
Rapid and Accurate Prediction of the Melting Point for Imidazolium-Based Ionic Liquids by Artificial Neural Network


 , and
, and
Abstract
:
1. Introduction
2. Data and Models
2.1. Data and Descriptor Selection
2.1.1. Database
2.1.2. Data Pre-Processing
- (1)
- If a particular melting point value for ILs with multiple melting temperature (Tm) values occurs three times or more in different experiments, then it is considered to be accurate.
- (2)
- If the variation in experimental melting point temperatures measured for a single IL falls within a range of less than 10 K, and no identical value occurs more than three times, then the mean value is chosen.
- (3)
- If there are data points that appear at least three times but differ from other data by no more than 10 K, the method of calculating the average is chosen.
- (4)
- If measurements of melting point temperatures for a single IL show variations exceeding 10 K across different literature sources, with no repeated occurrence of the same value more than three times, or if multiple instances of a value appear more than three times but their differences exceed 10 K, then these discrepancies imply a debatable nature of the melting point of the IL, warranting the utilization of the model for verification purposes.
2.1.3. Selection of Descriptors
2.1.4. The Calculation of the Descriptors
2.2. Model
2.3. Validation
3. Results and Discussions
3.1. Data Processing Results
3.2. Descriptor Importance
3.3. Model Training Results

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Descriptor Count | Descriptor Type | Model | R2 | R | MAE | RMSE | Ref. |
|---|---|---|---|---|---|---|---|---|
| 126 pyridinium bromides | 1085 | Constitutional, 2D and 3D | CPG NNs | 0.748 | —— | 18.07 | 23.41 | [28] |
| 126 pyridinium bromides | —— | Positional trees | RNN | —— | Training 0.9782 Test 0.8725 | Training 7.63 Test 19.37 | Training 10.08 Test 23.78 | [29] |
| 711 ILs | 2837 | Fragment, Fragment property | BPNN | Training 0.77 Test 0.58 | —— | Test 31.50 | Training 30.00 Test 39.90 | [66] |
| 667 ILs | 55 | Group contribution | ANN | —— | —— | Training %MAE 3.70 Test %MAE 14.60 | —— | [30] |
| 2212 ILs | —— | SMILES | Transformer-CNN | Training 0.63 Test 0.55 | —— | —— | Training 50.00 Test 45.00 | [64] |
| 1253 ILs | 137 | Constitutional, 2D and 3D | RNN | 0.90 | —— | —— | 32.88 | [34] |
| 280 imidazolium ILs | 12 | PM7 | MLP (ANN) | 0.75 | 0.87 | 25.03 | 33.75 | This work |
3.4. Model Validation Result
3.5. Challenges, Research Gaps, and Future Directions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Torrecilla, J.S.; Rodriguez, F.; Bravo, J.L.; Rothenberg, G.; Seddon, K.R.; Lopez-Martin, I. Optimising an artificial neural network for predicting the melting point of ionic liquids. Phys. Chem. Chem. Phys. 2008, 10, 5826–5831. [Google Scholar] [CrossRef] [PubMed]
- Dong, K.; Liu, X.; Dong, H.; Zhang, X.; Zhang, S. Multiscale Studies on Ionic Liquids. Chem. Rev. 2017, 117, 6636–6695. [Google Scholar] [CrossRef] [PubMed]
- Nordness, O.; Brennecke, J.F. Ion Dissociation in Ionic Liquids and Ionic Liquid Solutions. Chem. Rev. 2020, 120, 12873–12902. [Google Scholar] [CrossRef] [PubMed]
- Amarasekara, A.S. Acidic Ionic Liquids. Chem. Rev. 2016, 116, 6133–6183. [Google Scholar] [CrossRef]
- Noorhisham, N.A.; Amri, D.; Mohamed, A.H.; Yahaya, N.; Ahmad, N.M.; Mohamad, S.; Kamaruzaman, S.; Osman, H. Characterisation techniques for analysis of imidazolium-based ionic liquids and application in polymer preparation: A review. J. Mol. Liq. 2021, 326, 115340. [Google Scholar] [CrossRef]
- Hayes, R.; Warr, G.G.; Atkin, R. Structure and nanostructure in ionic liquids. Chem. Rev. 2015, 115, 6357–6426. [Google Scholar] [CrossRef]
- Singh, S.K.; Savoy, A.W. Ionic liquids synthesis and applications: An overview. J. Mol. Liq. 2020, 297, 112038. [Google Scholar] [CrossRef]
- Zhang, Y.; Chan, J.Y.G. Sustainable chemistry: Imidazolium salts in biomass conversion and CO2 fixation. Energy Environ. Sci. 2010, 3, 408–417. [Google Scholar] [CrossRef]
- Werner, S.; Haumann, M.; Wasserscheid, P. Ionic liquids in chemical engineering. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 203–230. [Google Scholar] [CrossRef]
- Greer, A.J.; Jacquemin, J.; Hardacre, C. Industrial Applications of Ionic Liquids. Molecules 2020, 25, 5207. [Google Scholar] [CrossRef]
- Martins, V.L.; Torresi, R.M. Ionic liquids in electrochemical energy storage. Curr. Opin. Electrochem. 2018, 9, 26–32. [Google Scholar] [CrossRef]
- Song, W.; Yan, J.; Ji, H. Tribological Performance of an Imidazolium Ionic Liquid-Functionalized SiO(2)@Graphene Oxide as an Additive. ACS Appl. Mater. Interfaces 2021, 13, 50573–50583. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.Q.; Bae, H.W.; Jeon, E.H.; Lee, J.S.; Cheong, M.; Kim, H.; Kim, H.S.; Lee, H. Zwitterionic imidazolium compounds with high cathodic stability as additives for lithium battery electrolytes. J. Power Source 2008, 183, 303–309. [Google Scholar] [CrossRef]
- Endo, T.; Sunada, K.; Sumida, H.; Kimura, Y. Origin of low melting point of ionic liquids: Dominant role of entropy. Chem. Sci. 2022, 13, 7560–7565. [Google Scholar] [CrossRef] [PubMed]
- Katritzky, A.R.; Jain, R.; Lomaka, A.; Petrukhin, R.; Maran, U.; Karelson, M. Perspective on the relationship between melting points and chemical structure. Cryst. Growth Des. 2001, 1, 261–265. [Google Scholar] [CrossRef]
- Abbott, A.P.; Ryder, K.S.; König, U. Electrofinishing of metals using eutectic based ionic liquids. Trans. IMF 2013, 86, 196–204. [Google Scholar] [CrossRef]
- Deyko, G.S.; Glukhov, L.M.; Kustov, L.M. Hydrogen storage in organosilicon ionic liquids. Int. J. Hydrogen Energy 2020, 45, 33807–33817. [Google Scholar] [CrossRef]
- Koutsoukos, S.; Philippi, F.; Malaret, F.; Welton, T. A review on machine learning algorithms for the ionic liquid chemical space. Chem Sci 2021, 12, 6820–6843. [Google Scholar] [CrossRef]
- Trohalaki, S.; Pachter, R. Prediction of Melting Points for Ionic Liquids. QSAR Comb. Sci. 2005, 24, 485–490. [Google Scholar] [CrossRef]
- Ambure, P.; Halder, A.K.; Gonzalez Diaz, H.; Cordeiro, M. QSAR-Co: An Open Source Software for Developing Robust Multitasking or Multitarget Classification-Based QSAR Models. J. Chem. Inf. Model. 2019, 59, 2538–2544. [Google Scholar] [CrossRef]
- Chen, S.; Xue, D.; Chuai, G.; Yang, Q.; Liu, Q. FL-QSAR: A federated learning-based QSAR prototype for collaborative drug discovery. Bioinformatics 2021, 36, 5492–5498. [Google Scholar] [CrossRef]
- Kutz, J.N. Machine learning for parameter estimation. Proc. Natl. Acad. Sci. USA 2023, 120, e2300990120. [Google Scholar] [CrossRef] [PubMed]
- Dobbelaere, M.R.; Plehiers, P.P.; Van de Vijver, R.; Stevens, C.V.; Van Geem, K.M. Machine Learning in Chemical Engineering: Strengths, Weaknesses, Opportunities, and Threats. Engineering 2021, 7, 1201–1211. [Google Scholar] [CrossRef]
- Chen, G.; Song, Z.; Qi, Z. Transformer-convolutional neural network for surface charge density profile prediction: Enabling high-throughput solvent screening with COSMO-SAC. Chem. Eng. Sci. 2021, 246, 117002. [Google Scholar] [CrossRef]
- Chen, G.; Song, Z.; Qi, Z.; Sundmacher, K. Neural recommender system for the activity coefficient prediction andUNIFACmodel extension of ionicliquid-solutesystems. AIChE J. 2021, 67, e17171. [Google Scholar] [CrossRef]
- Hashim, F.H.; Yu, F.; Izgorodina, E.I. Appropriate clusterset selection for the prediction of thermodynamic properties of liquid water with QCE theory. Phys. Chem. Chem. Phys. 2023, 25, 9846–9858. [Google Scholar] [CrossRef]
- Tan, T.; Cheng, H.; Chen, G.; Song, Z.; Qi, Z. Prediction of infinite-dilution activity coefficients with neural collaborative filtering. AIChE J. 2022, 68, e17789. [Google Scholar] [CrossRef]
- Carrera, G.A.; Aires-de-Sousa, J.O. Estimation of melting points of pyridinium bromide ionic liquids with decision trees and neural networks. Green Chem. 2005, 7, 20–27. [Google Scholar] [CrossRef]
- Bini, R.; Chiappe, C.; Duce, C.; Micheli, A.; Solaro, R.; Starita, A.; Tiné, M.R. Ionic liquids: Prediction of their melting points by a recursive neural network model. Green Chem. 2008, 10, 306–309. [Google Scholar] [CrossRef]
- Valderrama, J.O.; Faúndez, C.A.; Vicencio, V.J. Artificial Neural Networks and the Melting Temperature of Ionic Liquids. Ind. Eng. Chem. Res. 2014, 53, 10504–10511. [Google Scholar] [CrossRef]
- Venkatraman, V.; Evjen, S.; Knuutila, H.K.; Fiksdahl, A.; Alsberg, B.K. Predicting ionic liquid melting points using machine learning. J. Mol. Liq. 2018, 264, 318–326. [Google Scholar] [CrossRef]
- Low, K.; Kobayashi, R.; Izgorodina, E.I. The effect of descriptor choice in machine learning models for ionic liquid melting point prediction. J. Chem. Phys. 2020, 153, 104101. [Google Scholar] [CrossRef] [PubMed]
- Paduszyński, K.; Kłębowski, K.; Królikowska, M. Predicting melting point of ionic liquids using QSPR approach: Literature review and new models. J. Mol. Liq. 2021, 344, 117631. [Google Scholar] [CrossRef]
- Acar, Z.; Nguyen, P.; Lau, K.C. Machine-Learning Model Prediction of Ionic Liquids Melting Points. Appl. Sci. 2022, 12, 2408. [Google Scholar] [CrossRef]
- Makarov, D.M.; Fadeeva, Y.A.; Shmukler, L.E.; Tetko, I.V. Machine learning models for phase transition and decomposition temperature of ionic liquids. J. Mol. Liq. 2022, 366, 120247. [Google Scholar] [CrossRef]
- Dhakal, P.; Shah, J.K. Developing machine learning models for ionic conductivity of imidazolium-based ionic liquids. Fluid Phase Equilibria 2021, 549, 113208. [Google Scholar] [CrossRef]
- Krossing, I.; Slattery, J.M.; Daguenet, C.; Dyson, P.J.; Oleinikova, A.; Weingärtner, H. Why Are Ionic Liquids Liquid? A Simple Explanation Based on Lattice and Solvation Energies [J. Am. Chem. Soc. 2006, 128, 13427–13434]. J. Am. Chem. Soc. 2007, 129, 11296. [Google Scholar] [CrossRef]
- Zhang, S.J.; Zhou, Q.; Lu, X.M.; Wang, X.X.; Lu, C.H. “IPE Ionic Liquid Database”, Institute of Process Engineering, Chinese Academy Sciences, Beijing, 100190. Available online: http://ildate.ilct.com.cn/ (accessed on 1 January 2023).
- Velardez, G.F.; Alavi, S.; Thompson, D.L. Molecular dynamics studies of melting and liquid properties of ammonium dinitramide. J. Chem. Phys. 2003, 119, 6698–6708. [Google Scholar] [CrossRef]
- Jenkins, H.D.B.; Tudela, D.; Glasser, L. Lattice Potential Energy Estimation for Complex Ionic Salts from Density Measurements. Inorg. Chem. 2002, 41, 2364–2367. [Google Scholar] [CrossRef]
- Zheng, H.; Xu, G.; Wu, K.; Feng, L.; Zhang, R.; Bao, Y.; Wang, H.; Wang, K.; Qu, Z.; Shi, J. Highly Intrinsic Thermally Conductive Electrospinning Film with Intermolecular Interaction. J. Phys. Chem. C 2021, 125, 21580–21587. [Google Scholar] [CrossRef]
- Zahn, S.; Uhlig, F.; Thar, J.; Spickermann, C.; Kirchner, B. Intermolecular forces in an ionic liquid ([Mmim][Cl]) versus those in a typical salt (NaCl). Angew. Chem. Int. Ed. Engl. 2008, 47, 3639–3641. [Google Scholar] [CrossRef] [PubMed]
- Peppel, T.; Roth, C.; Fumino, K.; Paschek, D.; Köckerling, M.; Ludwig, R. The Influence of Hydrogen-Bond Defects on the Properties of Ionic Liquids. Angew. Chem. Int. Ed. 2011, 50, 6661–6665. [Google Scholar] [CrossRef] [PubMed]
- Slattery, J.M.; Daguenet, C.; Dyson, P.J.; Schubert, T.J.; Krossing, I. How to predict the physical properties of ionic liquids: A volume-based approach. Angew. Chem. Int. Ed. Engl. 2007, 46, 5384–5388. [Google Scholar] [CrossRef] [PubMed]
- Rabideau, B.D.; Soltani, M.; Parker, R.A.; Siu, B.; Salter, E.A.; Wierzbicki, A.; West, K.N.; Davis, J.H., Jr. Tuning the melting point of selected ionic liquids through adjustment of the cation’s dipole moment. Phys. Chem. Chem. Phys. 2020, 22, 12301–12311. [Google Scholar] [CrossRef]
- Kanagathara, N.; Usha, R.; Natarajan, V.; Marchewka, M.K. Molecular geometry, vibrational, NBO, HOMO–LUMO, first order hyper polarizability and electrostatic potential studies on anilinium hydrogen oxalate hemihydrate—An organic crystalline salt. Inorg. Nano-Met. Chem. 2021, 52, 226–233. [Google Scholar] [CrossRef]
- Pratuangdejkul, J.; Jaudon, P.; Ducrocq, C.; Nosoongnoen, W.; Guerin, G.-A.; Conti, M.; Loric, S.; Launay, J.-M.; Manivet, P. Cation-π Interactions in Serotonin: Conformational, Electronic Distribution, and Energy Decomposition Analysis. J. Chem. Theory Comput. 2006, 2, 746–760. [Google Scholar] [CrossRef]
- Sıdır, İ.; Sarı, T.; Gülseven Sıdır, Y.; Berber, H. Synthesis, solvatochromism and dipole moment in the ground and excited states of substitute phenol derivative fluorescent Schiff base compounds. J. Mol. Liq. 2022, 346, 117075. [Google Scholar] [CrossRef]
- Li, L.; Mayer, P.; Stephenson, D.S.; Ofial, A.R.; Mayer, R.J.; Mayr, H. An Overlooked Pathway in 1,3-Dipolar Cycloadditions of Diazoalkanes with Enamines. Angew. Chem. Int. Ed. 2022, 61, e202117047. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Rezac, J.; Hobza, P. Advanced Corrections of Hydrogen Bonding and Dispersion for Semiempirical Quantum Mechanical Methods. J. Chem. Theory Comput. 2012, 8, 141–151. [Google Scholar] [CrossRef]
- Rasspe-Lange, L.; Hoffmann, A.; Gertig, C.; Heck, J.; Leonhard, K.; Herres-Pawlis, S. Geometrical benchmarking and analysis of redox potentials of copper(I/II) guanidine-quinoline complexes: Comparison of semi-empirical tight-binding and DFT methods and the challenge of describing the entatic state (part III). J. Comput. Chem. 2023, 44, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Khetan, A.; Er, S. Comparison of computational chemistry methods for the discovery of quinone-based electroactive compounds for energy storage. Sci. Rep. 2020, 10, 22149. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Liu, J.; Cao, X.; Wei, C.; Liang, H.; Gong, C.; Ju, Z. Structures and hydrogen bonds of -SO3H functionalized acid ionic liquids. J. Mol. Liq. 2024, 406, 125129. [Google Scholar] [CrossRef]
- Bernardino, K.; Goloviznina, K.; Gomes, M.C.; Padua, A.A.H.; Ribeiro, M.C.C. Ion pair free energy surface as a probe of ionic liquid structure. J. Chem. Phys. 2020, 152, 014103. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Holder, A.J.; Dennington, I.R.D.; Liotard, D.A.; Truhlar, D.G.; Keith, T.A.; Millam, J.M.; Harris, C.D. AMPAC 10; Semichem, Inc.: Shawnee, KS, USA, 2016. [Google Scholar]
- Du, K.-L.; Leung, C.-S.; Mow, W.H.; Swamy, M.N.S. Perceptron: Learning, Generalization, Model Selection, Fault Tolerance, and Role in the Deep Learning Era. Mathematics 2022, 10, 4730. [Google Scholar] [CrossRef]
- Kohavi, R. A study of cross-validation and bootstrap for accuracy estimation and model selection. In Proceedings of the 14th International Joint Conference on Artificial Intelligence—Volume 2, Montreal, QC, Canada, 20–25 August 1995; pp. 1137–1143. [Google Scholar]
- Refaeilzadeh, P.; Tang, L.; Liu, H. Cross-Validation. Encycl. Database Syst. 2009, 1, 532–538. [Google Scholar]
- Pelegrina, G.D.; Duarte, L.T.; Grabisch, M. A k-additive Choquet integral-based approach to approximate the SHAP values for local interpretability in machine learning. Artif. Intell. 2023, 325, 104014. [Google Scholar] [CrossRef]
- Barber, C.; Lamontagne, J.R.; Vogel, R.M. Improved estimators of correlation and R2 for skewed hydrologic data. Hydrol. Sci. J. 2019, 65, 87–101. [Google Scholar] [CrossRef]
- Alexander, D.L.J.; Tropsha, A.; Winkler, D.A. Beware of R2: Simple, Unambiguous Assessment of the Prediction Accuracy of QSAR and QSPR Models. J. Chem. Inf. Model. 2015, 55, 1316–1322. [Google Scholar] [CrossRef]
- Baskin, I.; Epshtein, A.; Ein-Eli, Y. Benchmarking machine learning methods for modeling physical properties of ionic liquids. J. Mol. Liq. 2022, 351, 118616. [Google Scholar] [CrossRef]
- Makarov, D.M.; Fadeeva, Y.A.; Shmukler, L.E.; Tetko, I.V. Beware of proper validation of models for ionic Liquids! J. Mol. Liq. 2021, 344, 117722. [Google Scholar] [CrossRef]
- Cerecedo-Cordoba, J.A.; Gonzalez Barbosa, J.J.; Frausto Solis, J.; Gallardo-Rivas, N.V. Melting Temperature Estimation of Imidazole Ionic Liquids with Clustering Methods. J. Chem. Inf. Model. 2019, 59, 3144–3153. [Google Scholar] [CrossRef] [PubMed]
- Varnek, A.; Kireeva, N.; Tetko, I.V.; Baskin, I.I.; Solov’ev, V.P. Exhaustive QSPR Studies of a Large Diverse Set of Ionic Liquids: How Accurately Can We Predict Melting Points? J. Chem. Inf. Model. 2007, 47, 1111–1122. [Google Scholar] [CrossRef]
- Wambugu, N.; Chen, Y.; Xiao, Z.; Tan, K.; Wei, M.; Liu, X.; Li, J. Hyperspectral image classification on insufficient-sample and feature learning using deep neural networks: A review. Int. J. Appl. Earth Obs. Geoinf. 2021, 105, 102603. [Google Scholar] [CrossRef]
- Kwon, S.W.; Kim, J.S.; Lee, H.M.; Lee, J.S. Physics-added neural networks: An image-based deep learning for material printing system. Addit. Manuf. 2023, 73, 103668. [Google Scholar] [CrossRef]
- Chen, L.; Bryantsev, V.S. A density functional theory based approach for predicting melting points of ionic liquids. Phys. Chem. Chem. Phys. 2017, 19, 4114–4124. [Google Scholar] [CrossRef]
- Ryoo, H.; Lee, S.G.; Kim, J.G.; Chung, S.Y. Effect of Chemical Bonding Characteristics on Ordering Structure in Li Spinel Oxides. Adv. Funct. Mater. 2018, 29, 1805972. [Google Scholar] [CrossRef]
- Yan, F.; Xia, S.; Wang, Q.; Yang, Z.; Ma, P. Predicting the melting points of ionic liquids by the Quantitative Structure Property Relationship method using a topological index. Chem. Thermodyn. 2013, 62, 196–200. [Google Scholar] [CrossRef]
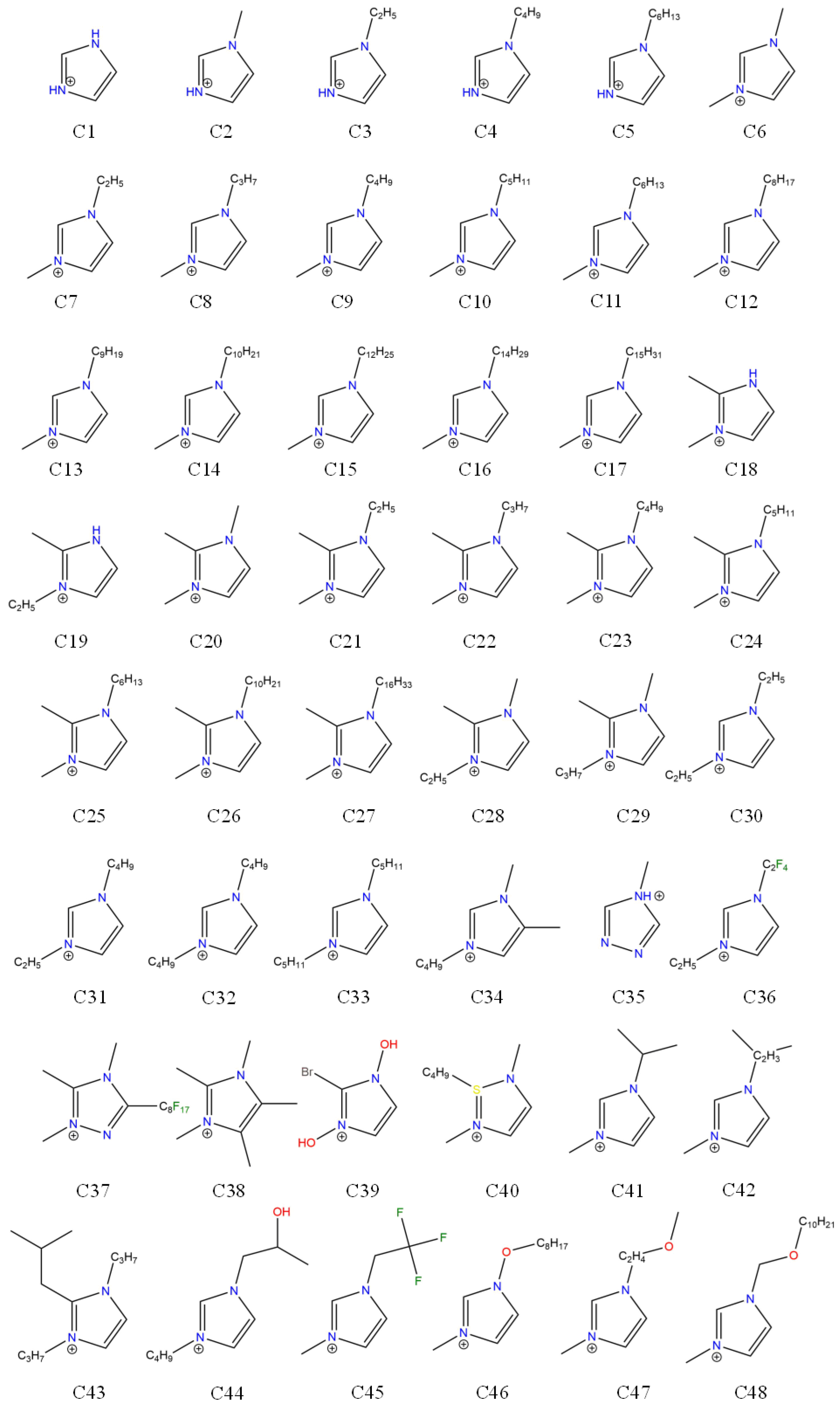
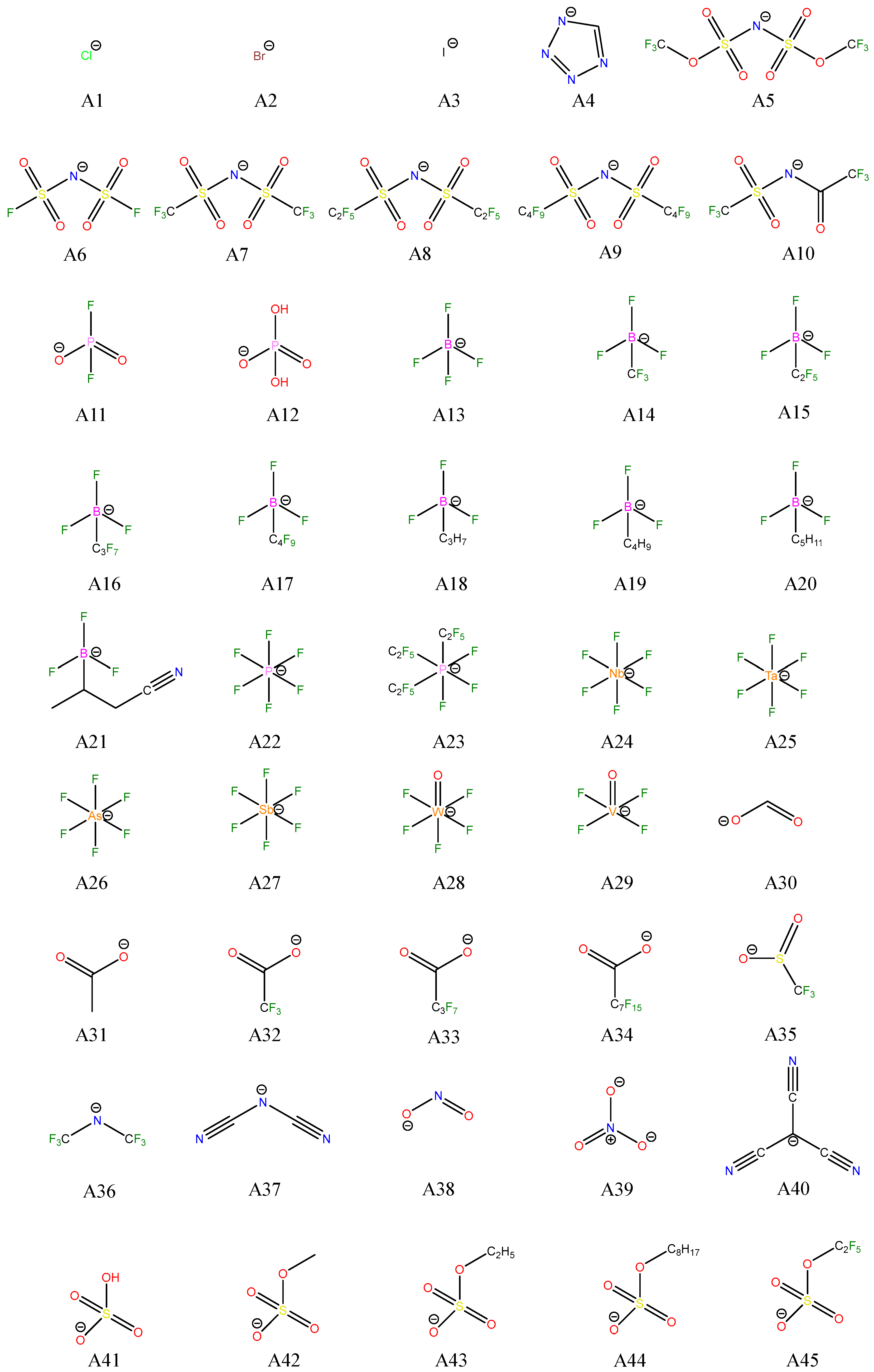




| Cations | Anions | ILs |
|---|---|---|
| Dipole moment | Dipole moment | Dipole moment |
| Enthalpy | Enthalpy | Enthalpy |
| Volume | Volume | |
| Mass | Mass | |
| LUMO | HOMO |
| Descriptors | Maximum | Minimum | |
|---|---|---|---|
| Cations | Dipole moment (Debye) | 36.93994 | 0.82669 |
| Enthalpy (Hartree) | 0.82448 | −0.76340 | |
| Volume (Bohr3/mol) | 6895.94200 | 1024.20200 | |
| Mass (amu) | 530.05250 | 69.04527 | |
| LUMO (Hartree) | −0.14256 | −0.23956 | |
| Anions | Dipole moment (Debye) | 27.691096 | 0 |
| Enthalpy (Hartree) | 0.37276 | −2.39248 | |
| Volume (Bohr3/mol) | 10195.328 | 50.77300 | |
| Mass (amu) | 935.33605 | 34.96885 | |
| HOMO (Hartree) | −0.10436 | −0.62145 | |
| ILs | Dipole moment (Debye) | 20.425138 | 0.44500 |
| Enthalpy (Hartree) | 0.700086 | −2.058234 |
| No. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Cations | C10 | C87 | C11 | C7 | C10 | C62 | C56 | C21 | ||
| Anions | A12 | A61 | A2 | A59 | A72 | A7 | A13 | A22 | ||
| Experimental values (K) | 438.15 | 386.69 | 221.00 | 209.00 | 337.15 | 230.65 | 192.05 | 469.00 | ||
| model 0 | R2 = 0.75 | Errors (K) | 81.76 | 99.79 | −88.32 | −104.53 | 118.01 | −85.26 | −104.22 | 80.51 |
| Dataset | Train | Train | Train | Validation | Validation | Validation | Validation | Validation | ||
| model 1 | R2 = 0.70 | Errors (K) | 75.73 | 29.45 | −106.39 | −14.11 | 83.85 | −43.06 | −82.84 | 48.28 |
| dataset | Train | Validation | Train | Train | Validation | Train | Train | Train | ||
| model 2 | R2 = 0.68 | Errors (K) | 118.38 | 24.23 | −99.47 | −48.68 | 41.38 | −65.48 | −97.39 | 85.13 |
| dataset | Train | Train | Train | Train | Train | Train | Train | Train | ||
| model 3 | R2 = 0.68 | Errors (K) | 80.34 | −0.99 | −77.66 | −19.03 | 55.76 | −49.99 | −82.64 | 51.91 |
| dataset | Train | Train | Train | Train | Train | Train | Train | Train | ||
| Cations | Anions | Errors (K) | Predicted Values (K) | Experimental Values (K) |
|---|---|---|---|---|
| C7 | A59 | −104.53 | 313.53 | 209 |
| A13 | 0.22 | 285.78 | 286.00 | |
| A7 | −2.23 | 258.23 | 256.00 | |
| C10 | A72 | 118.01 | 219.14 | 337.15 |
| A65 | −5.48 | 215.63 | 210.15 | |
| A51 | −7.92 | 219.87 | 211.95 | |
| C56 | A13 | −104.22 | 296.27 | 192.05 |
| C16 | 5.29 | 307.33 | 312.62 | |
| C65 | 3.74 | 186.81 | 190.55 | |
| C11 | A2 | −88.32 | 309.32 | 221.00 |
| C32 | −0.33 | 328.48 | 328.15 | |
| C92 | 1.63 | 445.52 | 447.15 |
| No. | Cations | Anions | Experimental Values (K) | Predicted Values (K) | Judged Values (K) |
|---|---|---|---|---|---|
| 1 | C2 | A2 | 314.15 345.15 | 393.99 | 345.15 |
| 2 | C2 | A13 | 310.15 325.55 | 345.28 | 325.55 |
| 3 | C6 | A2 | 382.65 398.15 449.00 | 398.42 | 398.15 |
| 4 | C7 | A9 | <298.15 304.00 | 294.88 | 294.88 |
| 5 | C7 | A31 | 228.15 259.15 <253.15 | 282.51 | 259.15 |
| 6 | C7 | A61 | 303.00 322.30 322.90 333.15 | 339.55 | 333.15 |
| 7 | C8 | A2 | 309.56 333.15 | 365.51 | 333.15 |
| 8 | C8 | A3 | 203.00 290.10 | 291.64 | 290.10 |
| 9 | C12 | A1 | 186.15 191.15 218.00 285.41 308.15 | 327.17 | 308.15 |
| 10 | C19 | A2 | 417.15 451.15 | 395.24 | 417.15 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Yin, J.; Zhang, X.; Qiu, W.; Jiang, W.; Zhang, M.; Zhu, L.; Li, H.; Li, H. Rapid and Accurate Prediction of the Melting Point for Imidazolium-Based Ionic Liquids by Artificial Neural Network. Chemistry 2024, 6, 1552-1571. https://doi.org/10.3390/chemistry6060094
Liu X, Yin J, Zhang X, Qiu W, Jiang W, Zhang M, Zhu L, Li H, Li H. Rapid and Accurate Prediction of the Melting Point for Imidazolium-Based Ionic Liquids by Artificial Neural Network. Chemistry. 2024; 6(6):1552-1571. https://doi.org/10.3390/chemistry6060094
Chicago/Turabian StyleLiu, Xinyu, Jie Yin, Xinmiao Zhang, Wenxiang Qiu, Wei Jiang, Ming Zhang, Linhua Zhu, Hongping Li, and Huaming Li. 2024. "Rapid and Accurate Prediction of the Melting Point for Imidazolium-Based Ionic Liquids by Artificial Neural Network" Chemistry 6, no. 6: 1552-1571. https://doi.org/10.3390/chemistry6060094
APA StyleLiu, X., Yin, J., Zhang, X., Qiu, W., Jiang, W., Zhang, M., Zhu, L., Li, H., & Li, H. (2024). Rapid and Accurate Prediction of the Melting Point for Imidazolium-Based Ionic Liquids by Artificial Neural Network. Chemistry, 6(6), 1552-1571. https://doi.org/10.3390/chemistry6060094