
Theoretical Study on One- and Two-Photon Absorption Properties of π-Stacked Multimer Models of Phenalenyl Radicals




Abstract

1. Introduction
2. Computational Details
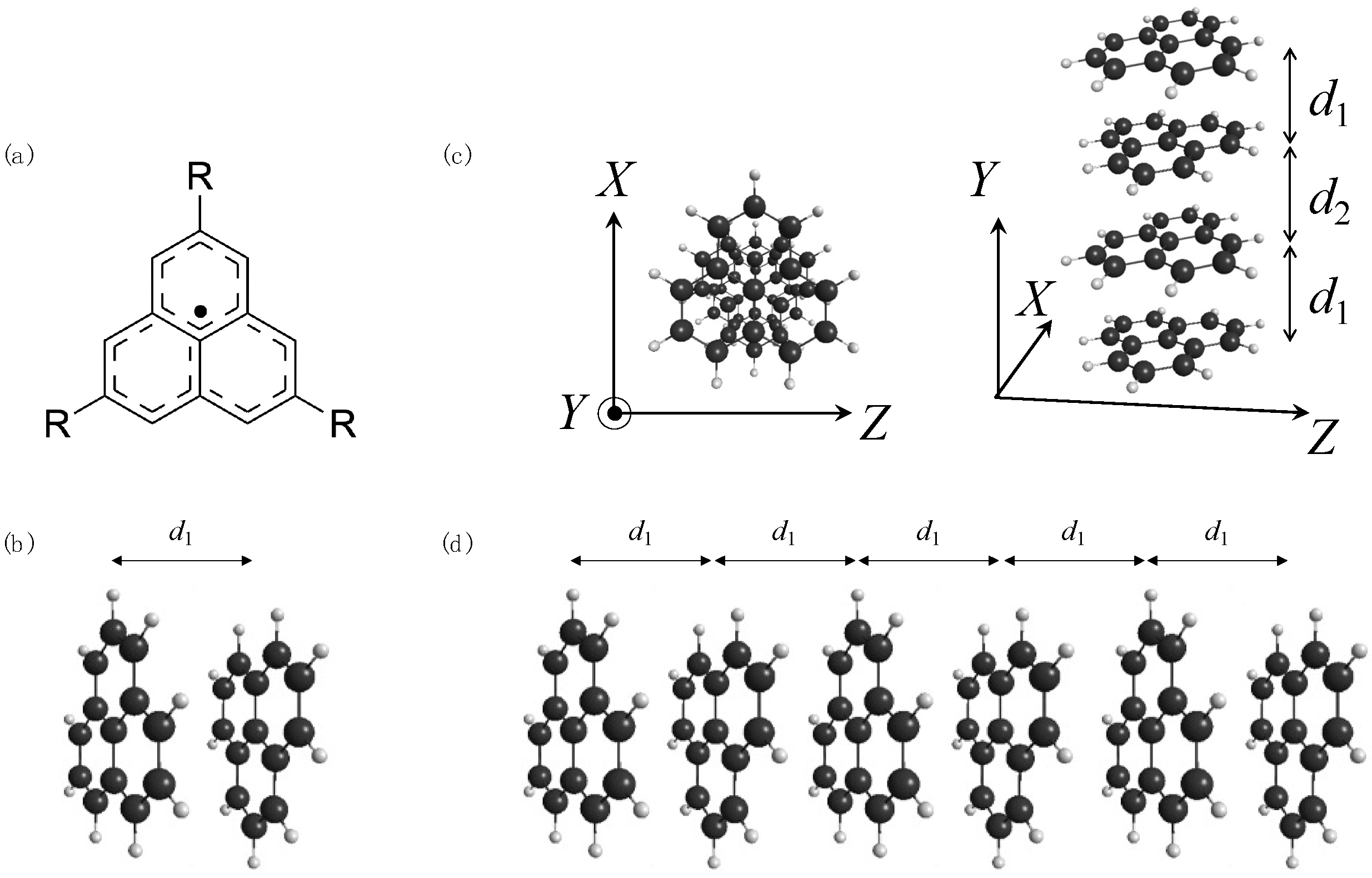
2.1. Calculated Models and Calculation Methods
2.2. Excited State Calculations and Spectrum Simulations
3. Results and Discussion
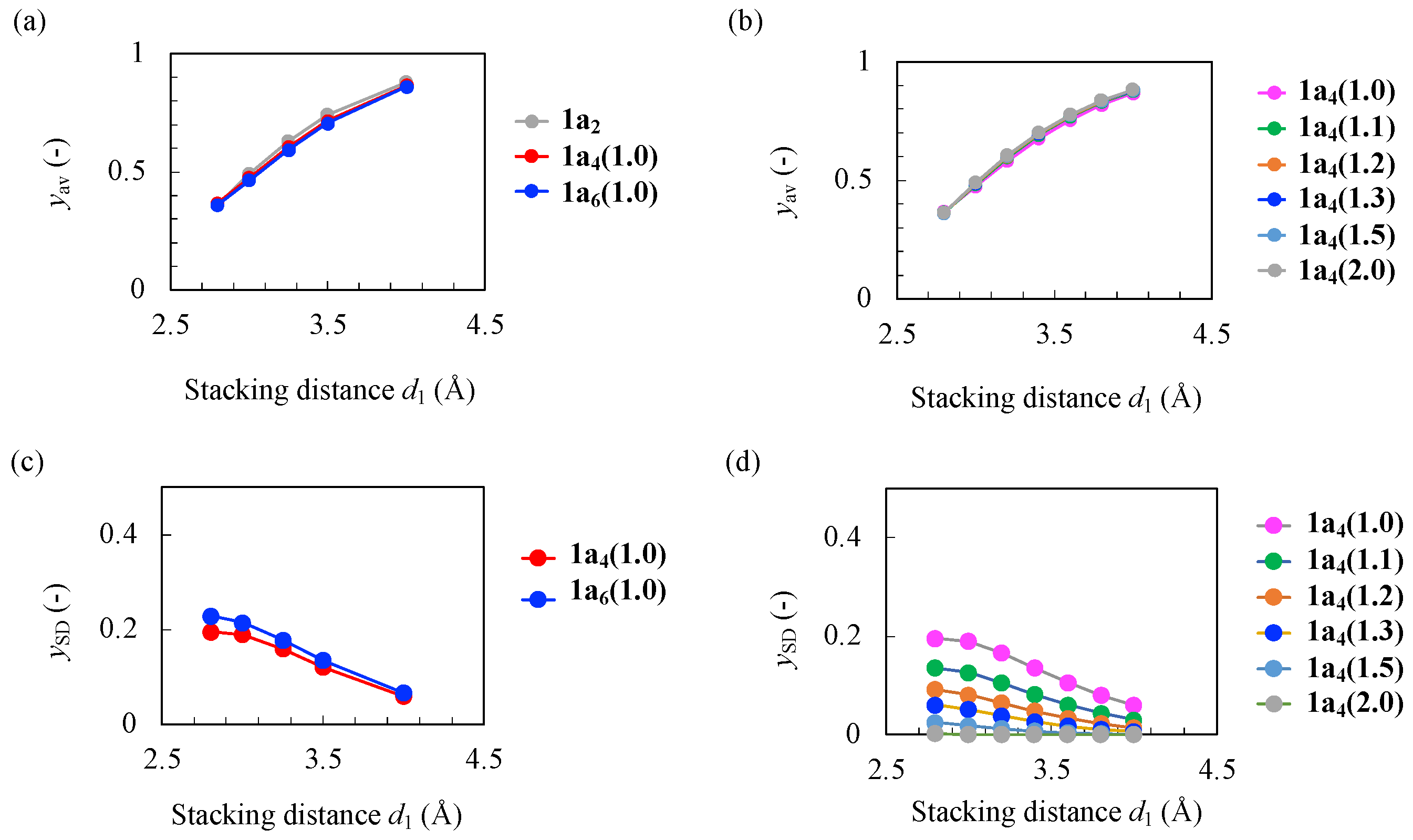
3.1. Correlation Between Stacking Distances and Diradical Characters
3.2. One-Photon and Two-Photon Absorption Properties of Tetramers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abe, M. Diradicals. Chem. Rev. 2013, 113, 7011–7088. [Google Scholar] [CrossRef] [PubMed]
- Wu, J. (Ed.) Diradicaloids, 1st ed.; Jenny Stanford Publishing: New York, NY, USA, 2022. [Google Scholar]
- Hinz, A.; Bresien, J.; Breher, F.; Schulz, A. Heteroatom-Based Diradical(Oid)s. Chem. Rev. 2023, 123, 10468–10526. [Google Scholar] [CrossRef] [PubMed]
- Ishigaki, Y.; Harimoto, T.; Shimajiri, T.; Suzuki, T. Carbon-Based Biradicals: Structural and Magnetic Switching. Chem. Rev. 2023, 123, 13952–13965. [Google Scholar] [CrossRef]
- Yamaguchi, K. The Electronic Structures of Biradicals in the Unrestricted Hartree-Fock Approximation. Chem. Phys. Lett. 1975, 33, 330–335. [Google Scholar] [CrossRef]
- Stuyver, T.; Chen, B.; Zeng, T.; Geerlings, P.; De Proft, F.; Hoffmann, R. Do Diradicals Behave Like Radicals? Chem. Rev. 2019, 119, 11291–11351. [Google Scholar] [CrossRef]
- Hayes, E.F.; Siu, A.K.Q. Electronic Structure of the Open Forms of Three-Membered Rings. J. Am. Chem. Soc. 1971, 93, 2090–2091. [Google Scholar] [CrossRef]
- Nakano, M.; Kishi, R.; Ohta, S.; Takahashi, H.; Kubo, T.; Kamada, K.; Ohta, K.; Botek, E.; Champagne, B. Relationship between Third-Order Nonlinear Optical Properties and Magnetic Interactions in Open-Shell Systems: A New Paradigm for Nonlinear Optics. Phys. Rev. Lett. 2007, 99, 033001. [Google Scholar] [CrossRef]
- Nakano, M.; Kishi, R.; Nitta, T.; Kubo, T.; Nakasuji, K.; Kamada, K.; Ohta, K.; Champagne, B.; Botek, E.; Yamaguchi, K. Second Hyperpolarizability (γ) of Singlet Diradical System: Dependence of γ on the Diradical Character. J. Phys. Chem. A 2005, 109, 885–891. [Google Scholar] [CrossRef]
- Nakano, M.; Kishi, R.; Ohta, S.; Takebe, A.; Takahashi, H.; Furukawa, S.-I.; Kubo, T.; Morita, Y.; Nakasuji, K.; Yamaguchi, K.; et al. Origin of the Enhancement of the Second Hyperpolarizability of Singlet Diradical Systems with Intermediate Diradical Character. J. Chem. Phys. 2006, 125, 074113. [Google Scholar] [CrossRef]
- Nakano, M. Open-Shell-Character-Based Molecular Design Principles: Applications to Nonlinear Optics and Singlet Fission. Chem. Rec. 2017, 17, 27–62. [Google Scholar] [CrossRef]
- Nakano, M.; Champagne, B. Nonlinear Optical Properties in Open-Shell Molecular Systems. WIREs Comput. Mol. Sci. 2016, 6, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Nakano, M.; Yoneda, K.; Kishi, R.; Takahashi, H.; Kubo, T.; Kamada, K.; Ohta, K.; Champagne, B.; Botek, E. One- and Two-Photon Absorptions in Open-Shell Singlet Systems. AIP Conf. Proc. 2012, 1504, 136–142. [Google Scholar]
- Nakano, M.; Yoneda, K.; Kishi, R.; Takahashi, H.; Kubo, T.; Kamada, K.; Ohta, K.; Botek, E.; Champagne, B. Remarkable Two-Photon Absorption in Open-Shell Singlet Systems. J. Chem. Phys. 2009, 131, 114316. [Google Scholar] [CrossRef] [PubMed]
- So, P.T.C.; French, T.; Yu, W.M.; Berland, K.M.; Dong, C.Y.; Gratton, E. Time-Resolved Fluorescence Microscopy Using Two-Photon Excitation. Bioimaging 1995, 3, 49–63. [Google Scholar] [CrossRef]
- Toriumi, A.; Kawata, S.; Gu, M. Reflection Confocal Microscope Readout System for Three-Dimensional Photochromic Optical Data Storage. Opt. Lett. 1998, 23, 1924–1926. [Google Scholar] [CrossRef]
- Ehrlich, J.E.; Wu, X.L.; Lee, I.-Y.S.; Hu, Z.-Y.; Röckel, H.; Marder, S.R.; Perry, J.W. Two-Photon Absorption and Broadband Optical Limiting with Bis-Donor Stilbenes. Opt. Lett. 1997, 22, 1843–1845. [Google Scholar] [CrossRef]
- Maruo, S.; Nakamura, O.; Kawata, S. Three-Dimensional Microfabrication with Two-Photon-Absorbed Photopolymerization. Opt. Lett. 1997, 22, 132–134. [Google Scholar] [CrossRef]
- He, G.S.; Xu, C.; Prasad, P.N.; Reinhardt, B.A.; Bhatt, J.C.; McKellar, R.; Dillard, A.G. Two-Photon Absorption and Optical-Limiting Properties of Novel Organic Compounds: Erratum. Opt. Lett. 1995, 20, 1930. [Google Scholar] [CrossRef]
- Cumpston, B.H.; Ananthavel, S.P.; Barlow, S.; Dyer, D.L.; Ehrlich, J.E.; Erskine, L.L.; Heikal, A.A.; Kuebler, S.M.; Lee, I.-Y.S.; McCord-Maughon, D.; et al. Two-Photon Polymerization Initiators for Three-Dimensional Optical Data Storage and Microfabrication. Nature 1999, 398, 51–54. [Google Scholar] [CrossRef]
- Denk, W.; Strickler, J.H.; Webb, W.W. Two-Photon Laser Scanning Fluorescence Microscopy. Science 1990, 248, 73–76. [Google Scholar] [CrossRef]
- Reinhardt, B.A.; Brott, L.L.; Clarson, S.J.; Dillard, A.G.; Bhatt, J.C.; Kannan, R.; Yuan, L.; He, G.S.; Prasad, P.N. Highly Active Two-Photon Dyes: Design, Synthesis, and Characterization toward Application. Chem. Mater. 1998, 10, 1863–1874. [Google Scholar] [CrossRef]
- Dvornikov, A.S.; Walker, E.P.; Rentzepis, P.M. Two-Photon Three-Dimensional Optical Storage Memory. J. Phys. Chem. A 2009, 113, 13633–13644. [Google Scholar] [CrossRef] [PubMed]
- Pawlicki, M.; Collins, H.A.; Denning, R.G.; Anderson, H.L. Two-Photon Absorption and the Design of Two-Photon Dyes. Angew. Chem. Int. Ed. 2009, 48, 3244–32667. [Google Scholar] [CrossRef] [PubMed]
- Terenziani, F.; Katan, C.; Badaeva, E.; Tretiak, S.; Blanchard-Desce, M. Enhanced Two-Photon Absorption of Organic Chromophores: Theoretical and Experimental Assessments. Adv. Mater. 2008, 20, 4641–4678. [Google Scholar] [CrossRef]
- Albota, M.; Beljonne, D.; Brédas, J.-L.; Ehrlich, J.E.; Fu, J.-Y.; Heikal, A.A.; Hess, S.E.; Kogej, T.; Levin, M.D.; Marder, S.R.; et al. Design of Organic Molecules with Large Two-Photon Absorption Cross Sections. Science 1998, 281, 1653–1656. [Google Scholar] [CrossRef]
- Reid, D.H. The Chemistry of the Phenalenes. Q. Rev. Chem. Soc. 1965, 19, 274–302. [Google Scholar] [CrossRef]
- Kubo, T. Syntheses and Properties of Open-Shell π-Conjugated Molecules. Bull. Chem. Soc. Jpn. 2021, 94, 2235–2244. [Google Scholar] [CrossRef]
- Kubo, T.; Shimizu, A.; Sakamoto, M.; Uruichi, M.; Yakushi, K.; Nakano, M.; Shiomi, D.; Sato, K.; Takui, T.; Morita, Y.; et al. Synthesis, Intermolecular Interaction, and Semiconductive Behavior of a Delocalized Singlet Biradical Hydrocarbon. Angew. Chem. Int. Ed. 2005, 44, 6564–6568. [Google Scholar] [CrossRef]
- Kubo, T.; Shimizu, A.; Uruichi, M.; Yakushi, K.; Nakano, M.; Shiomi, D.; Sato, K.; Takui, T.; Morita, Y.; Nakasuji, K. Singlet Biradical Character of Phenalenyl-Based Kekulé Hydrocarbon with Naphthoquinoid Structure. Org. Lett. 2007, 9, 81–84. [Google Scholar] [CrossRef]
- Kamada, K.; Ohta, K.; Kubo, T.; Shimizu, A.; Morita, Y.; Nakasuji, K.; Kishi, R.; Ohta, S.; Furukawa, S.-I.; Takahashi, H.; et al. Strong Two-Photon Absorption of Singlet Diradical Hydrocarbons. Angew. Chem. Int. Ed. 2007, 46, 3544–3546. [Google Scholar] [CrossRef]
- Kamada, K.; Ohta, K.; Shimizu, A.; Kubo, T.; Kishi, R.; Takahashi, H.; Botek, E.; Champagne, B.; Nakano, M. Singlet Diradical Character from Experiment. J. Phys. Chem. Lett. 2010, 1, 937–940. [Google Scholar] [CrossRef]
- Kamada, K.; Ohta, K.; Iwase, Y.; Kondo, K. Two-Photon Absorption Properties of Symmetric Substituted Diacetylene: Drastic Enhancement of the Cross Section near the One-Photon Absorption Peak. Chem. Phys. Lett. 2003, 372, 386–393. [Google Scholar] [CrossRef]
- Goto, K.; Kubo, T.; Yamamoto, K.; Nakasuji, K.; Sato, K.; Shiomi, D.; Takui, T.; Kubota, M.; Kobayashi, T.; Yakusi, K.; et al. A Stable Neutral Hydrocarbon Radical: Synthesis, Crystal Structure, and Physical Properties of 2,5,8-Tri-Tert-Butyl-Phenalenyl. J. Am. Chem. Soc. 1999, 121, 1619–1620. [Google Scholar] [CrossRef]
- Suzuki, S.; Morita, Y.; Fukui, K.; Sato, K.; Shiomi, D.; Takui, T.; Nakasuji, K. Aromaticity on the Pancake-Bonded Dimer of Neutral Phenalenyl Radical as Studied by MS and NMR Spectroscopies and NICS Analysis. J. Am. Chem. Soc. 2006, 128, 2530–2531. [Google Scholar] [CrossRef] [PubMed]
- Mou, Z.; Uchida, K.; Kubo, T.; Kertesz, M. Evidence of σ- and π-Dimerization in a Series of Phenalenyls. J. Am. Chem. Soc. 2014, 136, 18009–18022. [Google Scholar] [CrossRef]
- Uchida, K.; Hirao, Y.; Kurata, H.; Kubo, T.; Hatano, S.; Inoue, K. Dual Association Modes of the 2,5,8-Tris(Pentafluorophenyl)Phenalenyl Radical. Chem.–Asian J. 2014, 9, 1823–1829. [Google Scholar] [CrossRef]
- Kertesz, M. Pancake Bonding: An Unusual Pi-Stacking Interaction. Chem.–Eur. J. 2019, 25, 400–416. [Google Scholar] [CrossRef]
- Yoneda, K.; Nakano, M.; Fukuda, K.; Matsui, H.; Takamuku, S.; Hirosaki, Y.; Kubo, T.; Kamada, K.; Champagne, B. Third-Order Nonlinear Optical Properties of One-Dimensional Open-Shell Molecular Aggregates Composed of Phenalenyl Radicals. Chem.–Eur. J. 2014, 20, 11129–11136. [Google Scholar] [CrossRef]
- Salustro, S.; Maschio, L.; Kirtman, B.; Rérat, M.; Dovesi, R. Third-Order Electric Field Response of Infinite Linear Chains Composed of Phenalenyl Radicals. J. Phys. Chem. C 2016, 120, 6756–6761. [Google Scholar] [CrossRef]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-Mechanical Condensed Matter Simulations with CRYSTAL. WIREs Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Matsui, H.; Nakano, M.; Champagne, B. Theoretical Study on the Spin State and Open-Shell Character Dependences of the Second Hyperpolarizability in Hydrogen Chain Models. Phys. Rev. A 2016, 94, 42515. [Google Scholar] [CrossRef]
- Matsui, H.; Yamane, M.; Tonami, T.; Nagami, T.; Watanabe, K.; Kishi, R.; Kitagawa, Y.; Nakano, M. Theoretical Study on the Gigantic Effect of External Static Electric Field Application on the Nonlinear Optical Properties of 1,2,3,5-Dithiadiazolyl p-Radical Dimers. Mater. Chem. Front. 2018, 2, 785–790. [Google Scholar] [CrossRef]
- Shoda, J.; Yokoyama, M.; Yoshida, W.; Matsui, H.; Sugimori, R.; Kishi, R.; Kitagawa, Y. Theoretical Study on the Correlation between Open-Shell Electronic Structures and Third-Order Nonlinear Optical Properties in One-Dimensional Chains of π-Radicals. J. Phys. Chem. A 2024, 128, 8473–8482. [Google Scholar] [CrossRef]
- Nakano, H. Quasidegenerate Perturbation Theory with Multiconfigurational Self-consistent-field Reference Functions. J. Chem. Phys. 1993, 99, 7983–7992. [Google Scholar] [CrossRef]
- Granovsky, A.A. Extended Multi-Configuration Quasi-Degenerate Perturbation Theory: The New Approach to Multi-State Multi-Reference Perturbation Theory. J. Chem. Phys. 2011, 134, 214113. [Google Scholar] [CrossRef] [PubMed]
- Nakano, M.; Takebe, A.; Kishi, R.; Ohta, S.; Nate, M.; Kubo, T.; Kamada, K.; Ohta, K.; Champagne, B.; Botek, E.; et al. Second Hyperpolarizabilities (γ) of Open-Shell Singlet One-Dimensional Systems: Intersite Interaction Effects on the Average Diradical Character and Size Dependences of γ. Chem. Phys. Lett. 2006, 432, 473–479. [Google Scholar] [CrossRef]
- Nakano, M.; Minami, T.; Fukui, H.; Kishi, R.; Shigeta, Y.; Champagne, B. Full Configuration Interaction Calculations of the Second Hyperpolarizabilities of the H4 Model Compound: Summation-over-States Analysis and Interplay with Diradical Characters. J. Chem. Phys. 2012, 136, 024315. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01.; Gaussian, Inc.: Wallingford CT, USA, 2013. [Google Scholar]
- Barca, G.M.J.; Bertoni, C.; Carrington, L.; Datta, D.; De Silva, N.; Deustua, J.E.; Fedorov, D.G.; Gour, J.R.; Gunina, A.O.; Guidez, E.; et al. Recent developments in the general atomic and molecular electronic structure system. J. Chem. Phys. 2020, 152, 154102. [Google Scholar] [CrossRef]
- Cui, Z.; Lischka, H.; Beneberu, H.Z.; Kertesz, M. Rotational Barrier in Phenalenyl Neutral Radical Dimer: Separating Pancake and van Der Waals Interactions. J. Am. Chem. Soc. 2014, 136, 5539–5542. [Google Scholar] [CrossRef]
- Shirai, S.; Iwata, S.; Tani, T.; Inagaki, S. Ab Initio Studies of Aromatic Excimers Using Multiconfiguration Quasi-Degenerate Perturbation Theory. J. Phys. Chem. A 2011, 115, 7687–7699. [Google Scholar] [CrossRef]
- Ohta, K.; Yamada, S.; Kamada, K.; Slepkov, A.D.; Hegmann, F.A.; Tykwinski, R.R.; Shirtcliff, L.D.; Haley, M.M.; Sałek, P.; Gel’mukhanov, F.; et al. Two-Photon Absorption Properties of Two-Dimensional π-Conjugated Chromophores: Combined Experimental and Theoretical Study. J. Phys. Chem. A 2011, 115, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Beerepoot, M.T.P.; Friese, D.H.; List, N.H.; Kongsted, J.; Ruud, K. Benchmarking Two-Photon Absorption cross Sections: Performance of CC2 and CAM-B3LYP. Phys. Chem. Chem. Phys. 2015, 17, 19306–19314. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | yav | ySD | |||||
|---|---|---|---|---|---|---|---|
| 1Bu | 2Ag | 3Ag | 4Ag | 5Ag | |||
| 1a4(1.0) | 0.474 | 0.189 | 1.921 | 1.120 | 2.375 | 2.708 | 3.164 |
| 1a4(1.1) | 0.482 | 0.126 | 2.076 | 1.273 | 2.369 | 2.727 | 2.999 |
| 1a4(1.2) | 0.486 | 0.081 | 2.213 | 1.400 | 2.379 | 2.713 | 3.030 |
| 1a4(1.3) | 0.488 | 0.051 | 2.298 | 1.465 | 2.388 | 2.760 | 3.063 |
| 1a4(1.5) | 0.490 | 0.018 | 2.357 | 1.500 | 2.400 | 2.927 | 3.087 |
| 1a4(2.0) | 0.490 | 0.001 | 2.381 | 1.501 | 2.410 | 3.091 | 3.271 |
| 1a2 | 0.490 | – | 2.573 | 3.452 | – | – | – |
| System | yav | ySD | |||||
|---|---|---|---|---|---|---|---|
| 1Ag→1Bu | 1Bu→2Ag | 1Bu→3Ag | 1Bu→4Ag | 1Bu→5Ag | |||
| 1a4(1.0) | 0.474 | 0.189 | 11.6 | 1.0 | 1.1 | 21.2 | 2.3 |
| 1a4(1.1) | 0.482 | 0.126 | 10.6 | 2.7 | 0.1 | 19.2 | 7.4 |
| 1a4(1.2) | 0.486 | 0.081 | 10.2 | 2.1 | 0.4 | 14.8 | 12.1 |
| 1a4(1.3) | 0.488 | 0.051 | 10.2 | 0.9 | 1.0 | 10.4 | 13.2 |
| 1a4(1.5) | 0.490 | 0.018 | 10.1 | 0.1 | 1.4 | 4.1 | 13.7 |
| 1a4(2.0) | 0.490 | 0.001 | 10.0 | 0.0 | 1.0 | 0.1 | 13.5 |
| 1a2 | 0.490 | – | 7.3 | 12.4 | – | – | – |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yokoyama, M.; Kishi, R.; Kitagawa, Y. Theoretical Study on One- and Two-Photon Absorption Properties of π-Stacked Multimer Models of Phenalenyl Radicals. Chemistry 2024, 6, 1427-1438. https://doi.org/10.3390/chemistry6060085
Yokoyama M, Kishi R, Kitagawa Y. Theoretical Study on One- and Two-Photon Absorption Properties of π-Stacked Multimer Models of Phenalenyl Radicals. Chemistry. 2024; 6(6):1427-1438. https://doi.org/10.3390/chemistry6060085
Chicago/Turabian StyleYokoyama, Masako, Ryohei Kishi, and Yasutaka Kitagawa. 2024. "Theoretical Study on One- and Two-Photon Absorption Properties of π-Stacked Multimer Models of Phenalenyl Radicals" Chemistry 6, no. 6: 1427-1438. https://doi.org/10.3390/chemistry6060085
APA StyleYokoyama, M., Kishi, R., & Kitagawa, Y. (2024). Theoretical Study on One- and Two-Photon Absorption Properties of π-Stacked Multimer Models of Phenalenyl Radicals. Chemistry, 6(6), 1427-1438. https://doi.org/10.3390/chemistry6060085