

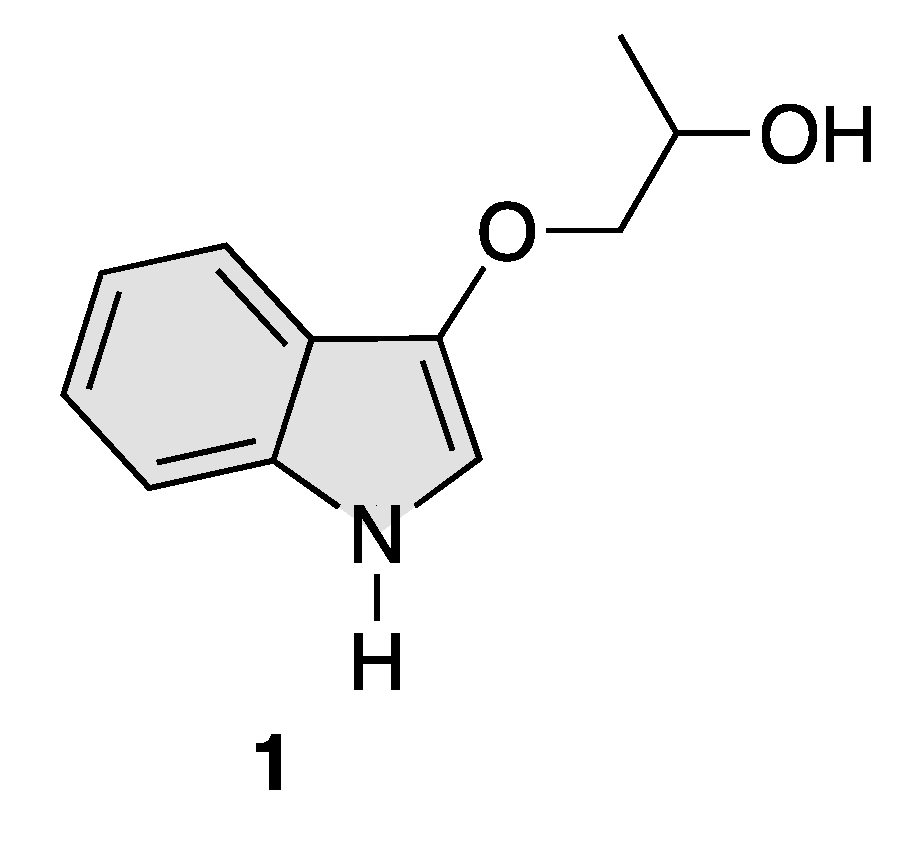
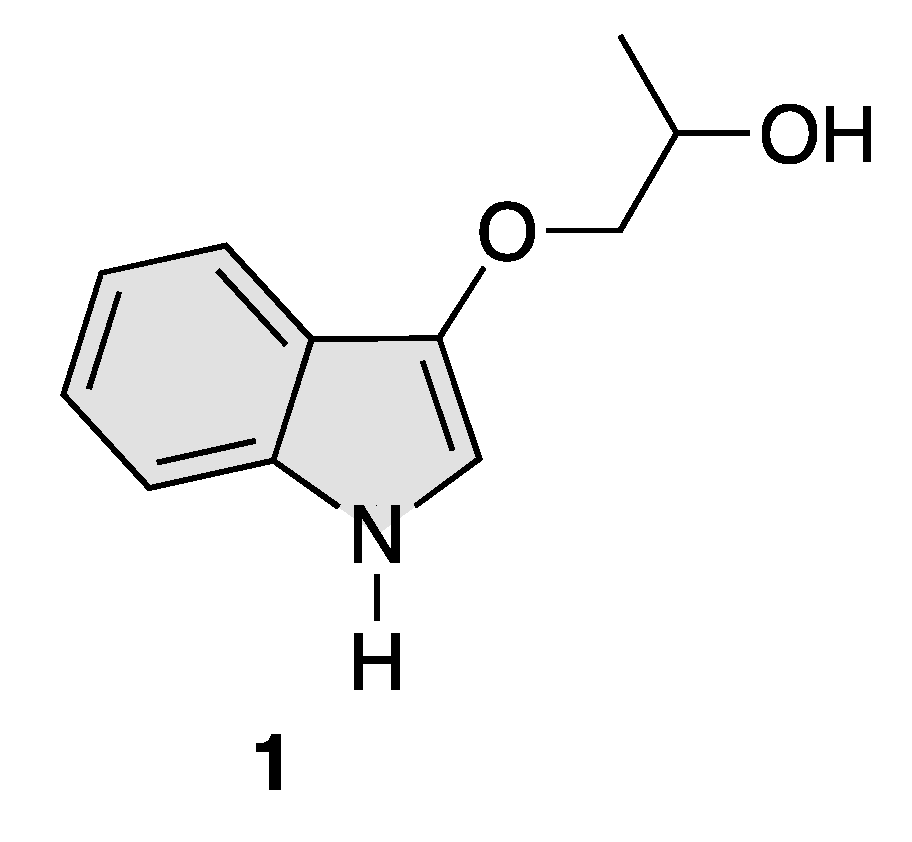
Total Synthesis of the Proposed Structure of Indolyl 1,2-Propanediol Alkaloid, 1-(1H-Indol-3-yloxy)propan-2-ol



Abstract
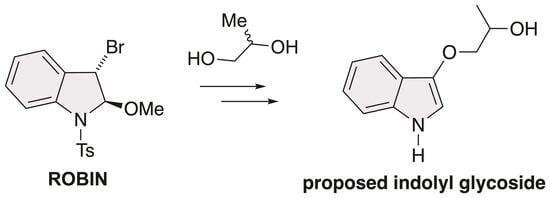
:
1. Introduction
2. Materials and Methods
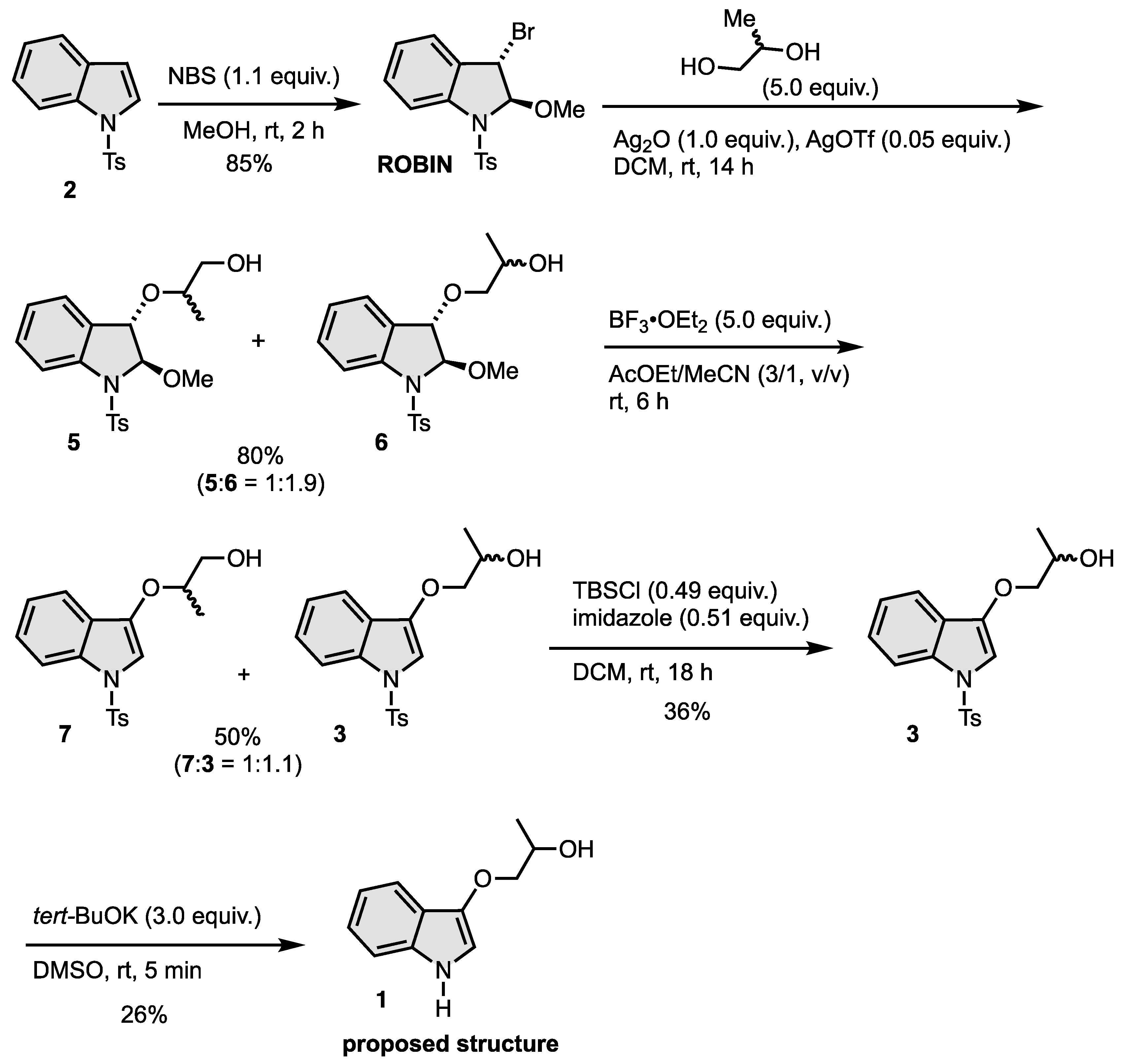
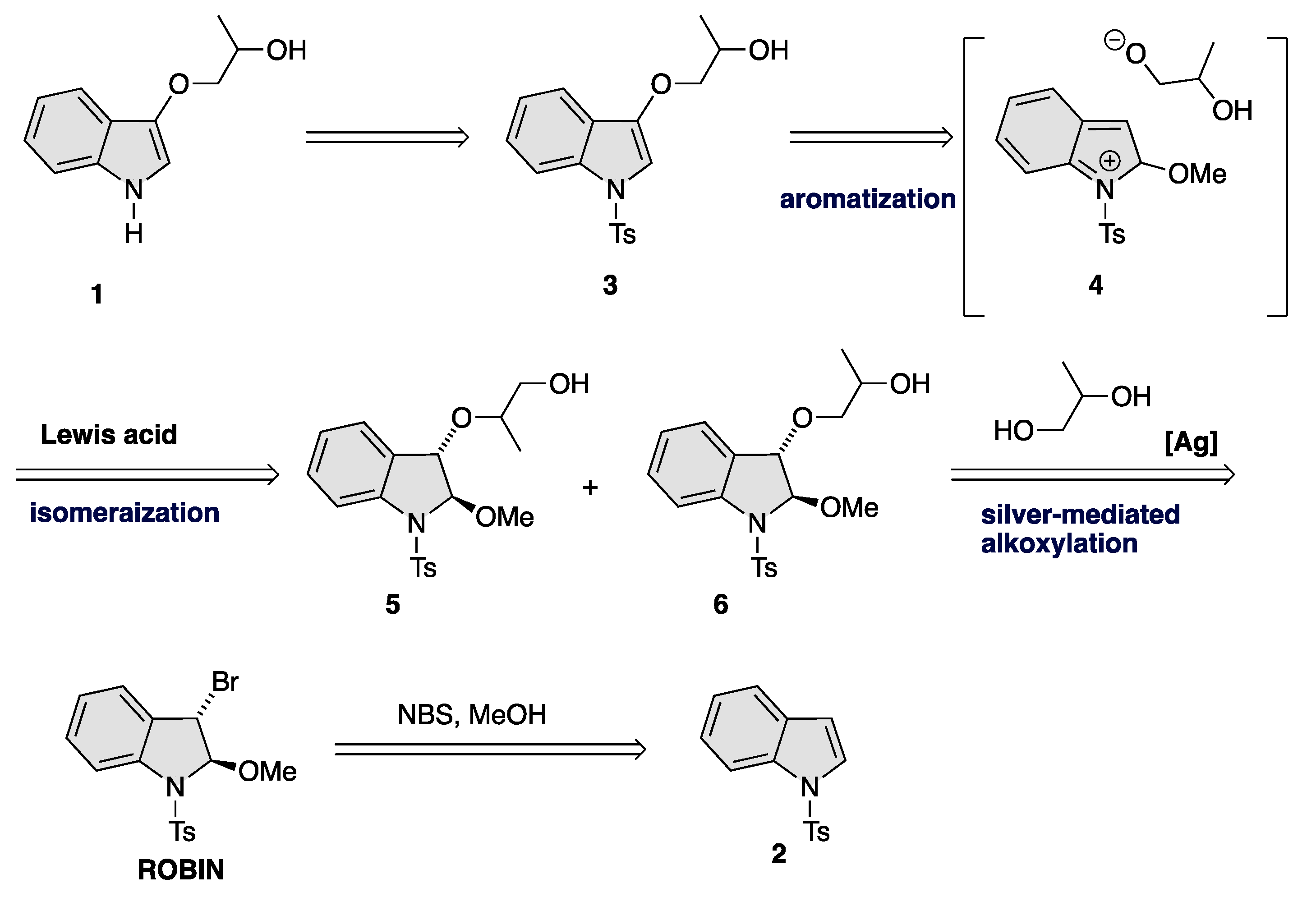
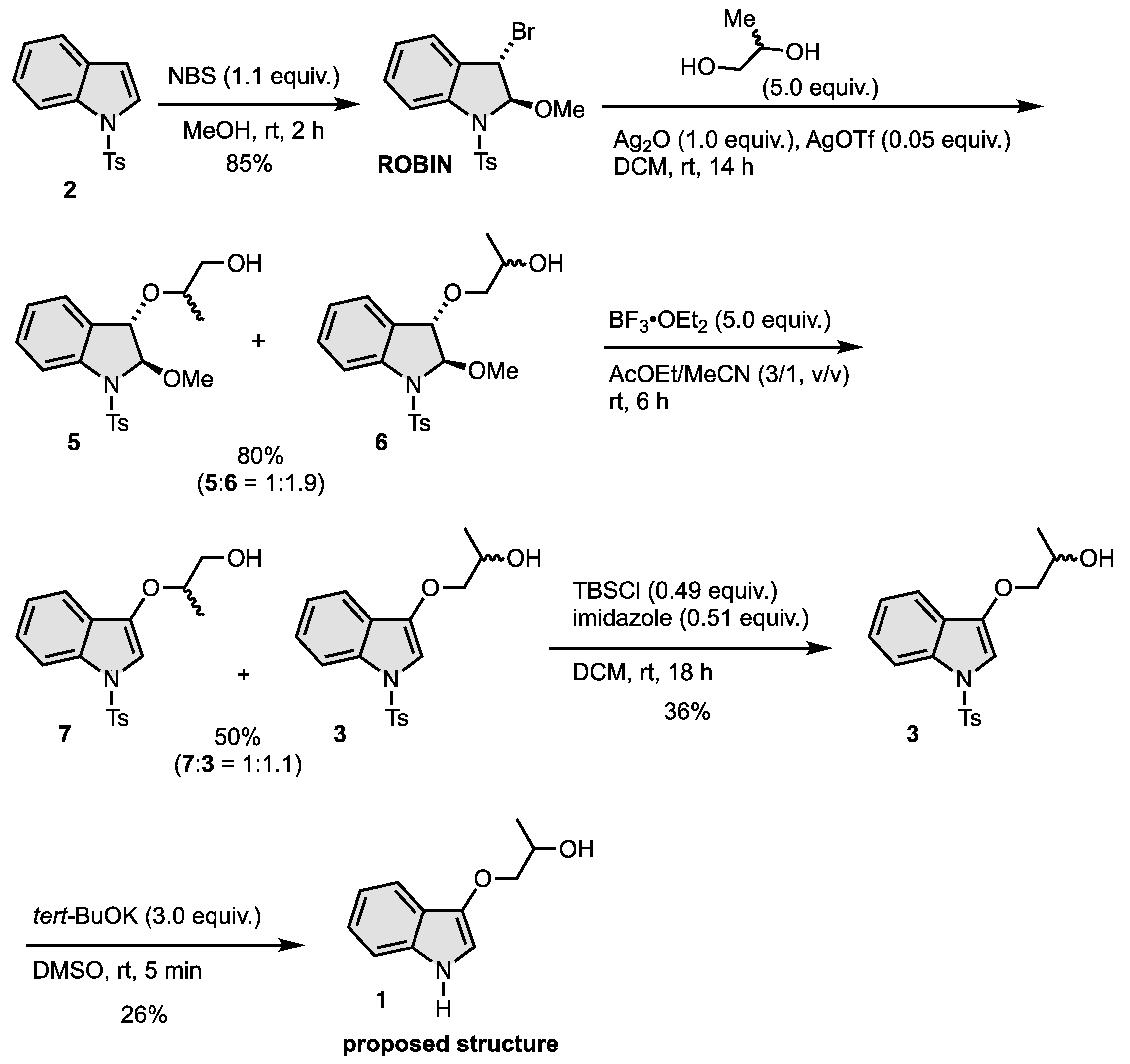
2.1. Synthesis of N-Tosylindoles (2) [51]: N-Tosylindoles (2) Was Prepared by a Reported Method [50]
2.2. Synthesis of Trans-3-Bromo-2-methoxy-1-tosylindoline (ROBIN) [48]

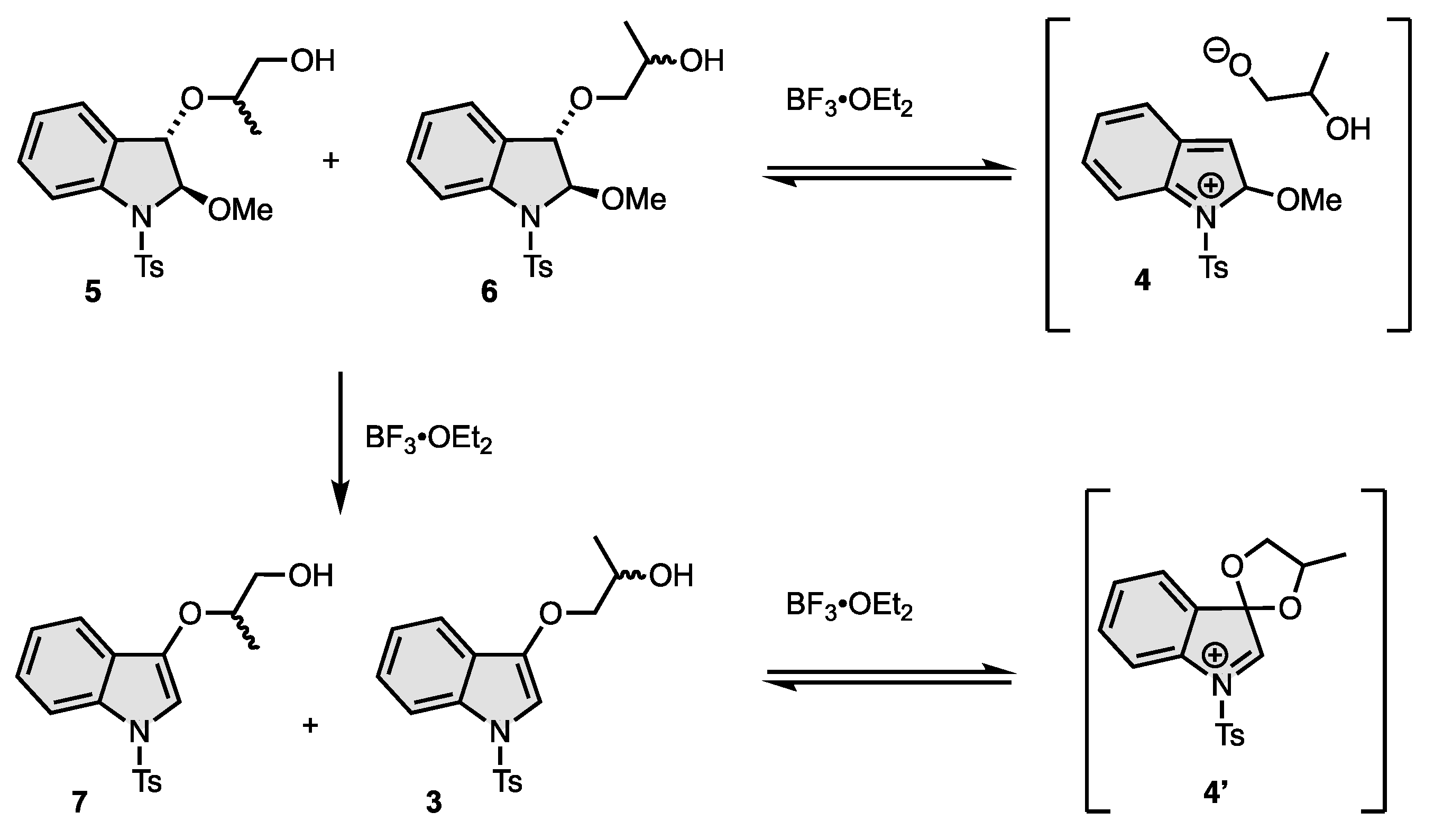
2.3. Synthesis of 5 and 6

2.3.1. Synthesis of Trans-3-((1-tert-Butyldimethylsilyloxy)propan-2-yloxy)-2-methoxy-1-tosylindoline (TBS-5)

2.3.2. Synthesis of 2-(Trans-2-methoxy-1-tosylindolin-3-yloxy)propan-1-ol (5)

2.3.3. Synthesis of 1-(Trans-2-Methoxy-1-tosylindolin-3-yloxy)propan-2-ol (TBS-6)

2.4. Synthesis of 7 and 3
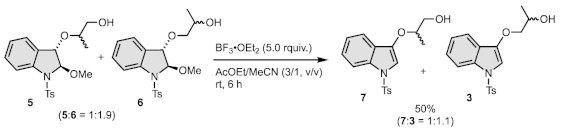
2.4.1. Synthesis of 3-((1-tert-Butyldimethylsilyloxy)propan-2-yloxy)-1-tosyl-1H-indole (TBS-7)
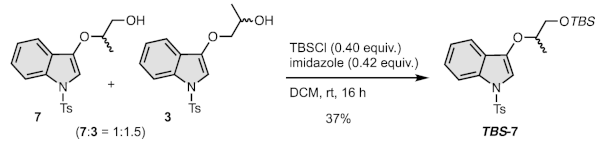
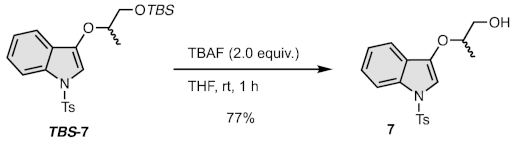
2.4.2. Synthesis of 1-(1-Tosyl-1H-indol-3-yloxy)propan-2-ol (7)
2.5. Synthesis of 2-(1-Tosyl-1H-indol-3-yloxy)propan-1-ol (3)

2.6. Synthesis of 1-(1H-Indol-3-yloxy)propan-2-ol (1)

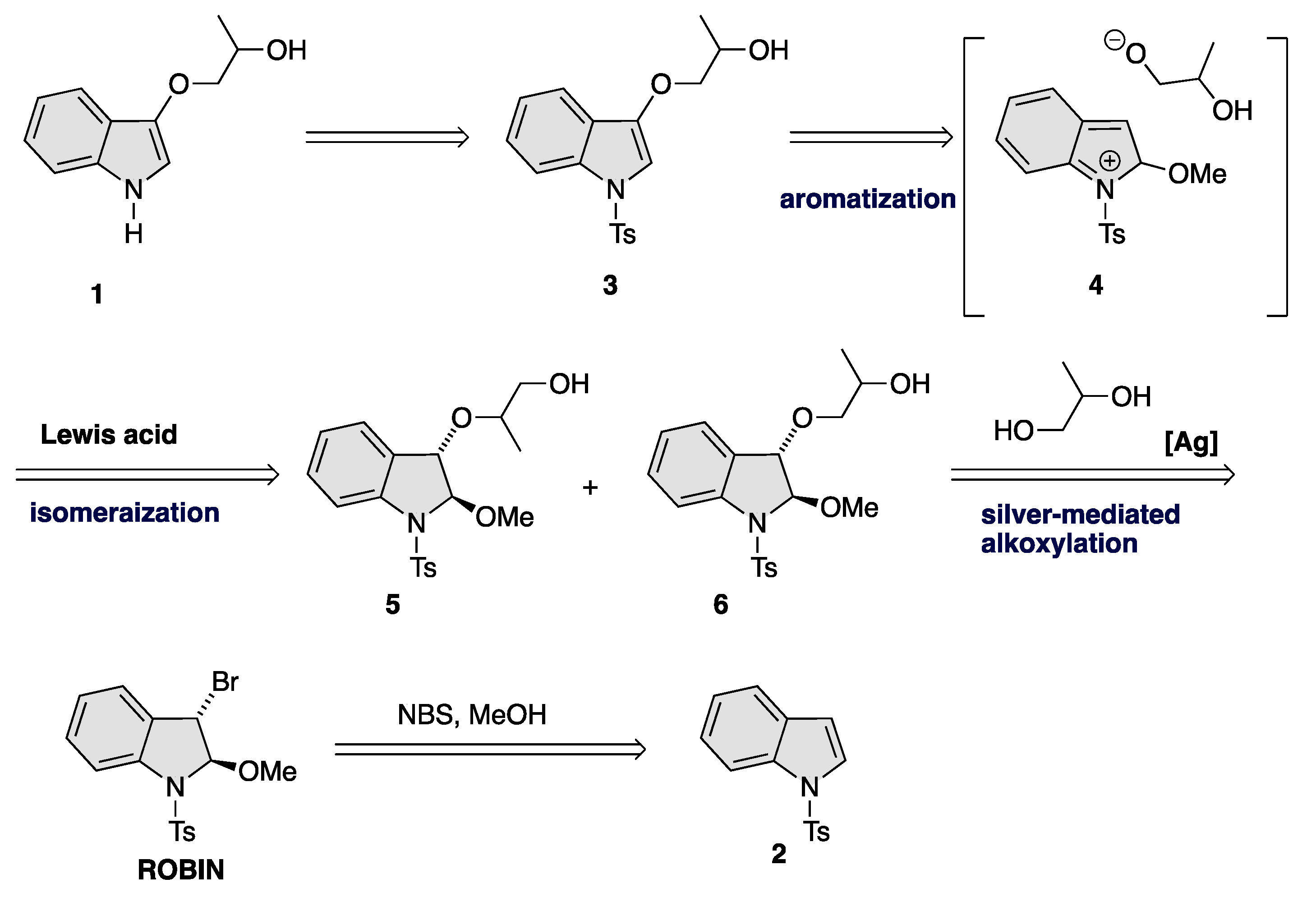
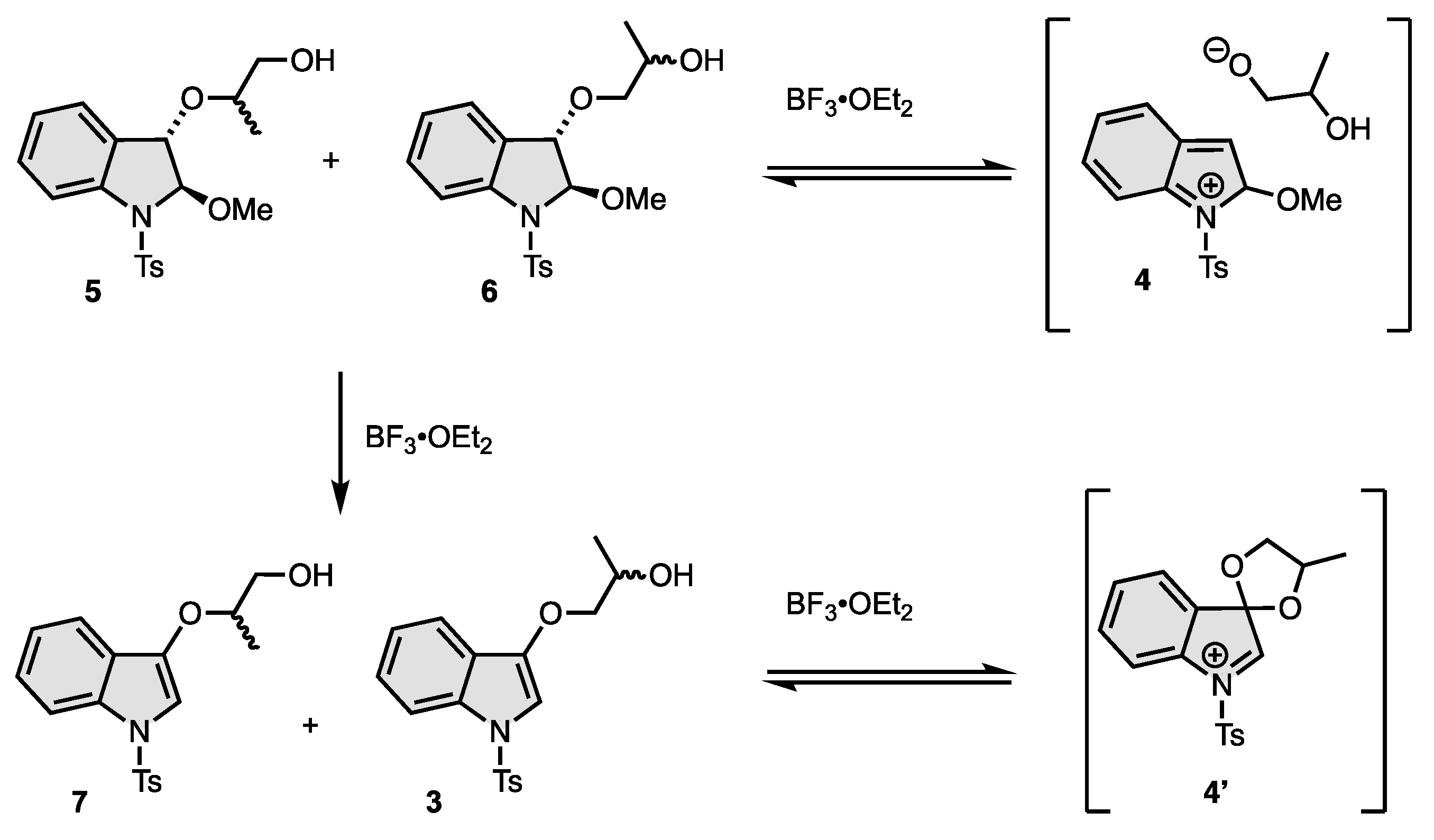
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nakase, K.; Nakajima, S.; Hirayama, M.; Kondo, H.; Kojiri, K.; Suda, H. Antitumor Substance BE-54017 and Its Production. JP Patent 2000178274, 27 June 2000. [Google Scholar]
- Grougnet, R.; Magiatis, P.; Fokialakis, N.; Mitaku, S.; Skaltsounis, A.-L.; Tillequin, F.; Sévenet, T.; Litaudon, M. Koniamborine, the First Pyrano[3,2-b]indole Alkaloid and Other Secondary Metabolites from Boronella koniambiensis. J. Nat. Prod. 2005, 68, 1083–1086. [Google Scholar] [CrossRef]
- Williams, D.E.; Davies, J.; Patrick, B.O.; Bottriell, H.; Tarling, T.; Roerge, M.; Anderson, R.J. Cladoniamides A–G, Tryptophan-Derived Alkaloids Produced in Culture by Streptomyces uncialis. Org. Lett. 2008, 10, 3501–3504. [Google Scholar] [CrossRef]
- Kitajima, M.; Mori, I.; Arai, K.; Kogure, N.; Takayama, H. Two New Typtamine-derived Alkaloids from Chimonanthus praecox f. concolor. Tetrahedron Lett. 2006, 47, 3199–3202. [Google Scholar] [CrossRef]
- Chen, W.-H.; Wu, Q.-X.; Wang, R.; Shi, Y.-P. Oxytrofalcatins A–F, N-Benzoylindole Analogues from the Roots of Oxytropis falcata (Leguminosae). Phytochemistry 2010, 71, 1002–1006. [Google Scholar] [CrossRef]
- Sugitate, K.; Yamashiro, T.; Takahashi, I.; Yamada, K.; Abe, T. Oxytrofalcatin Puzzle: Total Synthesis and Structural Revision of Oxytrofalcatins B and C. J. Org. Chem. 2023, 88, 9920–9926. [Google Scholar] [CrossRef]
- Du, Y.-L.; Ding, T.; Patrick, B.O.; Ryan, K.S. Xenocladoniamide F, minimal indolotryptoline from the cladoniamide pathway. Tetrahedron Lett. 2013, 54, 5635–5638. [Google Scholar] [CrossRef]
- Wardle, K.A.; Bingham, S.; Ellis, E.S.; Gaster, L.M.; Rushant, B.; Smith, M.I.; Sanger, G.J. Selective and functional 5-hydroxytryptamine4 receptor antagonism by SB 207266. Br. J. Pharmacol. 1996, 118, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.-Y.; Brady, S.F. Discovery of indolotryptoline antiproliferative agents by homology-guided metagenomic screening. Proc. Natl. Acad. Sci. USA 2013, 110, 2478–2483. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.-Y.; Kawashima, S.A.; Brady, S.F. Mutations in the Proteolipid Subunits of the Vacuolar H+-ATPase Provide Resistance to Indolotryptoline Natural Products. Biochemistry 2014, 53, 7123–7131. [Google Scholar] [CrossRef] [PubMed]
- Gowan, M.; Caillé, A.S.; Lau, C.K. Synthesis of 3-Alkoxyindoles via Palladium-Catalyzed Intramolecular Cyclization of N-Alkyl ortho-Siloxyallylanilines. Synlett 1997, 1997, 1312–1314. [Google Scholar] [CrossRef]
- Bös, M.; Jenck, F.; Martin, J.L.; Moreau, J.L.; Mutel, V.; Sleight, A.J.; Widmer, U. Synthesis, pharmacology and therapeutic potential of 10-methoxypyrazino[1,2-a]indoles, partial agonists at the 5HT2c receptor. Eur. J. Med. Chem. 1997, 32, 253–261. [Google Scholar] [CrossRef]
- Clawson, R.W., Jr.; Deavers III, R.E.; Akhmedov, N.G.; Söderberg, B.C.G. Palladium-catalyzed synthesis of 3-alkoxysubstituted indoles. Tetrahedron 2006, 62, 10829–10834. [Google Scholar] [CrossRef]
- Clawson, R.W., Jr.; Söderberg, B.C.G. A short synthesis of koniamborine, a naturally occurring pyrano[3,2-b]indole. Tetrahedron Lett. 2007, 48, 6019–6021. [Google Scholar] [CrossRef]
- Shi, Q.-Q.; Fu, L.-P.; Shi, Y.; Ding, H.-Q.; Luo, J.-J.; Jiang, B.; Tu, S.-J. Three-component synthesis of poly-substituted terahydroindoles through p-TsOH promoted alkoxylation. Tetrahedron Lett. 2013, 54, 3176–3179. [Google Scholar] [CrossRef]
- Li, T.-R.; Cheng, B.-Y.; Wang, Y.-N.; Zhang, M.-M.; Lu, L.-Q.; Xiao, W.-J. A Copper-Catalyzed Decarboxylative Amination/Hydroamination Sequence: Switchable Synthesis of Functionalized Indoles. Angew. Chem. Int. Ed. 2016, 55, 12422–12426. [Google Scholar] [CrossRef] [PubMed]
- Nykaza, T.V.; Ramirez, A.; Harrison, T.S.; Luzung, M.R.; Radosevich, A.T. Biphilic Organophosphorus-Catalyzed Intramolecular Csp2–H Amination: Evidence for a Nitrenoid in Catalytic Cadogan Cyclizations. J. Am. Chem. Soc. 2018, 140, 3103–3113. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Fan, H.; Yang, F.; Xu, S.; Zhao, X.; Liao, X.; Zhang, X. PPh3-Mediated Cascade Reaction of 2-Alkylnitrobenzenes and Thioureas for the Construction of Imidazo[4,5-b]indole-2-thiones. J. Org. Chem. 2023, 88, 2801–2808. [Google Scholar] [CrossRef]
- Kanaoka, Y.; Aiura, M.; Hariya, S. Direct Coversion of N-Methylindoles into Indoxyl, Oxindole, and Dioxindole O-Benzoates. J. Org. Chem. 1971, 36, 458–460. [Google Scholar] [CrossRef]
- Kwon, S.; Kuroki, N. Reaction of 1-Substituted indoles with Carboxylic Acids and N-Iodosuccinimide. Chem. Lett. 1980, 9, 237–238. [Google Scholar] [CrossRef]
- Vice, S.F.; Dmitrienko, G.L. The Bromination-Methanolysis of N-Acetyl-2,3-Dimethylindole. Can. J. Chem. 1982, 60, 1233–1237. [Google Scholar] [CrossRef]
- Kawasaki, T.; Chien, C.-S.; Sakamoto, M. Oxidation of 1-Acetylindoles with Molybdenum Peroxo Complex (MoO5•HMPA): Preparation of 1-Acetyl-trans- and cis-2,3-Dihydroxyindoline Derivatives. Chem. Lett. 1983, 12, 855–858. [Google Scholar] [CrossRef]
- Kettle, J.G.; Faull, A.W.; Fillery, S.M.; Flynn, A.P.; Hoyle, M.A.; Hudson, J.A. Facile synthesis of 3-alkoxyindoles via rhodium(II)-catalysed diazoindole O–H insertion reactions. Tetrahedron Lett. 2000, 41, 6905–6907. [Google Scholar] [CrossRef]
- Liang, Z.; Zhao, J.; Zhang, Y. Palladium-Catalyzed Regioselective Oxidative Coupling of Indoles and One-pot Synthesis of Acetoxylated Biindolyls. J. Org. Chem. 2010, 75, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.F., Jr.; Craveriro, M.V.; Gambardella, M.T.P. Synthesis of Polyalkylated Indoles Using a Thallium(III)-Mediated Ring-Construction Reaction. Synthesis 2007, 24, 3851–3857. [Google Scholar] [CrossRef]
- Mutule, I.; Suna, E.; Olofsson, K.; Pelcman, B. Catalytic Direct Acetoxylation of Indoles. J. Org. Chem. 2009, 74, 7195–7198. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.-S.; Coeffard, V.; Chataigner, I.; Vincent, G.; Moreau, X. Iron-Mediated Domino Interrupted Iso-Nazarov/Dearomatizative (3 + 2)-Cycloaddition of Electrophilic Indoles. Org. Lett. 2016, 18, 5296–5299. [Google Scholar] [CrossRef] [PubMed]
- Yamashiro, T.; Yamada, K.; Yoshida, H.; Tomisaka, Y.; Nishi, T.; Abe, T. Silver-Mediated Intramolecular Friedel–Crafts-Type Cyclizations of 2-Benzyloxy-3-bromoindolines: Synthesis of Isochromeno[3,4-b]indolines and 3-Arylindoles. Synlett 2019, 30, 2247–2252. [Google Scholar] [CrossRef]
- Abe, T.; Kosaka, Y.; Asano, M.; Harasawa, N.; Mishina, A.; Nagasue, M.; Sugimoto, Y.; Katakawa, K.; Sueki, S.; Anada, M.; et al. Direct C4-Benzylation of Indoles via Tandem Benzyl Claisen/Cope Rearrangements. Org. Lett. 2019, 21, 826–829. [Google Scholar] [CrossRef]
- Peng, H.; Ma, J.; Duan, L.; Zhang, G.; Yin, B. CuH-Catalyzed Synthesis of 3-Hydroxyindolines and 2-Aryl-3H-indol-3-ones from o-Alkynylnitroarenes, Using Nitro as Both the Nitrogen and Oxygen Source. Org. Lett. 2019, 21, 6194–6198. [Google Scholar] [CrossRef]
- Ryzhakov, D.; Jarret, M.; Baltaze, J.-P.; Guillot, R.; Kouklovsky, C.; Vincent, G. Synthesis of 3,3-Spirocyclic 2-Phosphonoindolines via a Dearomative Addition of Phosphonyl Radicals to Indoles. Org. Lett. 2019, 21, 4986–4990. [Google Scholar] [CrossRef]
- Xu, M.-M.; Cao, W.-B.; Ding, R.; Li, H.-Y.; Xu, X.-P.; Ji, S.-J. Dearomatization of Indoles via Azido Radical Addition and Dioxygen Trapping to Access 2-Azidoindolin-3-ols. Org. Lett. 2019, 21, 6217–6220. [Google Scholar] [CrossRef]
- Ren, H.; Song, J.-R.; Li, Z.-Y.; Pan, W.-D. Oxazoline-/Copper-Catalyzed Alkoxyl Radical Generation: Solvent-Switched to Access 3a,3a’-Bisfuroindoline and 3-Alkoxyl Furoindoline. Org. Lett. 2019, 21, 6774–6778. [Google Scholar] [CrossRef]
- Hirao, S.; Yamashiro, T.; Kohira, K.; Mishima, N.; Abe, T. 2,3-Dimethoxyindolines: A latent electrophile for SNAr reactions triggered by indium catalysts. Chem. Commun. 2020, 56, 5139–5142. [Google Scholar] [CrossRef] [PubMed]
- Ali, K.; Bera, M.; Cho, E.J. [3,3]-Rearrangements of N-Oxyindoles. Synlett 2023, 34, 1019–1022. [Google Scholar]
- Takayama, H.; Misawa, K.; Okada, N.; Ishikawa, H.; Kitajima, M.; Hatori, Y.; Murayama, T.; Wongseripipatana, S.; Tashima, K.; Matsumoto, K.; et al. New Procedure to Mask the 2,3-p Bond of the Indole Nucleus and Its Application to the Preparation of Potent Opioid Receptor Agonists with a Corynanthe Skeleton. Org. Lett. 2006, 25, 5705–5708. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhao, Q.Y.; Liu, J.; Wu, P.; Yi, H.; Lei, A. A trans Diacyloxylation of Indoles. Chem. Commun. 2012, 48, 3239–3241. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; You, S.-L. Asymmetric Chlorocyclization of indole-3-yl-benzamides for the Construction of Fused Indolines. Org. Lett. 2014, 16, 2426–2429. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, A.A.; Chisholm, J.D. Lewis Acid Catalyzed Displacement of Trichloroacetamidates in the Synthesis of Functionalized Pyrroloindolines. Org. Lett. 2016, 18, 4100–4103. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Dou, Y.; Guillot, R.; Kouklovsky, C.; Vincent, G. Electrochemical Dearomative 2,3-Difunctionalization of Indoles. J. Am. Chem. Soc. 2019, 141, 2832–2837. [Google Scholar] [CrossRef]
- Zhang, S.; Li, L.; Wu, P.; Gong, P.; Liu, R.; Xu, K. Substrate-Depedent Electrochemical Dimethoxylation of Olefins. Adv. Synth. Catal. 2019, 361, 485–489. [Google Scholar] [CrossRef]
- Chen, N.; Deng, T.-T.; Li, J.-Q.; Cui, X.-Y.; Sun, W.-W.; Wu, B. Hypervalent Iodine(III)-Mediated Umpolung Dialkoxylation of N-Substituted Indoles. J. Org. Chem. 2022, 87, 12759–12771. [Google Scholar] [CrossRef] [PubMed]
- Yamashiro, T.; Abe, T.; Sawada, D. Synthesis of 2-monosubstituted indolin-3-ones by cine-substitution of 3-azido-2-methoxyindolines. Org. Chem. Front. 2022, 9, 1897–1903. [Google Scholar] [CrossRef]
- Bera, M.; Hwang, H.S.; Um, T.-W.; Oh, S.M.; Shin, S.; Cho, E.J. Energy Transfer Photocatalytic Radical Rearrangement in N-Indolyl Carbonates. Org. Lett. 2022, 24, 1774–1779. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, D.; Liu, Z.; Wang, Y.; Liu, Y.; Wang, S.; Wang, P.; Cong, H.; Chen, Y.-H.; Lu, L.; et al. Unraveling the Structure and Reactivity Patterns of the Indole Radical Cation in Regioselective Electrochemical Oxidative Annulations. J. Am. Chem. Soc. 2023, 145, 3175–3186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Duan, B.; Zhou, L.; Song, X.; Song, Z. Metal-Free Oxidative Dearomatization-Alkoxylation/Acyloxylation of Indoles: Synthesis of 2-Monosubstituted Indolin-3-ones. Org. Lett. 2023, 25, 7678–7682. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Guo, J.; Bao, Y.; Yuan, Y.; Hu, M.; Li, X.; Yan, H.; Cai, Y.; Xia, Q. KI-Catalyzed C(sp3)–H Amination and Acyloxylation of Indolin-3-ones Using Air as the Oxidants. Org. Lett. 2023, 25, 8162–8167. [Google Scholar] [CrossRef]
- Abe, T.; Kosaka, Y.; Kawasaki, T.; Ohata, Y.; Yamashiro, T.; Yamada, K. Revisiting 2-Alkoxy-3-bromoindolines: Control C-2 vs. C-3 Elimination for Regioselective Synthesis of Alkoxyindoles. Chem. Pharm. Bull. 2020, 68, 555–558. [Google Scholar] [CrossRef]
- Al-Massarani, S.M.; El-Gamal, A.A.; Al-Said, M.S.; Abdel-Kader, M.S.; Ashour, A.E.; Kumar, A.; Abdel-Mageed, W.M.; Al-Rehaily, A.J.; Ghabbour, H.A.; Fun, H.-K. Studies on the Red Sea Sponge Haliclona sp. for its Chemical and Cytotoxic Properties. Pharmacogn. Mag. 2016, 12, 114–119. [Google Scholar] [CrossRef]
- Iwasaki, T.; Uchiyama, R.; Nosaka, K. Difference in Anti-microbial Activity of Propan-1,3-diol and Propylene Glycol. Chem. Pharm. Bull. 2023, 71, 74–77. [Google Scholar] [CrossRef]
- Yoo, H.-D.; Nam, S.-J.; Chin, Y.-W.; Kim, M.-S. Misassigned natural products and their revised structures. Arch. Pharmacal Res. 2016, 39, 143–153. [Google Scholar] [CrossRef]
- Last, K.; Hoffmann, H.M.R. Vicinal Bromopropargoxylation of Cyclic Olefins and Cobaloxime-Mediated Heteroannulation to Functionalized 3-Methyleneoxacyclopentanes. Synthesis 1989, 1989, 901–905. [Google Scholar] [CrossRef]
- Johny, M.; Philip, R.M.; Rajendar, G. Highly Regio- and Stereoselective Intramolecular Rearrangement of Glycidol Acetal to Alkoxy Cyclic Acetals. Org. Lett. 2022, 24, 6165–6170. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, H.; Okitsu, T.; Sawama, Y.; Murata, N.; Li, R.; Kita, Y. Reaction of the Acetals with TESOTf-Base Combination; Speculation of the Intermediates and Efficient Mixed Acetal Formation. J. Am. Chem. Soc. 2006, 128, 5930–5938. [Google Scholar] [CrossRef] [PubMed]
- Hamel, P. Mechanism of the Second Sulfenylation of Indole. J. Org. Chem. 2002, 67, 2854–2858. [Google Scholar] [CrossRef] [PubMed]
- Heredia, M.D.; Walter, D.G.; Barolo, S.M.; Fornasier, S.J.; Rossi, R.A.; Budén, M.E. Transition-Metal-Free and Visible-Light-Mediated Desulfonylation and Dehalogenation Reactions: Hantzsch Ester Anion as Electron and Hydrogen Atom Donor. J. Org. Chem. 2020, 85, 13481–13494. [Google Scholar] [CrossRef] [PubMed]
- Banzragchgarav, O.; Murata, T.; Odontuya, G.; Buyankhishing, B.; Suganuma, K.; Davaapuurev, B.-O.; Inoue, N.; Batkhuu, J.; Sasaki, K. Trypanocidal Activity of 2,5-Diphenyloxazoles Isolated from the Roots of Oxytropis Lanata. J. Nat. Prod. 2016, 79, 2933–2940. [Google Scholar] [CrossRef]
- Suyama, T.L.; Gerwick, W.H.; McPhail, K.L. Survey of marine natural product structure revisions: A synergy of spectroscopy and chemical synthesis. Bioorg. Med. Chem. 2011, 19, 6675–6701. [Google Scholar] [CrossRef]
- Chhetri, B.K.; Lavoie, S.; Sweeney-Jones, A.M.; Kubanek, J. Recent trends in the structural revision of natural products. Nat. Prod. Rep. 2018, 35, 514–531. [Google Scholar] [CrossRef]
- Costa, F.L.P.; de Albuquerque, A.C.F.; Fiorot, R.G.; Liao, L.M.; Martorano, L.H.; Mota, G.V.S.; Carneiro, J.W.M.; dos Santos Junior, F.M. Structural characterization of natural products by means of quantum chemical calculations of NMR parameters: New insights. Org. Chem. Front. 2021, 8, 2019–2058. [Google Scholar] [CrossRef]
- Elyashberg, M.; Novitskiy, I.M.; Bates, R.W.; Kutateladze, A.G.; Williams, C.M. Reassignment of Improbable Natural Products Identified through Chemical Principle Screening. Eur. J. Org. Chem. 2022, 34, e202200572. [Google Scholar] [CrossRef]
- Shen, S.-M.; Appendino, G.; Guo, Y.-W. Pitfalls in the structural elucidation of small molecules. A critical analysis of a decade of structural misassignments of marine natural products. Nat. Prod. Rep. 2022, 39, 1803–1832. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.W.; Kim, J.; Paek, S.-M. Recent Achievements in Total Synthesis for Integral Structural Revisions of Marine Natural Products. Mar. Drugs 2022, 20, 171. [Google Scholar] [CrossRef] [PubMed]
- Maier, M.E. Structural revisions of natural products by total synthesis. Nat. Prod. Rep. 2009, 26, 1105–1124. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Hu, W.; He, D.; He, C.; Zhang, L.; Shi, Q. Machine-learning assisted molecular formula assignment to high-resolution mass spectrometry data of dissolved organic matter. Talanta 2023, 259, 124484. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Snyder, S.A. Chasing Molecules That Were Never There: Misassigned Natural Products and the Role of Chemical Synthesis in Modern Structure Elucidation. Angew. Chem. Int. Ed. 2005, 44, 1012–1044. [Google Scholar] [CrossRef] [PubMed]
- Willoughby, P.H.; Jansma, M.J.; Hoye, T.R. A guide too small-molecule structure assignment through computation of (1H and 13C) NMR chemical shifts. Nat. Protoc. 2014, 9, 643–660. [Google Scholar] [CrossRef]
- Novitskity, I.M.; Kutateladze, A.G. DU8ML: Machine Learning-Augmented Density Functional Theory Nuclear Magnetic Resonance Computations for High–Throughput in Silico Solution Structure Validation and Revision of complex Alkaloids. J. Org. Chem. 2022, 87, 4818–4828. [Google Scholar] [CrossRef]
- Novitskiy, I.M.; Kutateladze, A.G. Brief Overview of Recently reported Misassigned Natural Products and Their in Silico Revisions Enabled by DU8ML, a machine Learning-Augmented DFT Computational NMR Method. Nat. Prod. Rep. 2022, 39, 2003–2007. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
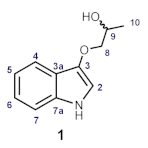
| Position | Synthetic 1 δH (mult, J in Hz) 600 MHz, methanol-d4 | Natural 1 δH (mult, J in Hz) 500 MHz, methanol-d4 |
|---|---|---|
| 2 | 6.80 (s) | 7.95 (br s) |
| 4 | 7.57 (ddd, 7.8, 1.2, 1.2) | 8.13 (br d, 7.8) |
| 5 | 6.95 (ddd, 8.0, 7.2, 1.2) | 7.16 (dt, 7.6, 1.3) |
| 6 | 7.08 (ddd, 8.1, 7.2, 1.2) | 7.20 (dt, 7.6, 1.3) |
| 7 | 7.25 (ddd, 8.4, 0.6, 0.6) | 7.45 (br d, 7.6) |
| 8 | 3.90 (dd, 5.7, 0.6) | 3.44 (m) |
| 9 | 4.17 (tq, 6.0, 5.4) | 3.80 (m) |
| 10 | 1.30 (d, 6.0) | 1.15 (d, 6.4) |
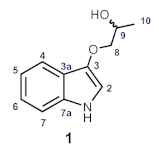
| Position | Synthetic 1 δC 151 MHz, methanol-d4 | Natural 1 δC 125 MHz, methanol-d4 |
| 2 | 105.5 | 133.6 |
| 3 | 119.4 | 110.0 |
| 3a | 140.1 | 128.0 |
| 4 | 117.1 | 122.2 |
| 5 | 117.7 | 122.1 |
| 6 | 121.6 | 123.4 |
| 7 | 110.9 | 112.8 |
| 7a | 134.5 | 138.2 |
| 8 | 76.2 | 68.5 |
| 9 | 65.9 | 69.2 |
| 10 | 18.4 | 19.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kimata, M.; Abe, T. Total Synthesis of the Proposed Structure of Indolyl 1,2-Propanediol Alkaloid, 1-(1H-Indol-3-yloxy)propan-2-ol. Chemistry 2023, 5, 2772-2784. https://doi.org/10.3390/chemistry5040177
Kimata M, Abe T. Total Synthesis of the Proposed Structure of Indolyl 1,2-Propanediol Alkaloid, 1-(1H-Indol-3-yloxy)propan-2-ol. Chemistry. 2023; 5(4):2772-2784. https://doi.org/10.3390/chemistry5040177
Chicago/Turabian StyleKimata, Momoko, and Takumi Abe. 2023. "Total Synthesis of the Proposed Structure of Indolyl 1,2-Propanediol Alkaloid, 1-(1H-Indol-3-yloxy)propan-2-ol" Chemistry 5, no. 4: 2772-2784. https://doi.org/10.3390/chemistry5040177
APA StyleKimata, M., & Abe, T. (2023). Total Synthesis of the Proposed Structure of Indolyl 1,2-Propanediol Alkaloid, 1-(1H-Indol-3-yloxy)propan-2-ol. Chemistry, 5(4), 2772-2784. https://doi.org/10.3390/chemistry5040177