.-Q.; Zheng, R.-H.; Liu, Y.-Y.; Zhao, T.-T.; Fang, W.-J.; Qin, Y.-D. Theoretical study of the mechanism of palladium-catalyzed hydroaminocarbonylation of styrene with ammonium chloride. Comput. Theor. Chem. 2020, 1191, 113040. [Google Scholar] [CrossRef]
- Cernak, T.A.; Lambert, T.H. Multicatalytic Synthesis of α-Pyrrolidinyl Ketones via a Tandem Palladium(II)/Indium(III)-Catalyzed Aminochlorocarbonylation/Friedel−Crafts Acylation Reaction. J. Am. Chem. Soc. 2009, 131, 3124–3125. [Google Scholar] [CrossRef]
- Malkov, A.V.; Barłóg, M.; Miller-Potucká, L.; Kabeshov, M.A.; Farrugia, L.J.; Kočovský, P. Stereoselective Palladium-Catalyzed Functionalization of Homoallylic Alcohols: A Convenient Synthesis of Di- and Trisubstituted Isoxazolidines and β-Amino-δ-Hydroxy Esters. Chem.–A Eur. J. 2012, 18, 6873–6884. [Google Scholar] [CrossRef]
- Malkov, A.V.; Lee, D.S.; Barłóg, M.; Elsegood, M.R.J.; Kočovský, P. Palladium-Catalyzed Stereoselective Intramolecular Oxidative Amidation of Alkenes in the Synthesis of 1,3- and 1,4-Amino Alcohols and 1,3-Diamines. Chem.–A Eur. J. 2014, 20, 4901–4905. [Google Scholar] [CrossRef]
- Wu, X.-F.; Neumann, H.; Beller, M. Synthesis of Heterocycles via Palladium-Catalyzed Carbonylations. Chem. Rev. 2013, 113, 1–35. [Google Scholar] [CrossRef]
- Kočovský, P.; Bäckvall, J. The syn/anti-Dichotomy in the Palladium-Catalyzed Addition of Nucleophiles to Alkenes. Chem.–A Eur. J. 2015, 21, 36–56. [Google Scholar] [CrossRef]
- Shen, C.; Dong, K. Assembling Chiral Center of Heterocycles by Palladium-Catalyzed Asymmetric Hydrocarbonylation. Synlett 2022, 33, 815–821. [Google Scholar] [CrossRef]
- Gallarati, S.; Dingwall, P.; Fuentes, J.A.; Bühl, M.; Clarke, M.L. Understanding Catalyst Structure–Selectivity Relationships in Pd-Catalyzed Enantioselective Methoxycarbonylation of Styrene. Organometallics 2020, 39, 4544–4556. [Google Scholar] [CrossRef]
- Jing, T.-H.; Zhuang, Y.-Y.; Zhang, X.-X.; Qian, J.-G.; Zhao, X.-L.; Lu, Y.; Wang, H.-J.; Liu, Y. Pinwheel-like tridentate phosphines for controlling divergent regioselectivity in Pd-catalyzed alkoxycarbonylation of alkenes. J. Catal. 2024, 432, 115406. [Google Scholar] [CrossRef]
- Xu, T.; Sha, F.; Alper, H. Highly Ligand-Controlled Regioselective Pd-Catalyzed Aminocarbonylation of Styrenes with Aminophenols. J. Am. Chem. Soc. 2016, 138, 6629–6635. [Google Scholar] [CrossRef]
- Li, H.; Dong, K.; Jiao, H.; Neumann, H.; Jackstell, R.; Beller, M. The scope and mechanism of palladium-catalysed Markovnikov alkoxycarbonylation of alkenes. Nat. Chem. 2016, 8, 1159–1166. [Google Scholar] [CrossRef]
- Aguirre, P.A.; Lagos, C.A.; Moya, S.A.; Zúñiga, C.; Vera-Oyarce, C.; Sola, E.; Peris, G.; Bayón, J.C. Methoxycarbonylation of olefins catalyzed by palladium complexes bearing P,N-donor ligands. Dalt. Trans. 2007, 5419. [Google Scholar] [CrossRef]
- Dai, M.; Wang, C.; Dong, G.; Xiang, J.; Luo, T.; Liang, B.; Chen, J.; Yang, Z. Development of Thiourea-Based Ligands for the Palladium-Catalyzed Bis(methoxycarbonylation) of Terminal Olefins. Eur. J. Org. Chem. 2003, 2003, 4346–4348. [Google Scholar] [CrossRef]
- Fekri, S.; Mansoori, Y. Supported Bis-(NHC)-Pd(II) complex: A phosphine-free catalyst supported on magnetic mesoporous silica for the CO-free carbonylation of aryl halides. J. Organomet. Chem. 2024, 1014, 123193. [Google Scholar] [CrossRef]
- Klingshirn, M.A.; Rogers, R.D.; Shaughnessy, K.H. Palladium-catalyzed hydroesterification of styrene derivatives in the presence of ionic liquids. J. Organomet. Chem. 2005, 690, 3620–3626. [Google Scholar] [CrossRef]
- Sims, H.S.; Dai, M. Palladium-Catalyzed Carbonylations: Application in Complex Natural Product Total Synthesis and Recent Developments. J. Org. Chem. 2023, 88, 4925–4941. [Google Scholar] [CrossRef]
- Zhang, B.; Yuan, H.; Liu, Y.; Deng, Z.; Douthwaite, M.; Dummer, N.F.; Lewis, R.J.; Liu, X.; Luan, S.; Dong, M.; et al. Ambient-pressure alkoxycarbonylation for sustainable synthesis of ester. Nat. Commun. 2024, 15, 7837. [Google Scholar] [CrossRef] [PubMed]
- Schelwies, M.; Paciello, R.; Pelzer, R.; Siegel, W.; Breuer, M. Palladium-Catalyzed Low Pressure Carbonylation of Allylic Alcohols by Catalytic Anhydride Activation. Chem.–A Eur. J. 2021, 27, 9263–9266. [Google Scholar] [CrossRef] [PubMed]
- Fortman, G.C.; Nolan, S.P. N-Heterocyclic carbene (NHC) ligands and palladium in homogeneous cross-coupling catalysis: A perfect union. Chem. Soc. Rev. 2011, 40, 5151. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.-A.; Ye, Z.-S.; Duan, Y.; Zhou, Y.-G. Homogeneous palladium-catalyzed asymmetric hydrogenation. Chem. Soc. Rev. 2013, 42, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Roberts, G.M.; Pierce, P.J.; Woo, L.K. Palladium Complexes with N-Heterocyclic Carbene Ligands As Catalysts for the Alkoxycarbonylation of Olefins. Organometallics 2013, 32, 2033–2036. [Google Scholar] [CrossRef]
- Demir, S.; Özdemir, İ.; Arslan, H.; VanDerveer, D. Butylene linked palladium N-heterocyclic carbene complexes: Synthesis and catalytic properties. J. Organomet. Chem. 2011, 696, 2589–2593. [Google Scholar] [CrossRef]
- Brennführer, A.; Neumann, H.; Beller, M. Palladium-Catalyzed Carbonylation Reactions of Aryl Halides and Related Compounds. Angew. Chemie Int. Ed. 2009, 48, 4114–4133. [Google Scholar] [CrossRef]
- Flahaut, A.; Roland, S.; Mangeney, P. Allylic alkylation and amination using mixed (NHC)(phosphine) palladium complexes under biphasic conditions. J. Organomet. Chem. 2007, 692, 5754–5762. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Lothschütz, C.; Döpp, R.; Rudolph, M.; Ramamurthi, T.D.; Rominger, F. Gold and Palladium Combined for Cross-Coupling. Angew. Chemie Int. Ed. 2009, 48, 8243–8246. [Google Scholar] [CrossRef]
- Amatore, C.; Carre, E.; Jutand, A.; M’Barki, M.A. Rates and Mechanism of the Formation of Zerovalent Palladium Complexes from Mixtures of Pd(OAc)2 and Tertiary Phosphines and Their Reactivity in Oxidative Additions. Organometallics 1995, 14, 1818–1826. [Google Scholar] [CrossRef]
- Tooze, R.P.; Whiston, K.; Malyan, A.P.; Taylor, M.J.; Wilson, N.W. Evidence for the hydride mechanism in the methoxycarbonylation of ethene catalysed by palladium–triphenylphosphine complexes. J. Chem. Soc. Dalt. Trans. 2000, 3441–3444. [Google Scholar] [CrossRef]
- Nguyen, D.H.; Laurenczy, G.; Urrutigoïty, M.; Kalck, P. Hydride Route for the Palladium-Catalysed Cyclocarbonylation of Monoterpenes. Eur. J. Inorg. Chem. 2005, 2005, 4215–4225. [Google Scholar] [CrossRef]
- Mehara, J.; Anania, M.; Kočovský, P.; Roithová, J. Competing Mechanisms in Palladium-Catalyzed Alkoxycarbonylation of Styrene. ACS Catal. 2024, 14, 5710–5719. [Google Scholar] [CrossRef] [PubMed]
- Seayad, A.; Jayasree, S.; Damodaran, K.; Toniolo, L.; Chaudhari, R.V. On the mechanism of hydroesterification of styrene using an in situ-formed cationic palladium complex. J. Organomet. Chem. 2000, 601, 100–107. [Google Scholar] [CrossRef]
- Eastham, G.R.; Tooze, R.P.; Heaton, B.T.; Iggo, J.A.; Whyman, R.; Zacchini, S. Synthesis and spectroscopic characterisation of all the intermediates in the Pd-catalysed methoxycarbonylation of ethene. Chem. Commun. 2000, 609–610. [Google Scholar] [CrossRef]
- Muñoz, B.K.; Santos Garcia, E.; Godard, C.; Zangrando, E.; Bo, C.; Ruiz, A.; Claver, C. HP-NMR Study of the Pd-Catalyzed Methoxycarbonylation of Styrene Using Monodentate and Bidentate Phosphane-Modified Systems. Eur. J. Inorg. Chem. 2008, 2008, 4625–4637. [Google Scholar] [CrossRef]
- Bianchini, C.; Meli, A.; Oberhauser, W.; Parisel, S.; Gusev, O.V.; Kal’sin, A.M.; Vologdin, N.V.; Dolgushin, F.M. Methoxycarbonylation of styrene to methyl arylpropanoates catalyzed by palladium(II) precursors with 1,1′-bis(diphenylphosphino)metallocenes. J. Mol. Catal. A Chem. 2004, 224, 35–49. [Google Scholar] [CrossRef]
- Clegg, W.; Eastham, G.R.; Elsegood, M.R.J.; Heaton, B.T.; Iggo, J.A.; Tooze, R.P.; Whyman, R.; Zacchini, S. Characterization and Dynamics of [Pd(L−L)H(solv)]+, [Pd(L−L)(CH2CH3)]+, and [Pd(L−L)(C(O)Et)(THF)]+ (L−L = 1,2-(CH2PBut2)2C6H4): Key Intermediates in the Catalytic Methoxycarbonylation of Ethene to Methylpropanoate. Organometallics 2002, 21, 1832–1840. [Google Scholar] [CrossRef]
- Naigre, R.; Chenal, T.; Ciprés, I.; Kalck, P.; Daran, J.-C.; Vaissermann, J. Carbon monoxide as a building block in organic synthesis. Part V. Involvement of palladium-hydride species in carbonylation reactions of monoterpenes. X-ray crystal structure of [Ph3PCH2CH=CHPh]4[PdCl6][SnCl6]. J. Organomet. Chem. 1994, 480, 91–102. [Google Scholar] [CrossRef]
- Eastham, G.R.; Tooze, R.P.; Kilner, M.; Foster, D.F.; Cole-Hamilton, D.J. Deuterium labelling evidence for a hydride mechanism in the formation of methyl propanoate from carbon monoxide, ethene and methanol catalysed by a palladium complex. J. Chem. Soc. Dalt. Trans. 2002, 1613–1617. [Google Scholar] [CrossRef]
- Liu, J.; Heaton, B.T.; Iggo, J.A.; Whyman, R.; Bickley, J.F.; Steiner, A. The Mechanism of the Hydroalkoxycarbonylation of Ethene and Alkene–CO Copolymerization Catalyzed by Pd II –Diphosphine Cations. Chem.–A Eur. J. 2006, 12, 4417–4430. [Google Scholar] [CrossRef] [PubMed]
- Amadio, E.; Cavinato, G.; Dolmella, A.; Toniolo, L. Catalytic Properties of [Pd(COOMe)nX2−n(PPh3)2] (n = 0, 1, 2; X = Cl, NO2, ONO2, OAc and OTs) in the Oxidative Carbonylation of MeOH. Inorg. Chem. 2010, 49, 3721–3729. [Google Scholar] [CrossRef] [PubMed]
- Malkov, A.V.; Derrien, N.; Barłóg, M.; Kočovský, P. Palladium-Catalyzed Alkoxycarbonylation of Terminal Alkenes To Produce α,β-Unsaturated Esters: The Key Role of Acetonitrile as a Ligand. Chem.–A Eur. J. 2014, 20, 4542–4547. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Bühl, M. Computational modelling of Pd-catalysed alkoxycarbonylation of alkenes and alkynes. Phys. Chem. Chem. Phys. 2021, 23, 15869–15880. [Google Scholar] [CrossRef]
- van Leeuwen, P.W.N.M.; Zuideveld, M.A.; Swennenhuis, B.H.G.; Freixa, Z.; Kamer, P.C.J.; Goubitz, K.; Fraanje, J.; Lutz, M.; Spek, A.L. Alcoholysis of Acylpalladium(II) Complexes Relevant to the Alternating Copolymerization of Ethene and Carbon Monoxide and the Alkoxycarbonylation of Alkenes: The Importance of Cis-Coordinating Phosphines. J. Am. Chem. Soc. 2003, 125, 5523–5539. [Google Scholar] [CrossRef]
- Clegg, W.; Eastham, G.R.; Elsegood, M.R.J.; Heaton, B.T.; Iggo, J.A.; Tooze, R.P.; Whyman, R.; Zacchini, S. Synthesis and reactivity of palladium hydrido-solvento complexes, including a key intermediate in the catalytic methoxycarbonylation of ethene to methyl propanoate. J. Chem. Soc. Dalt. Trans. 2002, 3300–3308. [Google Scholar] [CrossRef]
- Ahmad, S.; Crawford, L.E.; Bühl, M. Palladium-catalysed methoxycarbonylation of ethene with bidentate diphosphine ligands: A density functional theory study. Phys. Chem. Chem. Phys. 2020, 22, 24330–24336. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew–Burke–Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Dolg, M.; Stoll, H.; Preuss, H.; Pitzer, R.M. Relativistic and correlation effects for element 105 (hahnium, Ha): A comparative study of M and MO (M = Nb, Ta, Ha) using energy-adjusted ab initio pseudopotentials. J. Phys. Chem. 1993, 97, 5852–5859. [Google Scholar] [CrossRef]
- Bergner, A.; Dolg, M.; Küchle, W.; Stoll, H.; Preuß, H. Ab initio energy-adjusted pseudopotentials for elements of groups 13–17. Mol. Phys. 1993, 80, 1431–1441. [Google Scholar] [CrossRef]
- Das, D.; Prakash, D.; Sarkar, P.; Sutradhar, S.; Patra, S.G.; Ghosh, B.N. Experimental and theoretical studies for instantaneous detection of l-cysteine and l-histidine using a simple Cu(II)-dppy complex. Inorganica Chim. Acta 2024, 573, 122346. [Google Scholar] [CrossRef]
- Sutradhar, S.; Suresh, G.K.; Sarma, S.; Das, D.; Sarkar, P.; Patra, S.G.; Ghosh, B.N. Sulphide and iodide anion recognition by a selective copper hydrogel of a chloro substituted terpyridine ligand. Inorg. Chem. Commun. 2024, 170, 113263. [Google Scholar] [CrossRef]
- Gogoi, B.; Borgohain, P.; Patra, S.G.; Borah, B.J.; Das, P. Pd-PEPPSI Catalyzed Dehydrogenative Oxidation of Alcohols to Carboxylic Acids: An Experimental and Theoretical Study. Organometallics 2024, 43, 2002–2015. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Martin, R.L.; Hay, P.J.; Pratt, L.R. Hydrolysis of Ferric Ion in Water and Conformational Equilibrium. J. Phys. Chem. A 1998, 102, 3565–3573. [Google Scholar] [CrossRef]
- Sparta, M.; Riplinger, C.; Neese, F. Mechanism of Olefin Asymmetric Hydrogenation Catalyzed by Iridium Phosphino-Oxazoline: A Pair Natural Orbital Coupled Cluster Study. J. Chem. Theory Comput. 2014, 10, 1099–1108. [Google Scholar] [CrossRef]
- Fantuzzi, F.; Nascimento, M.A.C.; Ginovska, B.; Bullock, R.M.; Raugei, S. Splitting of multiple hydrogen molecules by bioinspired diniobium metal complexes: A DFT study. Dalt. Trans. 2021, 50, 840–849. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Gopal Patra, S.; Kumar Chattaraj, P. Three coordinated first row-transition metal complexes, [M{N(SiMe3)2}3]−1/0: Structure, bonding, and magnetic properties. Polyhedron 2024, 256, 116990. [Google Scholar] [CrossRef]
- Pal, R.; Patra, S.G.; Chattaraj, P.K. Can a chemical bond be exclusively covalent or ionic? J. Chem. Sci. 2022, 134, 108. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Simple, Efficient, and Universal Energy Decomposition Analysis Method Based on Dispersion-Corrected Density Functional Theory. J. Phys. Chem. A 2023, 127, 7023–7035. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Mondal, H.; Patra, S.G.; Chattaraj, P.K. Small molecule activation and dehydrogenation of an amine–borane system using frustrated Lewis pairs. Struct. Chem. 2022, 33, 1853–1865. [Google Scholar] [CrossRef]
- Patra, S.G.; Jha, R.; Mondal, H.; Chattaraj, P.K. Fischer and Schrock carbene complexes in the light of global and local electrophilicity-based descriptors. J. Phys. Org. Chem. 2022, 2, e4337. [Google Scholar] [CrossRef]
- Mondal, H.; Patra, S.G.; Chattaraj, P.K. Unveiling novel reactivity of P/Al frustrated Lewis pair: Ring size-dependent activation of cyclic ethers/thioethers and CO2 insertion therein. J. Chem. Sci. 2022, 0123456789, 39–42. [Google Scholar] [CrossRef]
- Walther, G.; Knöpke, L.R.; Rabeah, J.; Chęciński, M.P.; Jiao, H.; Bentrup, U.; Brückner, A.; Martin, A.; Köckritz, A. From sunflower oil toward 1,19-diester: Mechanistic elucidation. J. Catal. 2013, 297, 44–55. [Google Scholar] [CrossRef]
- Roesle, P.; Caporaso, L.; Schnitte, M.; Goldbach, V.; Cavallo, L.; Mecking, S. A Comprehensive Mechanistic Picture of the Isomerizing Alkoxycarbonylation of Plant Oils. J. Am. Chem. Soc. 2014, 136, 16871–16881. [Google Scholar] [CrossRef]
- Dong, K.; Sang, R.; Wei, Z.; Liu, J.; Dühren, R.; Spannenberg, A.; Jiao, H.; Neumann, H.; Jackstell, R.; Franke, R.; et al. Cooperative catalytic methoxycarbonylation of alkenes: Uncovering the role of palladium complexes with hemilabile ligands. Chem. Sci. 2018, 9, 2510–2516. [Google Scholar] [CrossRef] [PubMed]
- Doming, L.R.; Ríos-Gutiérrez, M. A Useful Classification of Organic Reactions Based on the Flux of the Electron Density. Sci. Radices 2023, 2, 1–24. [Google Scholar] [CrossRef]
- Domingo, L.R. A new C–C bond formation model based on the quantum chemical topology of electron density. RSC Adv. 2014, 4, 32415–32428. [Google Scholar] [CrossRef]
- Patra, S.G.; Drew, M.G.B.; Datta, D. δ-Acidity of benzene in [(benzene)RuII(N-N)Cl]+. Crystal structures, nuclear magnetic resonance spectra and nucleus independent chemical shifts. Inorganica Chim. Acta 2018, 471, 228–233. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Essén, H. The characterization of atomic interactions. J. Chem. Phys. 1984, 80, 1943–1960. [Google Scholar] [CrossRef]
- Bader, R.F.W. A Bond Path: A Universal Indicator of Bonded Interactions. J. Phys. Chem. A 1998, 102, 7314–7323. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules, A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
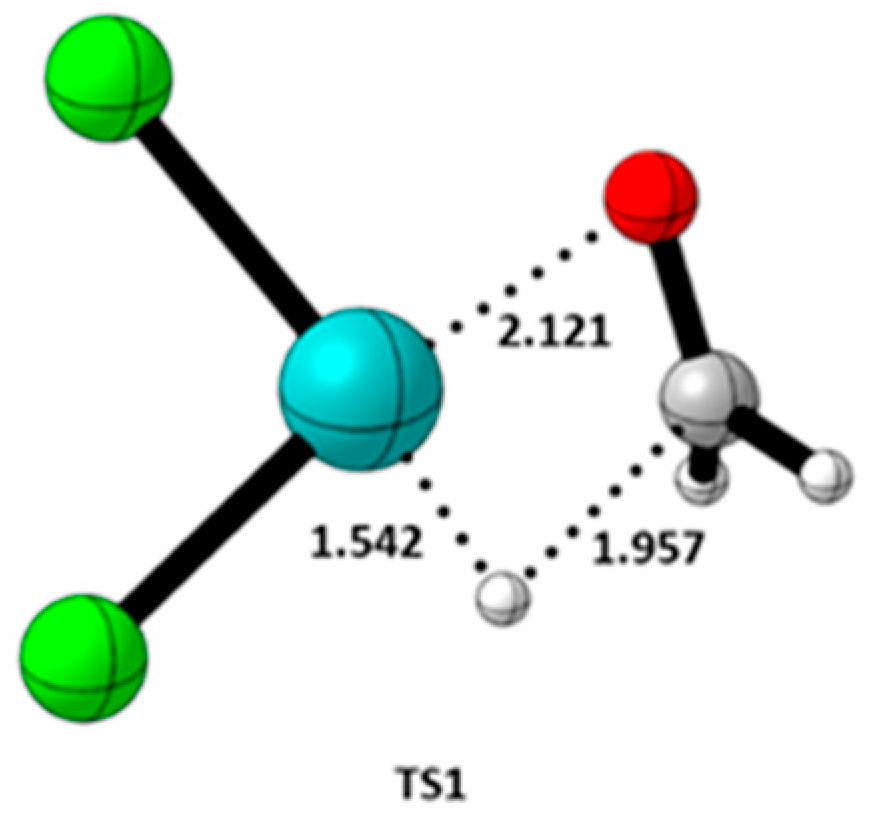
| TS | Fragment 1 (Charge, Spin Multiplicity) | Fragment 2 (Charge, Spin Multiplicity) |
|---|---|---|
| TS1 | HCHO (0, 1) | [PdCl2H]– (−1, 1) |
| TS2 | styrene (0, 1) | [PdCl2H]– (−1, 1) |
| TS2a | styrene (0, 1) | [PdCl2H]– (−1, 1) |
| TS3 | PhCH2CH2 (−1, 1) | [PdCl2CO] (1, 1) |
| TS3a | PhCH3CH (−1, 1) | [PdCl2CO] (1, 1) |
| TS4 | CH3O (−1, 1) | [PdCl2H(PhCH2CH2CO)] (−1, 1) |
| TS4a | CH3O (−1, 1) | [PdCl2H(Ph(CH3)CHCO)] (0, 1) |
| TS5 | CH3O (−1, 1) | [PdCl2H(PhCH2CH2CO)] (0, 1) |
| TS5a | CH3O (−1, 1) | [PdCl2H(Ph(CH3)CHCO)] (0, 1) |
| Species | ∆Eels | ∆Eex | ∆Epauli | ∆Eorb | ∆Ecor | ∆Edisp | ∆Etot |
|---|---|---|---|---|---|---|---|
| TS1 | −80.6 | −74.2 | 237.0 | −96.9 | −13.9 | −3.3 | −32.1 |
| (15.9) | (14.6) | (46.8) | (19.1) | (2.7) | (0.6) | ||
| TS2 | −114.2 | −110.5 | 334.2 | −120.8 | −18.4 | −7.4 | −37.3 |
| (16.1) | (15.6) | (47.3) | (17.1) | (2.6) | (1.0) | ||
| TS2a | −139.1 | −115.3 | 339.2 | −198.1 | −20.9 | −7.6 | −142.0 |
| (16.9) | (14.0) | (41.3) | (24.1) | (2.5) | (0.9) | ||
| TS3 | −192.6 | −127.7 | 428.8 | −217.3 | −17.7 | −7.3 | −134.1 |
| (19.4) | (12.8) | (43.2) | (21.9) | (1.7) | (0.7) | ||
| TS3a | −150.1 | −104.5 | 349.3 | −182.2 | −17.3 | −9.9 | −114.8 |
| (18.4) | (12.8) | (42.9) | (22.4) | (2.1) | (1.2) | ||
| TS4 | −117.1 | −74.3 | 224.6 | −123.0 | −14.6 | −5.1 | −109.5 |
| (20.9) | (13.3) | (40.2) | (22.0) | (2.6) | (0.9) | ||
| TS4a | −118.4 | −77.0 | 232.3 | −125.9 | −15.4 | −5.6 | −110.1 |
| (20.6) | (13.4) | (40.4) | (21.9) | (2.6) | (0.9) | ||
| TS5 | −132.3 | −81.5 | 253.0 | −122.4 | −16.5 | −6.0 | −105.8 |
| (21.6) | (13.3) | (41.3) | (20.0) | (2.7) | (0.9) | ||
| TS5a | −130.0 | −81.3 | 252.2 | −121.7 | −16.8 | −6.4 | −104.2 |
| (21.3) | (13.3) | (41.4) | (20.0) | (2.7) | (1.0) |
| TS | Distance (Å) | Bond | ρ(rc) | ∇2ρ(rc) | G(rc) | V(rc) | H(rc) | ELF | λ |
|---|---|---|---|---|---|---|---|---|---|
| TS1 | 1.957 | C-H | 0.3008 | 0.1226 | 0.1235 | −0.2163 | −0.0928 | 0.6343 | −0.3008 |
| TS1 | 1.542 | Pd-H | 0.1369 | 0.0699 | 0.0985 | −0.1795 | −0.0810 | 0.5295 | −0.1369 |
| TS2/ | 1.683/ | C-H | 0.3091/ | 0.0907/ | 0.1239 | −0.2253/ | −0.1013/ | 0.6532/ | −0.3091/ |
| TS2a | 1.704 | 0.0823 | 0.0419 | 0.0359 | −0.0613 | −0.0254 | 0.6076 | −0.0823 | |
| TS2/ | 1.558 | Pd-H | 0.1418/ | 0.0625/ | 0.1022/ | −0.1889/ | −0.8664/ | 0.5397/ | −0.1418/ |
| TS2a | 1.553 | 0.1442 | 0.0513 | 0.102 | −0.1912 | −0.0892 | 0.5548 | −0.1442 | |
| TS2/ | 1.421 | C-C | 0.2896/ | −0.7424 | 0.0984/ | −0.3825 | −0.2840 | 0.9317 | −0.2896 |
| TS2a | 1.417 | 0.2923 | −0.7560 | 0.0981 | −0.3855 | −0.2873 | 0.9341 | −0.2923 | |
| TS2a | 2.164 | Pd-C | 0.089 | 0.254 | 0.0862 | −0.1090 | −0.0227 | 0.2586 | −0.0890 |
| TS2/ | 2.076 | Pd-C | 0.1045/ | 0.2010/ | 0.0865/ | −0.1229/ | −0.0363/ | 0.3713/ | −0.1045/ |
| TS2a | 2.122 | 0.0947 | 0.2143 | 0.0825 | −0.1114 | −0.0289 | 0.3197 | −0.0947 | |
| TS3 | 3.321 | Cl-H(Ph) | 0.0456 | 0.11813 | 0.0241 | −0.0187 | 0.01962 | 0.146 | −0.0456 |
| TS3/ | 1.911/ | C-C | 0.0405/ | −0.0663/ | 0.1452/ | −0.3070/ | −0.1618/ | 0.7724/ | −0.1102/ |
| TS3a | 1.951 | 0.1026 | −0.1231 | 0.1335 | −0.2754 | −0.1419 | 0.7599 | −0.1026 | |
| TS4 | 2.801/ | Cl-H(Ph) | 0.1091/ | 0.0843/ | 0.0628/ | −0.0482/ | 0.0145/ | 0.1421/ | −0.1091/ |
| TS4a | 2.847 | 0.052 | 0.2001 | 0.0433 | −0.1345 | 0.0245 | 0.1438 | −0.0520 | |
| TS4 | 2.820/ | Cl-H(CH3) | 0.0377/ | 0.1351/ | 0.1008/ | −0.0777/ | 0.0231/ | 0.1230/ | −0.0377/ |
| TS4a | 2.852 | 0.1131 | 0.0987 | 0.073 | −0.0555 | 0.0175 | 0.1193 | −0.1131 | |
| TS4a | 2.315 | H-O | 0.0377 | 0.1413 | 0.1052 | −0.0808 | 0.0243 | 0.113 | −0.0377 |
| TS5 | 2.343/ | O-H(Ph) | 0.0444 | 0.1488 | 0.1179 | −0.0993 | 0.0185 | 0.1541 | −0.0445 |
| TS5a | 2.561 | 0.1228 | 0.1277 | 0.0972 | −0.0774 | 0.0198 | 0.0893 | −0.1228 | |
| TS5 | 2.011 | H-H | 0.0419 | 0.1381 | 0.1063 | −0.0859 | 0.0204 | 0.1553 | −0.0419 |
| TS5a | 2.031 | 0.0429 | 0.1381 | 0.1064 | −0.0862 | 0.0202 | 0.167 | −0.0429 | |
| TS5 | 2.006/ | C-O | 0.2567 | 0.1706 | 0.1907 | −0.2245 | −0.1237 | 0.3001 | −0.2567 |
| TS5a | 2.011 | 0.2537 | 0.1724 | 0.1905 | −0.2223 | −0.1168 | 0.2924 | −0.2537 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patra, S.G.; Saha, A.; Chattaraj, P.K. DFT Investigation of the Mechanism of Methoxycarbonylation of Styrene by Palladium Chloride. Chemistry 2024, 6, 1593-1607. https://doi.org/10.3390/chemistry6060096
Patra SG, Saha A, Chattaraj PK. DFT Investigation of the Mechanism of Methoxycarbonylation of Styrene by Palladium Chloride. Chemistry. 2024; 6(6):1593-1607. https://doi.org/10.3390/chemistry6060096
Chicago/Turabian StylePatra, Shanti Gopal, Aritra Saha, and Pratim Kumar Chattaraj. 2024. "DFT Investigation of the Mechanism of Methoxycarbonylation of Styrene by Palladium Chloride" Chemistry 6, no. 6: 1593-1607. https://doi.org/10.3390/chemistry6060096
APA StylePatra, S. G., Saha, A., & Chattaraj, P. K. (2024). DFT Investigation of the Mechanism of Methoxycarbonylation of Styrene by Palladium Chloride. Chemistry, 6(6), 1593-1607. https://doi.org/10.3390/chemistry6060096