
py.Aroma: An Intuitive Graphical User Interface for Diverse Aromaticity Analyses


Abstract
:
1. Introduction
2. Methods
3. Usage
3.1. Compute HOMA and HOMER Index
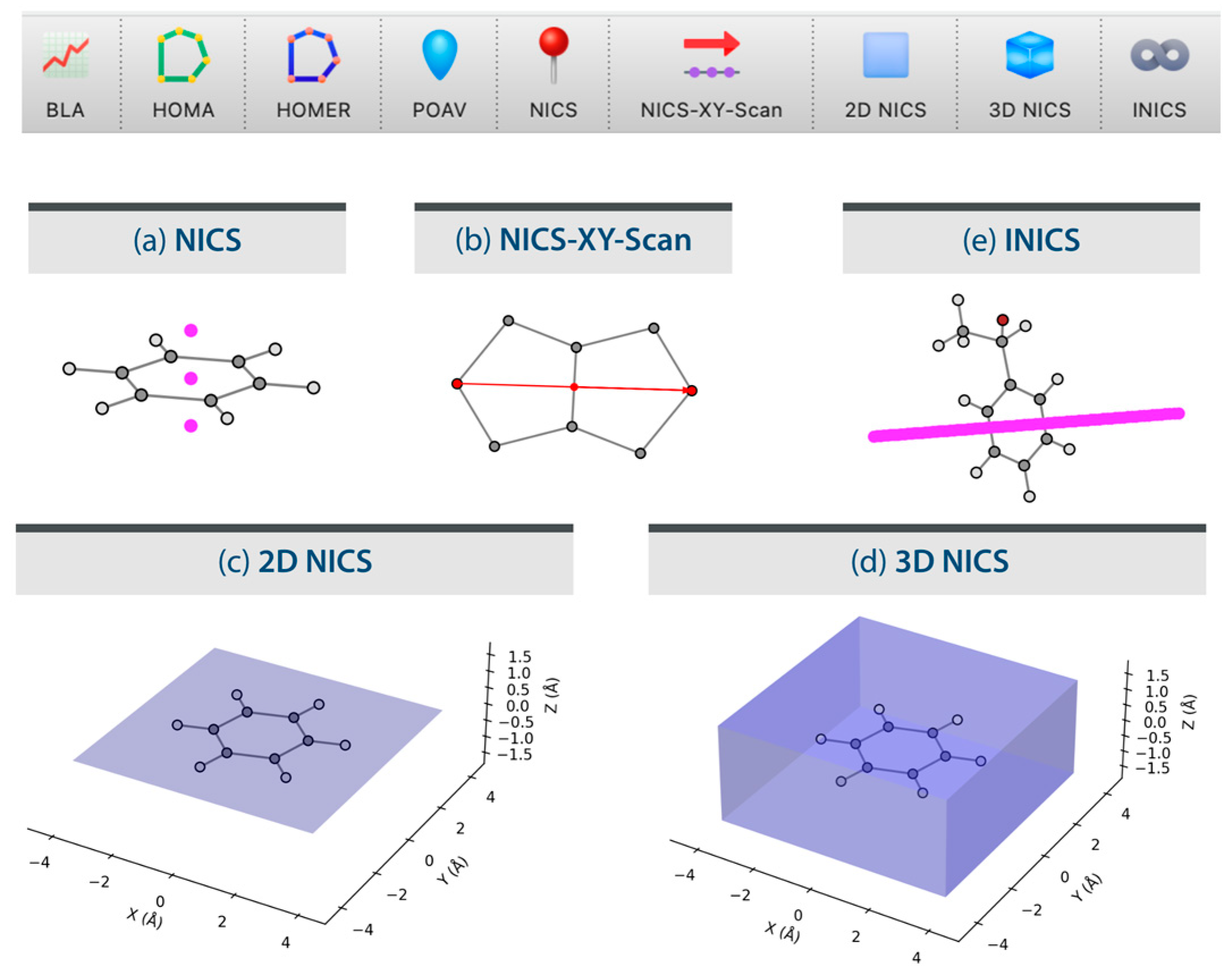
3.2. Generate Gaussian Input Files for NICS Calculations
3.3. Process Gaussian Output Files for NICS Calculations
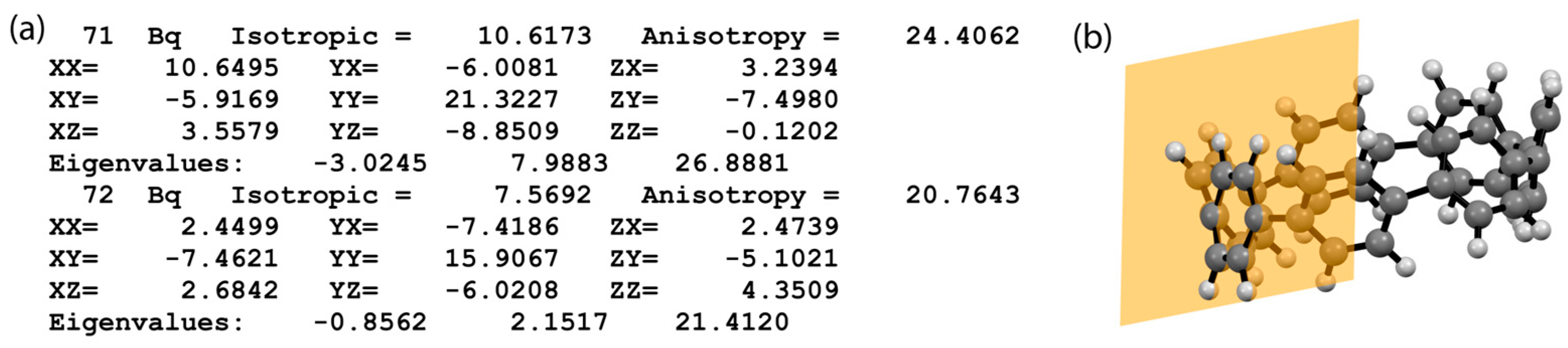
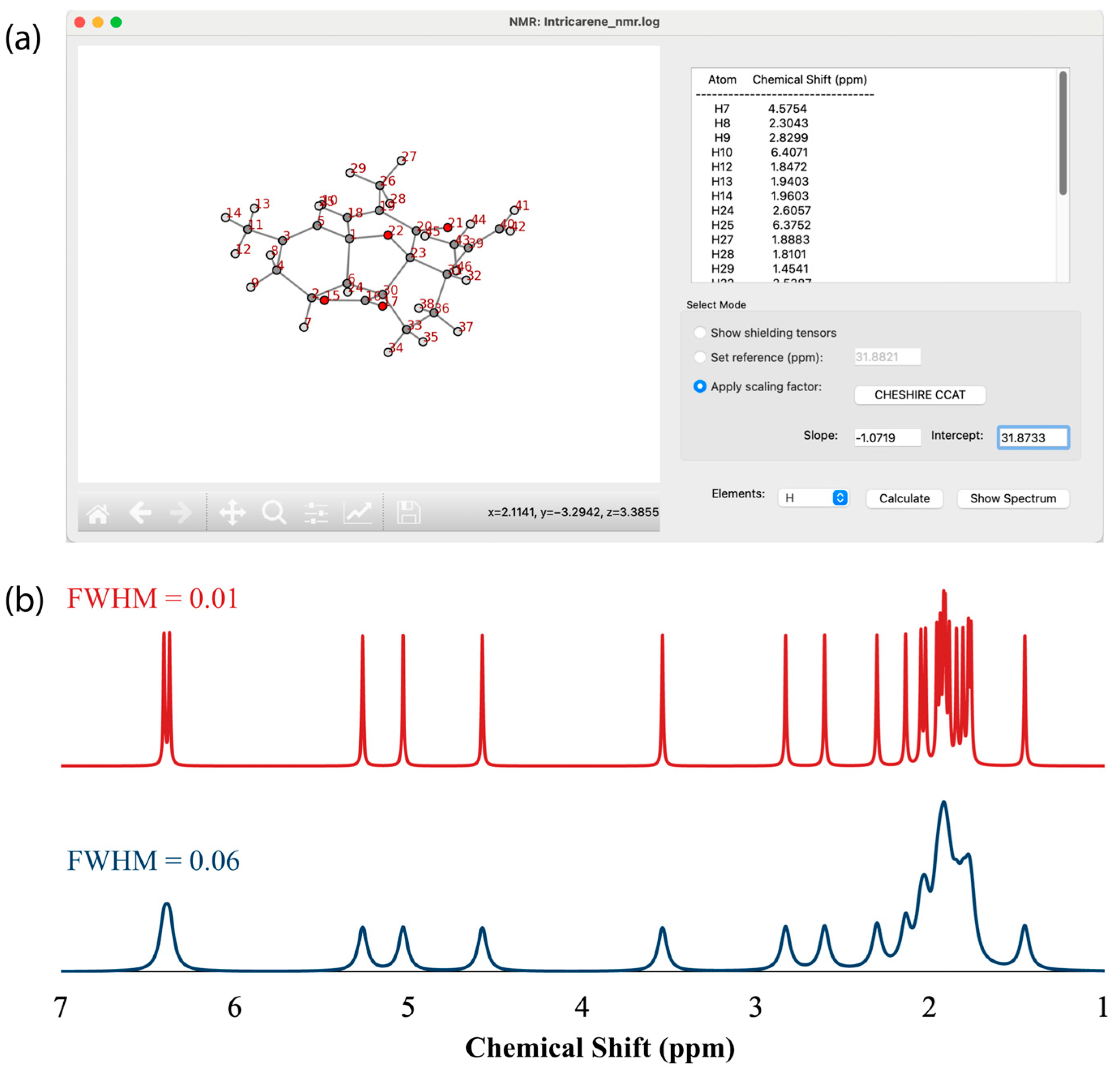
3.4. NMR Spectrum
3.5. Generate Computational Supporting Information
- Routine section: identifies the computational method and basis set employed.
- Charge and spin multiplicity: specifies the electronic configuration of the system.
- Point group: characterizes the molecule’s symmetry.
- Electronic energy: provides the optimized ground-state energy in hartree units.
- Cartesian coordinates: compiles the atomic positions of the optimized geometry.
- Number of imaginary frequencies (freq jobs only): indicates vibrational stability or instability.
- Thermal parameters (freq jobs only): summarizes relevant thermodynamic properties, including zero-point energy, thermal energy, enthalpy, and free energy.
4. Conclusions
Supplementary Materials
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kekulé, F.A. Sur la constitution des substances aromatiques. Bull. Soc. Chim. Paris 1865, 3, 98–110. [Google Scholar]
- Cossío, F.P. Aromaticity in molecules and transition structures: From atomic and molecular orbitals to simple ring current models. In Aromaticity. Modern Computational Methods and Applications; Fernández, I., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2021; pp. 1–40. [Google Scholar]
- Hückel, E. Quantentheoretische beiträge zum benzolproblem. I. Die elektronenkonfiguration des benzols und verwandter beziehungen. Z. Phys. 1931, 70, 204–286. [Google Scholar] [CrossRef]
- Breslow, R. Aromaticity. Acc. Chem. Res. 1973, 6, 393–398. [Google Scholar] [CrossRef]
- Heilbronner, E. Hückel molecular orbitals of Möbius-type conformation of annulenes. Tetrahedron Lett. 1964, 5, 1923–1928. [Google Scholar] [CrossRef]
- Rzepa, H.S. Möbius aromaticity and delocalization. Chem. Rev. 2005, 105, 3697–3715. [Google Scholar] [CrossRef] [PubMed]
- Krygowski, T.M.; Cyrański, M.K. Structural Aspects of Aromaticity. Chem. Rev. 2001, 101, 1385–14120. [Google Scholar] [CrossRef] [PubMed]
- Krygowski, T.M.; Szatylowicz, H.; Stasyuk, O.A.; Dominikowska, J.; Palusiak, M. Aromaticity from the Viewpoint of Molecular Geometry: Application to Planar Systems. Chem. Rev. 2014, 114, 6383–6422. [Google Scholar] [CrossRef]
- Schleyer, P.v.R.; Pühlhofer, F. Recommendations for the Evaluation of Aromatic Stabilization Energies. Org. Lett. 2002, 4, 2873–2876. [Google Scholar] [CrossRef] [PubMed]
- Gershoni-Poranne, R.; Stanger, A. Magnetic criteria of aromaticity. Chem. Soc. Rev. 2015, 44, 6597–6615. [Google Scholar] [CrossRef] [PubMed]
- Poater, J.; Fradera, X.; Duran, M.; Solà, M. The delocalization index as an electronic aromaticity criterion: Application to a series of planar polycyclic aromatic hydrocarbons. Chem. Eur. J. 2003, 9, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Arpa, E.M.; Durbeej, B. HOMER: A reparameterization of the harmonic oscillator model of aromaticity (HOMA) for excited states. Phys. Chem. Chem. Phys. 2023, 25, 16763–16771. [Google Scholar] [CrossRef]
- Schleyer, P.v.R.; Maerker, C.; Dransfeld, A.; Jiao, H.; van Eikema Hommes, N.J.R. Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [Google Scholar] [CrossRef]
- Chen, Z.; Wannere, C.S.; Corminboeuf, C.; Puchta, R.; Schleyer, P.v.R. Nucleus-Independent Chemical Shifts (NICS) as an Aromaticity Criterion. Chem. Rev. 2005, 105, 3842–3888. [Google Scholar] [CrossRef] [PubMed]
- Stanger, A. NICS—Past and Present. Eur. J. Org. Chem. 2020, 21, 3120–3127. [Google Scholar] [CrossRef]
- Stanger, A. Nucleus Independent Chemical Shift (NICS) at Small Distances from the Molecular Plane: The Effect of Electron Density. ChemPhysChem 2023, 24, e202300080. [Google Scholar] [CrossRef]
- Stanger, A. Nucleus-Independent Chemical Shifts (NICS): Distance Dependence and Revised Criteria for Aromaticity and Antiaromaticity. J. Org. Chem. 2006, 71, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Gershoni-Poranne, R.; Stanger, A. The NICS-XY-Scan: Identification of Local and Global Ring Current in Multi-Ring Systems. Chem. Eur. J. 2014, 20, 5673–5688. [Google Scholar] [CrossRef] [PubMed]
- Stanger, A.; Monaco, G.; Zanasi, R. NICS-XY-Scan Predictions of Local, Semi-Global, and Global Ring Currents in Annulated Pentalene and s-Indacene Cores Compared to First-Principles Current Density Maps. ChemPhysChem 2020, 21, 65–82. [Google Scholar] [CrossRef] [PubMed]
- Gershoni-Poranne, R. Piecing it Together: An Additivity Scheme for Aromaticity using NICS-XY Scans. Chem. Eur. J. 2018, 24, 4165–4172. [Google Scholar] [CrossRef]
- Liu, C.; Ni, Y.; Lu, X.; Li, G.; Wu, J. Global Aromaticity in Macrocyclic Polyradicaloids: Hückel’s Rule or Baird’s Rule? Acc. Chem. Res. 2019, 52, 2309–2321. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Gopalakrishna, T.Y.; Phan, H.; Kim, T.; Herng, T.S.; Han, Y.; Tao, T.; Ding, J.; Kim, D.; Wu, J. 3D global aromaticity in a fully conjugated diradicaloid cage at different oxidation states. Nat. Chem. 2020, 12, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Gopalakrishna, T.Y.; Phan, H.; Herng, T.S.; Jiang, Q.; Liu, C.; Li, G.; Ding, J.; Wu, J. Global Aromaticity in Macrocyclic Cyclopenta-Fused Tetraphenanthrenylene Tetraradicaloid and Its Charged Species. Angew. Chem. Int. Ed. 2018, 57, 13052–13056. [Google Scholar] [CrossRef] [PubMed]
- Peeks, M.D.; Claridge, T.D.W.; Anderson, H.L. Aromatic and antiaromatic ring currents in a molecular nanoring. Nature 2017, 541, 200–203. [Google Scholar] [CrossRef]
- Peeks, M.D.; Gong, J.Q.; McLoughlin, K.; Kobatake, T.; Haver, R.; Herz, L.M.; Anderson, H.L. Aromaticity and Antiaromaticity in the Excited States of Porphyrin Nanorings. J. Phys. Chem. Lett. 2019, 10, 2017–2022. [Google Scholar] [CrossRef]
- Rickhaus, M.; Jirasek, M.; Tejerina, L.; Gotfredsen, H.; Peeks, M.D.; Haver, R.; Jiang, H.-W.; Claridge, T.D.W.; Anderson, H.L. Global aromaticity at the nanoscale. Nat. Chem. 2020, 12, 236–241. [Google Scholar] [CrossRef]
- Peeks, M.D.; Jirasek, M.; Claridge, T.D.W.; Anderson, H.L. Global Aromaticity and Antiaromaticity in Porphyrin Nanoring Anions. Angew. Chem. Int. Ed. 2019, 58, 15717–15720. [Google Scholar] [CrossRef]
- Klod, S.; Koch, A.; Kleinpeter, E. Ab-initio quantum-mechanical GIAO calculation of the anisotropic effect of C–C and X–C single bonds—Application to the 1H NMR spectrum of cyclohexane. J. Chem. Soc. Perkin Trans. 2 2002, 9, 1506–1509. [Google Scholar] [CrossRef]
- Klod, S.; Kleinpeter, E. Ab initio calculation of the anisotropy effect of multiple bonds and the ring current effect of arenes—Application in conformational and configurational analysis. J. Chem. Soc. Perkin Trans. 2 2001, 10, 1893–1898. [Google Scholar]
- Stanger, A. Reexamination of NICSπ,ZZ: Height Dependence, Off-Center Values, and Integration. J. Phys. Chem. A 2019, 123, 3922–3927. [Google Scholar] [CrossRef]
- Dudek, W.M.; Ostrowski, S.; Dobrowolski, J.C. On Aromaticity of the Aromatic α-Amino Acids and Tuning of the NICS Indices to Find the Aromaticity Order. J. Phys. Chem. A 2022, 126, 3433–3444. [Google Scholar] [CrossRef]
- Aroma. Available online: https://chemistry.technion.ac.il/en/team/amnon-stanger/ (accessed on 20 November 2024).
- Wahab, A.; Fleckenstein, F.; Feusi, S.; Gershoni-Poranne, R. Predi-XY: A python program for automated generation of NICS-XY-scans based on an additivity scheme. Electron. Struct. 2020, 2, 047002. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifuncitonal Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Haddon, R.C.; Scott, L.T. π-Orbital conjugation and rehybridization in bridged annulenes and deformed molecules in general: π-orbital axis vector analysis. Pure Appl. Chem. 1986, 58, 137–142. [Google Scholar] [CrossRef]
- Haddon, R.C. Comment on the Relationship of the Pyramidalization Angle at a Conjugated Carbon Atom to the σ Bond Angles. J. Phys. Chem A 2001, 105, 4164–4165. [Google Scholar] [CrossRef]
- Haddon, R.C. Hybridization and the Orientation and Alignment of π-Orbitals in Nonplanar Conjugated Organic Molecules: π-Orbital Axis Vector Analysis (POAV2). J. Am. Chem. Soc. 1986, 108, 2837–2842. [Google Scholar] [CrossRef]
- PyQt6. Available online: https://pypi.org/project/PyQt6 (accessed on 20 November 2024).
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Hagberg, A.A.; Schult, D.A.; Swart, P.J. Exploring network structure, dynamics, and function using NetworkX. In Proceedings of the 7th Python in Science Conference (SciPy2008), Pasadena, CA, USA, 19–24 August 2008. [Google Scholar]
- OpenPyxl. Available online: https://foss.heptapod.net/openpyxl/openpyxl (accessed on 20 November 2024).
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li−F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Szatylowicz, H.; Wieczorkiewicz, P.A.; Krygowski, T.M. Molecular geometry as a source of electronic structure of π-electron systems and their physicochemical properties. In Aromaticity. Modern Computational Methods and Applications; Fernández, I., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2021; Chapter 3; pp. 71–98. [Google Scholar]
- Zborowski, K.K.; Alkorta, I.; Elguero, J.; Proniewicz, L.M. HOMA parameters for the boron–boron bond: How the introduction of a bb bond influences the aromaticity of selected hydrocarbons. Struct. Chem. 2013, 24, 543–548. [Google Scholar] [CrossRef]
- Zborowski, K.K.; Alkorta, I.; Elguero, J.; Proniewicz, L.M. Calculation of the HOMA model parameters for the carbon–boron bond. Struct. Chem. 2012, 23, 595–600. [Google Scholar] [CrossRef]
- Frizzo, C.P.; Martins, M.A.P. Aromaticity in heterocycles: New HOMA index parametrization. Struct. Chem. 2012, 23, 375–380. [Google Scholar] [CrossRef]
- Dobrowolski, J.C.; Ostrowski, S. HOMA Index Establishes Similarity to a Reference Molecule. J. Chem. Inf. Model. 2023, 63, 7744–7754. [Google Scholar] [CrossRef]
- Gershoni-Poranne, R.; Stanger, A. NICS–Nucleus-independent Chemical Shift. In Aromaticity. Modern Computational Methods and Applications; Fernández, I., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2021; pp. 99–154. [Google Scholar]
- Fallah-Bagher-Shaidaei, H.; Wannere, C.S.; Corminboeuf, C.; Puchta, R.; Schleyer, P.v.R. Which NICS Aromaticity Index for Planar π Rings Is Best? Org. Lett. 2006, 8, 863–866. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Dobrowolski, J.C.; Lipiński, P.F.J. On splitting of the NICS(1) magnetic aromaticity index. RCS Adv. 2016, 6, 23900–23904. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Sundholm, D. On the Aromaticity of the Planar Hydrogen-Bonded (HF)3 Trimer. J. Chem. Theory Comput. 2006, 2, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Buzsáki, D.; Kovács, M.B.; Hümpfner, E.; Harcsa-Pintér, Z.; Kelemen, Z. Conjugation between 3D and 2D aromaticity: Does it really exist? The case of carborane-fused heterocycles. Chem. Sci. 2022, 13, 11388–11393. [Google Scholar] [CrossRef]
- Zhao, L.; Grande-Aztatzi, R.; Foroutan-Nejad, C.; Ugalde, J.M.; Frenking, G. Aromaticity, the Hückel 4n+2 Rule and Magnetic Current. ChemistrySelect 2017, 2, 863–870. [Google Scholar] [CrossRef]
- Buzsáki, D.; Gál, D.; Harcsa-Pintér, Z.; Kalabay, L.; Kelemen, Z. The Possible Aromatic Conjugation via the Different Edges of (Car)Boran Clusters: Can the Relationship Between 3D and 2D Aromatic Systems Be Reconciled? Chem. Eur. J. 2024, 30, e202402970. [Google Scholar] [CrossRef] [PubMed]
- Damme, S.V.; Acke, G.; Havenith, R.W.A.; Bultinck, P. Can the current density map topology be extracted from the nucleus independent chemical shifts? Phys. Chem. Chem. Phys. 2016, 18, 11746–11755. [Google Scholar] [CrossRef] [PubMed]
- Foroutan-Nejad, C. Is NICS a reliable aromaticity index for transition metal clusters? Theor. Chem. Acc. 2015, 134, 8. [Google Scholar] [CrossRef]
- Zarate, X.; MacLeod-Carey, D.; Muñoz-Castro, A.; Schott, E. Understanding the aromaticity of C6X6 (X = H, F, Cl, Br, I). Insights from different theoretical criteria. Chem. Phys. Lett. 2019, 720, 52–57. [Google Scholar] [CrossRef]
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Computational Prediction of 1H and 13C Chemical Shifts: A Useful Tool for Nature Product, Mechanistic, and Synthetic Organic Chemistry. Chem. Rev. 2012, 112, 1839–1862. [Google Scholar] [CrossRef] [PubMed]
- Chemical Shift Repository with Coupling Constants Added Too. Available online: http://cheshirenmr.info/index.htm (accessed on 20 November 2024).
- Adamo, C.; Barone, V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models. J. Chem. Phys. 1998, 108, 664–675. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Pedregal, J.R.-G.; Gómez-Orellana, P.; Maréchal, J.-D. ESIgen: Electronic Supporting Information Generator for Computational Chemistry Publications. J. Chem. Inf. Model. 2018, 58, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://insilichem.com/esigen-loading (accessed on 20 November 2024).
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system–Version 5.0. WIREs Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Program | Detect Cycle? | BLA | HOMA, HOMER | POAV | NICS | NMR | GUI | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SP | XY-Scan | 2D | 3D | INICS | |||||||
| Aroma | — | — | — | — | — | ○ | — | — | — | — | ○ |
| Predi-XY | — | — | — | — | — | ○ | — | — | — | — | — |
| Multiwfn | — | ○ | ○ 1 | — | ○ 2 | — | ○ | ○ | ○ | ○ | — |
| py.Aroma | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z. py.Aroma: An Intuitive Graphical User Interface for Diverse Aromaticity Analyses. Chemistry 2024, 6, 1692-1703. https://doi.org/10.3390/chemistry6060103
Wang Z. py.Aroma: An Intuitive Graphical User Interface for Diverse Aromaticity Analyses. Chemistry. 2024; 6(6):1692-1703. https://doi.org/10.3390/chemistry6060103
Chicago/Turabian StyleWang, Zhe. 2024. "py.Aroma: An Intuitive Graphical User Interface for Diverse Aromaticity Analyses" Chemistry 6, no. 6: 1692-1703. https://doi.org/10.3390/chemistry6060103
APA StyleWang, Z. (2024). py.Aroma: An Intuitive Graphical User Interface for Diverse Aromaticity Analyses. Chemistry, 6(6), 1692-1703. https://doi.org/10.3390/chemistry6060103