
The dn Number in Transition Metal Chemistry: Its Utility and Limitations


Abstract
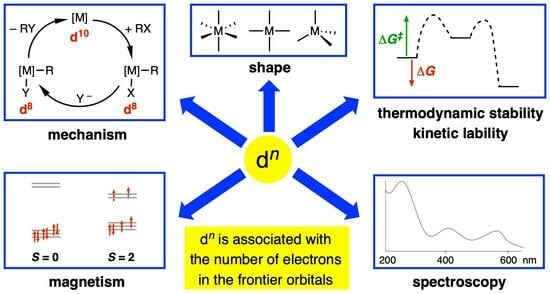
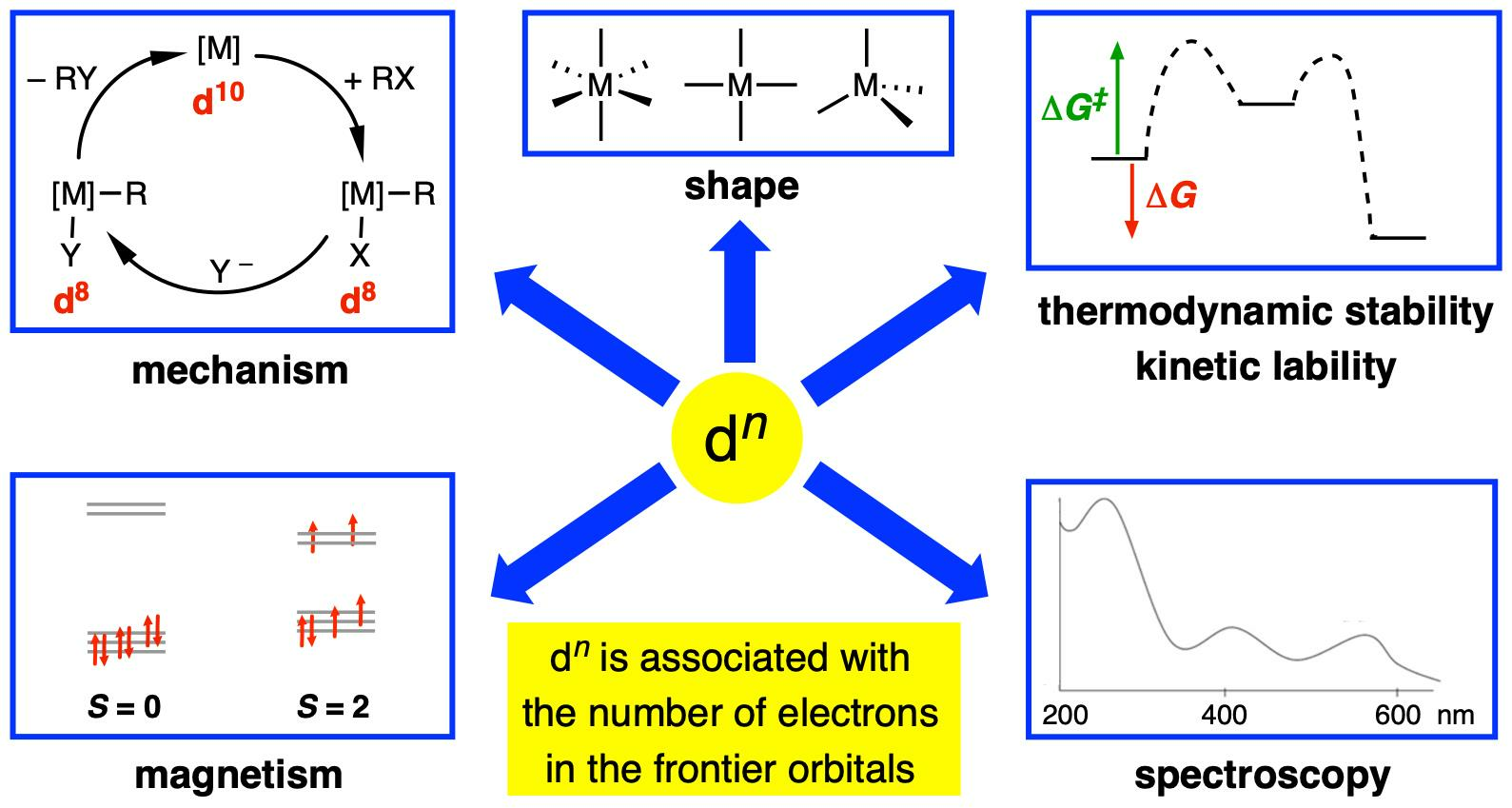
:
1. Introduction
2. A Definition of the dn Number and Why It Is a Useful Parameter
- ‘The dn configuration is the number of electrons that are housed in metal d orbitals that are either primarily non-bonding or have metal–ligand antibonding character.’ (Appendix A Note 3) [6,7].
- ‘The number of d-electrons generally describes the number of electrons not involved in the primary metal–ligand bonding interactions.’ [8].
- ‘…in the dn configuration using the definition that this quantity refers to electrons in d-based molecular orbitals that are not considered to be bonding.’ [9].
- ‘…the dn configuration corresponds to the number of electrons in d-based molecular orbitals that are formally localised on the metal after bond formation, i.e., those that are not components of metal–ligand bonding orbitals.’ [9].
- (i)
- The magnetic properties of complexes based on the number of unpaired electrons present, which includes a consideration of whether the complex is low spin or high spin according to the relative magnitude of the crystal field splitting energy (Δ) vs. the electron pairing energy (P) (Appendix A Note 6).
- (ii)
- The expectation that d0 complexes should not be susceptible to oxidation at the metal centre.
- (iii)
- The observation that many four-coordinate d8 metal complexes are square planar, whereas four-coordinate d10 and d0 complexes are almost invariably tetrahedral (Appendix A Note 7) [18].
- (iv)
- The observation that octahedral coordination is particularly favourable for metals with a d6 or a d3 configuration.
- (v)
- Whether or not complexes are kinetically labile or inert with respect to ligand substitution reactions according to the magnitude of the crystal/ligand field stabilisation energy (CFSE/LFSE) and the derived crystal/ligand field activation energy.
- (vi)
- The rationalisation of trends in lattice energies and hydration energies according to the magnitude of the CFSE/LFSE.
- (vii)
- The mechanisms of homogeneous catalysis by late transition metal complexes, which frequently involve interchange between d6, d8 and d10 species.
- (viii)
- Jahn-Teller distortions from idealised geometries for particular dn configurations; for example, d9 octahedral complexes.
- (ix)
- The interpretation of the electronic spectra of complexes using qualitative Orgel or more quantitative Tanabe–Sugano diagrams.
- (x)
- The isolobal principle and the fragment orbital analysis from which it is derived [19].
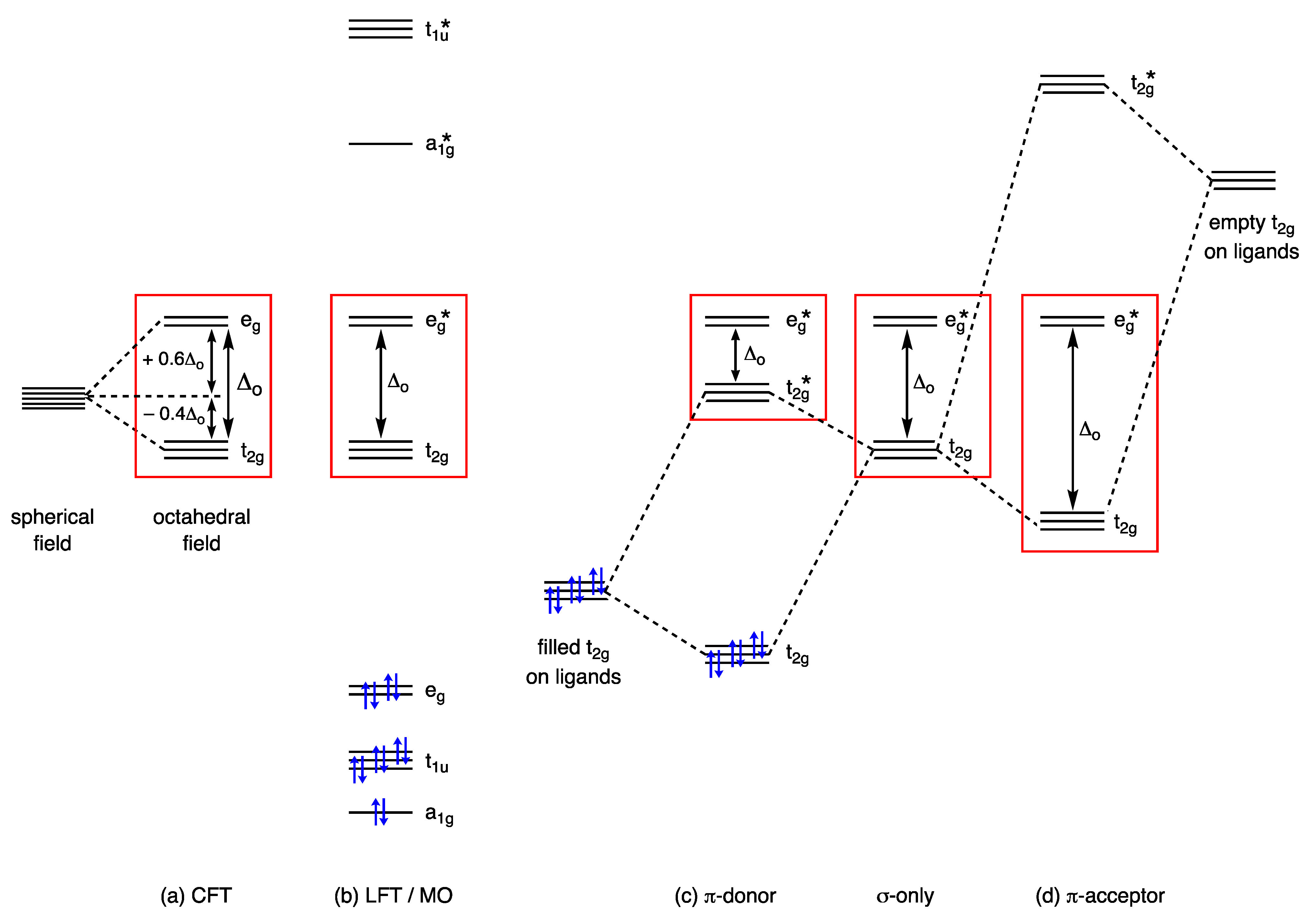
3. Crystal Field Theory and Ligand Field Theory
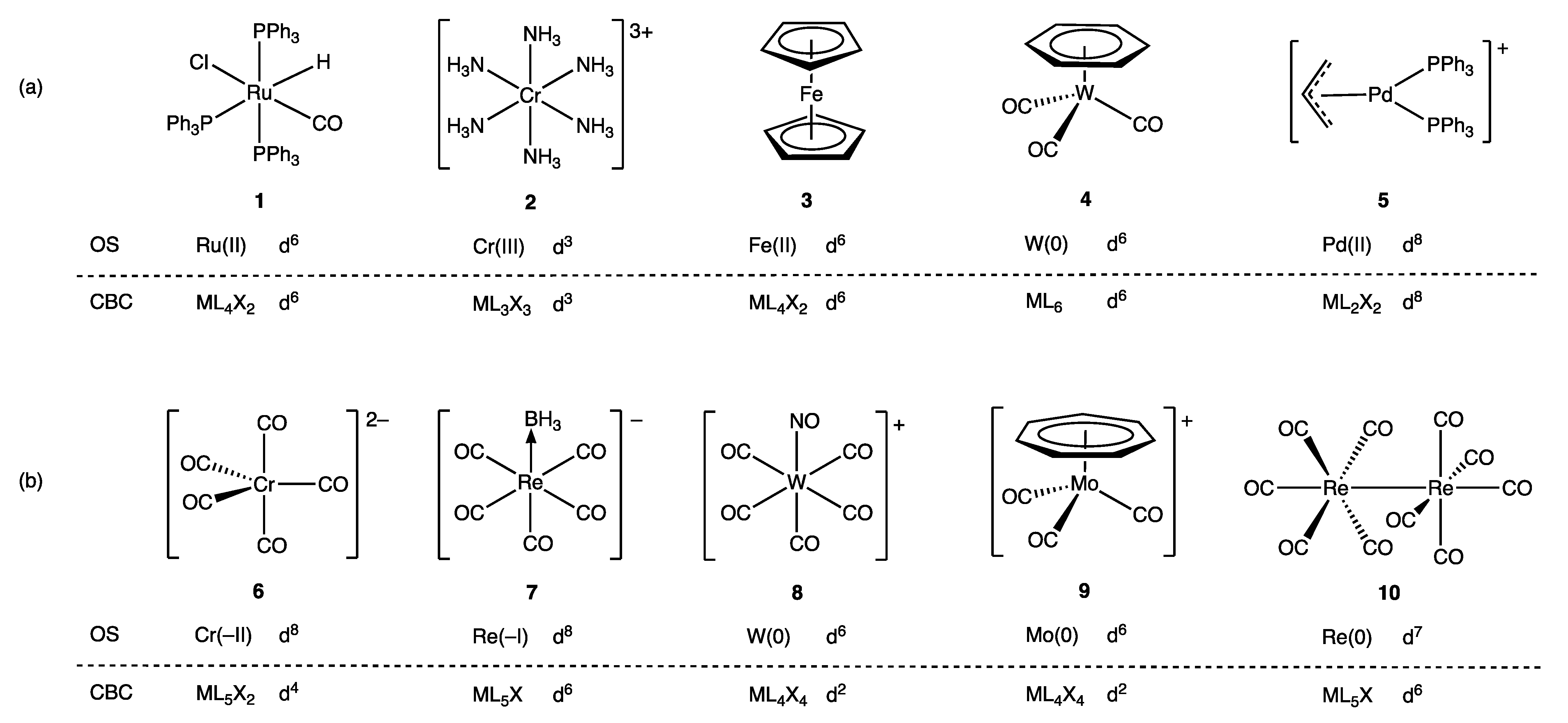
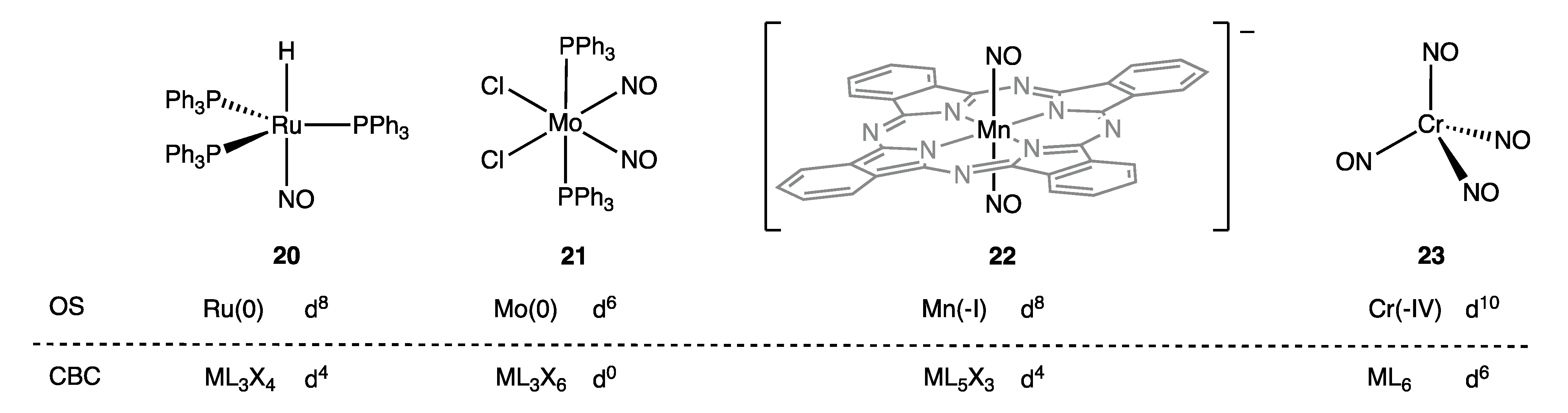
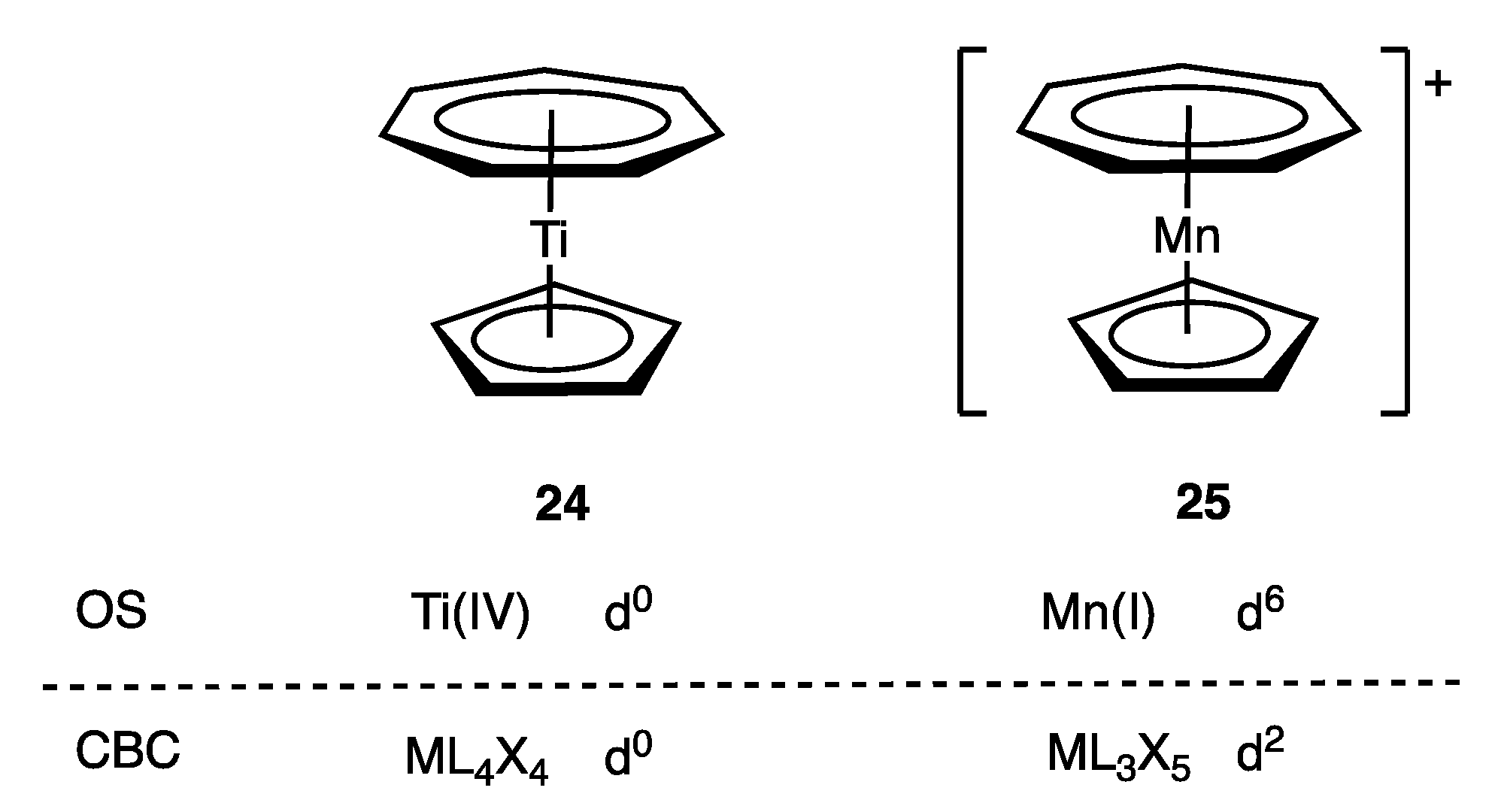
4. Covalent Bond Classification
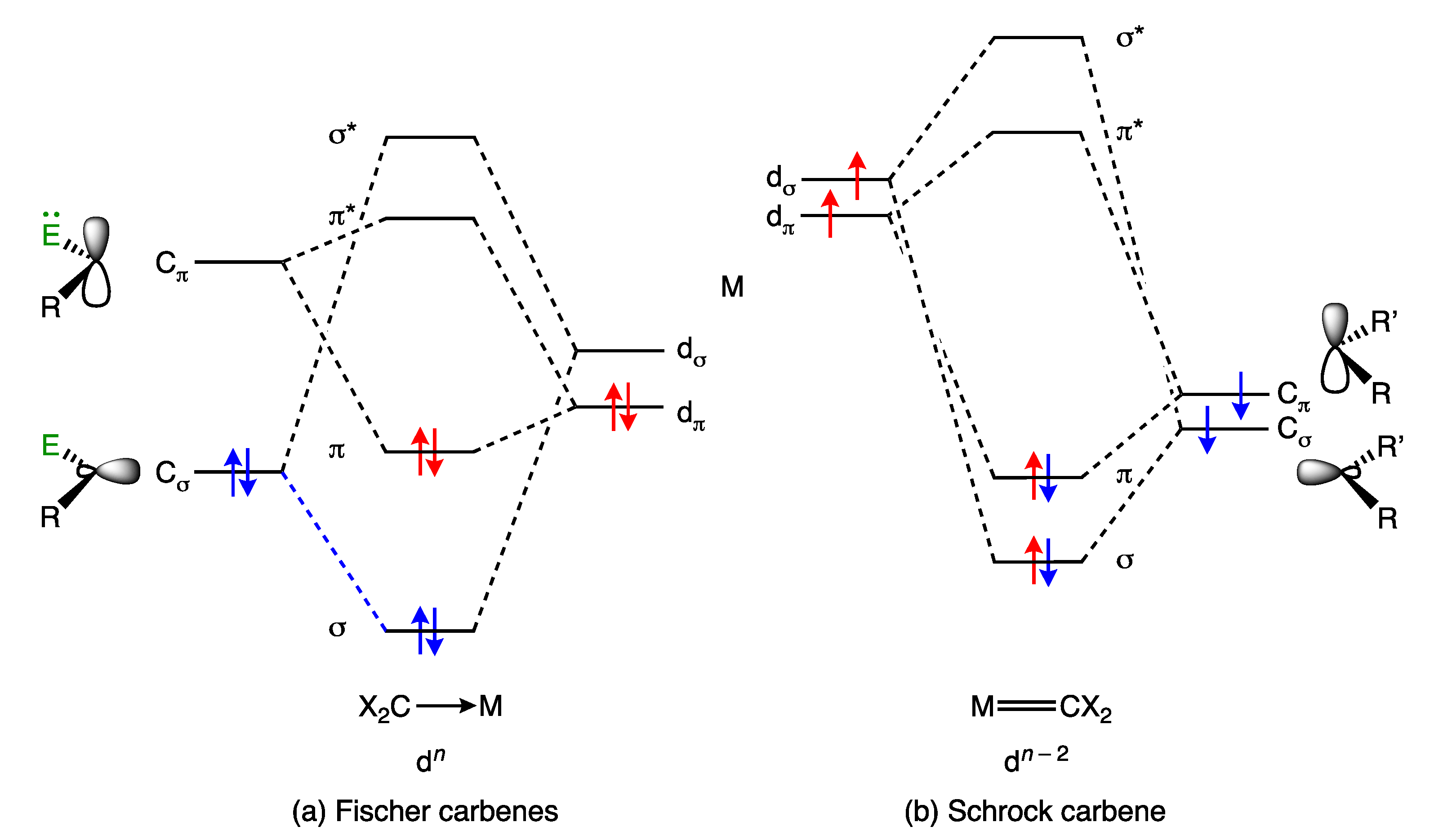
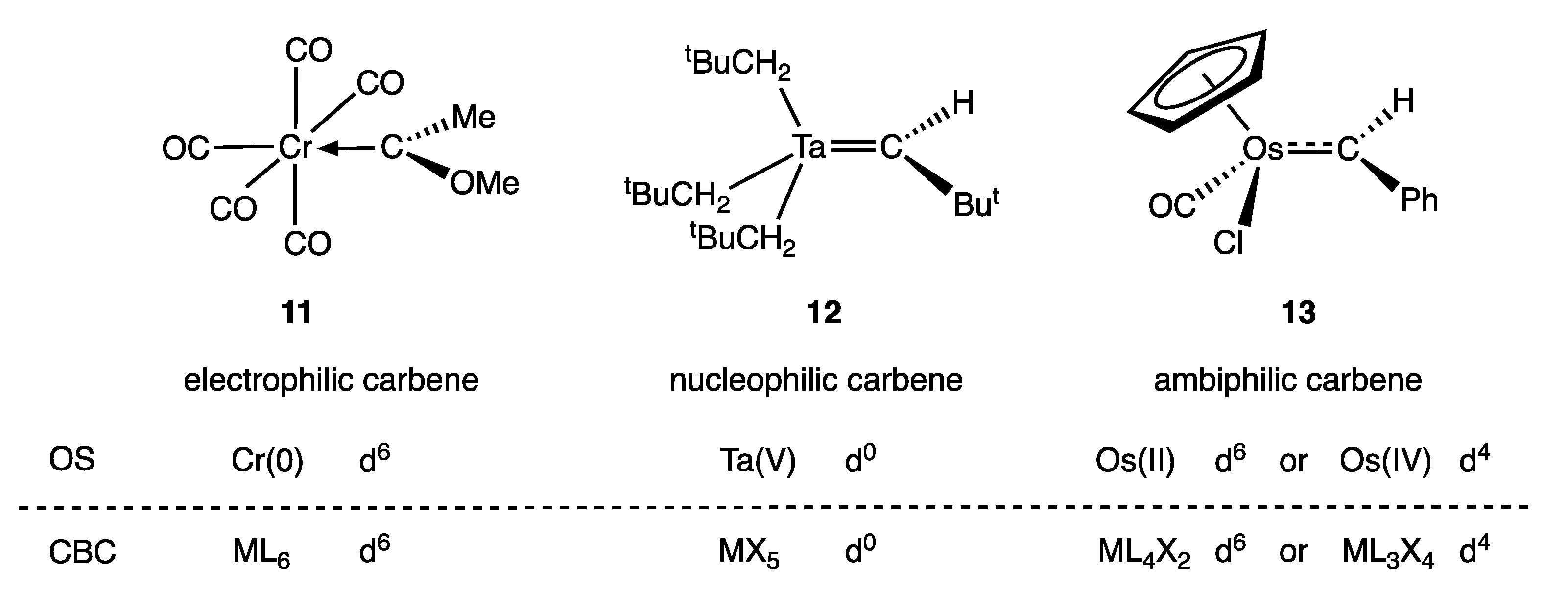
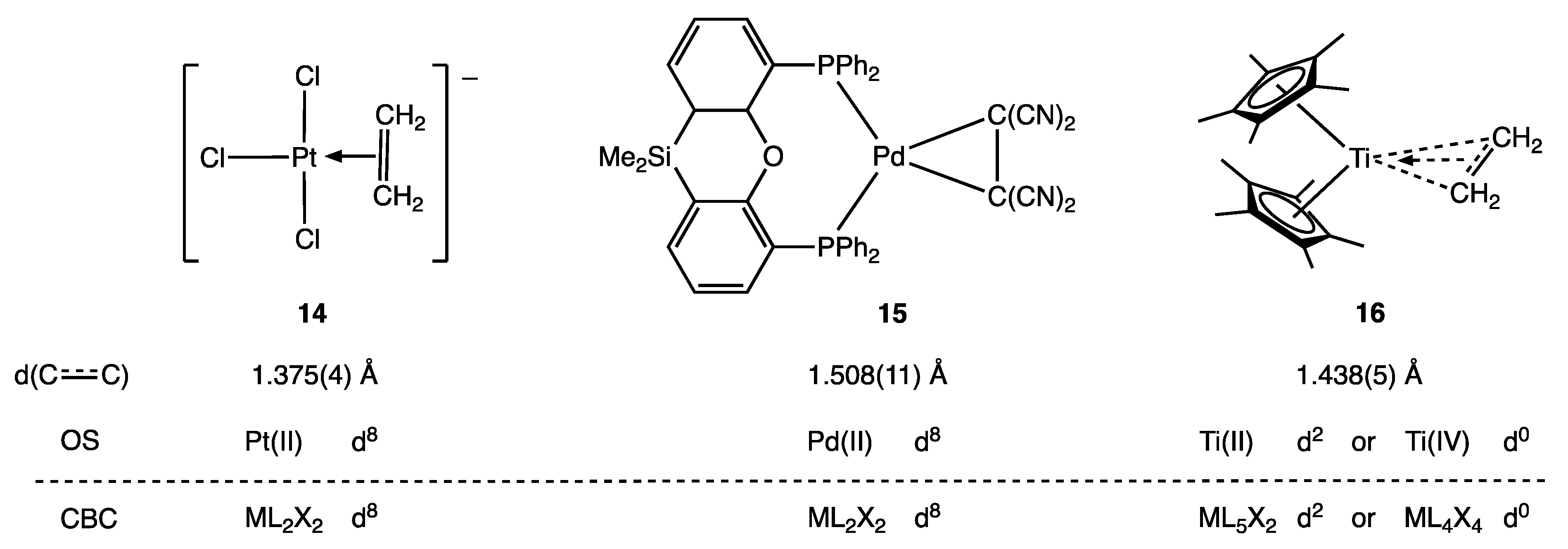
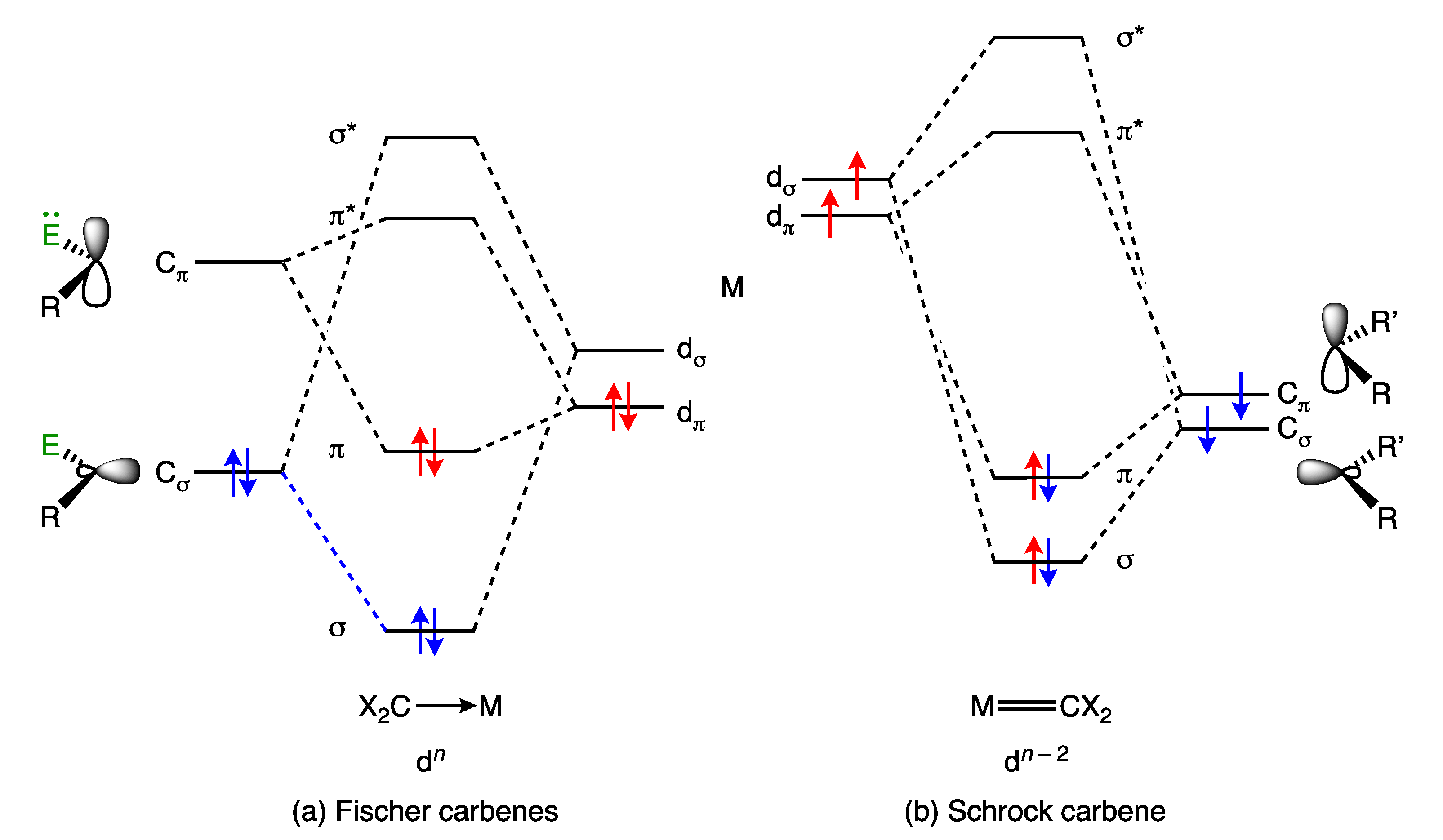
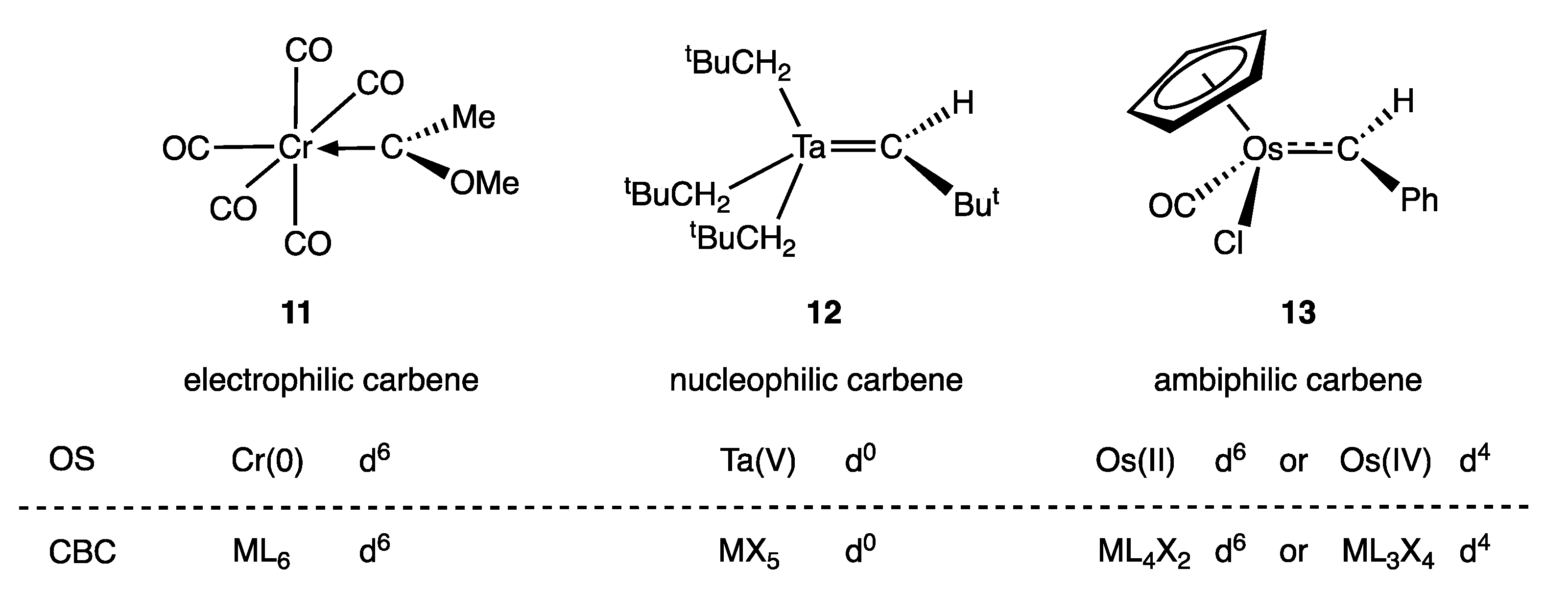
5. Carbene and Alkene Complexes
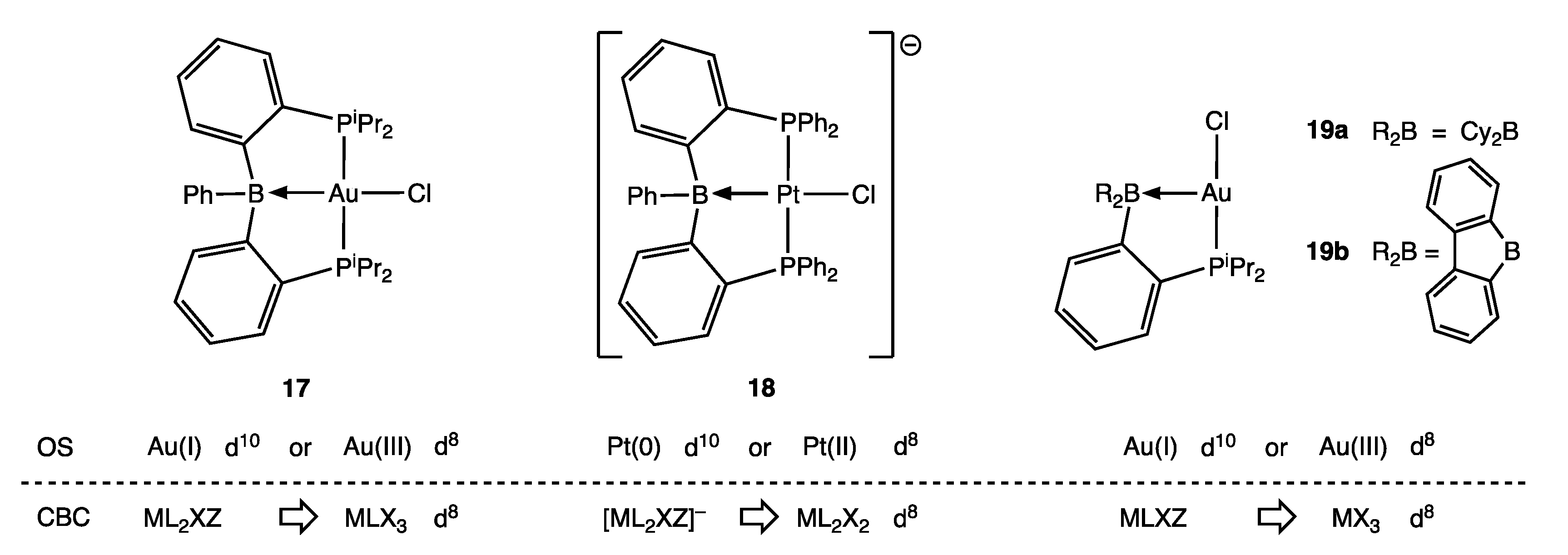
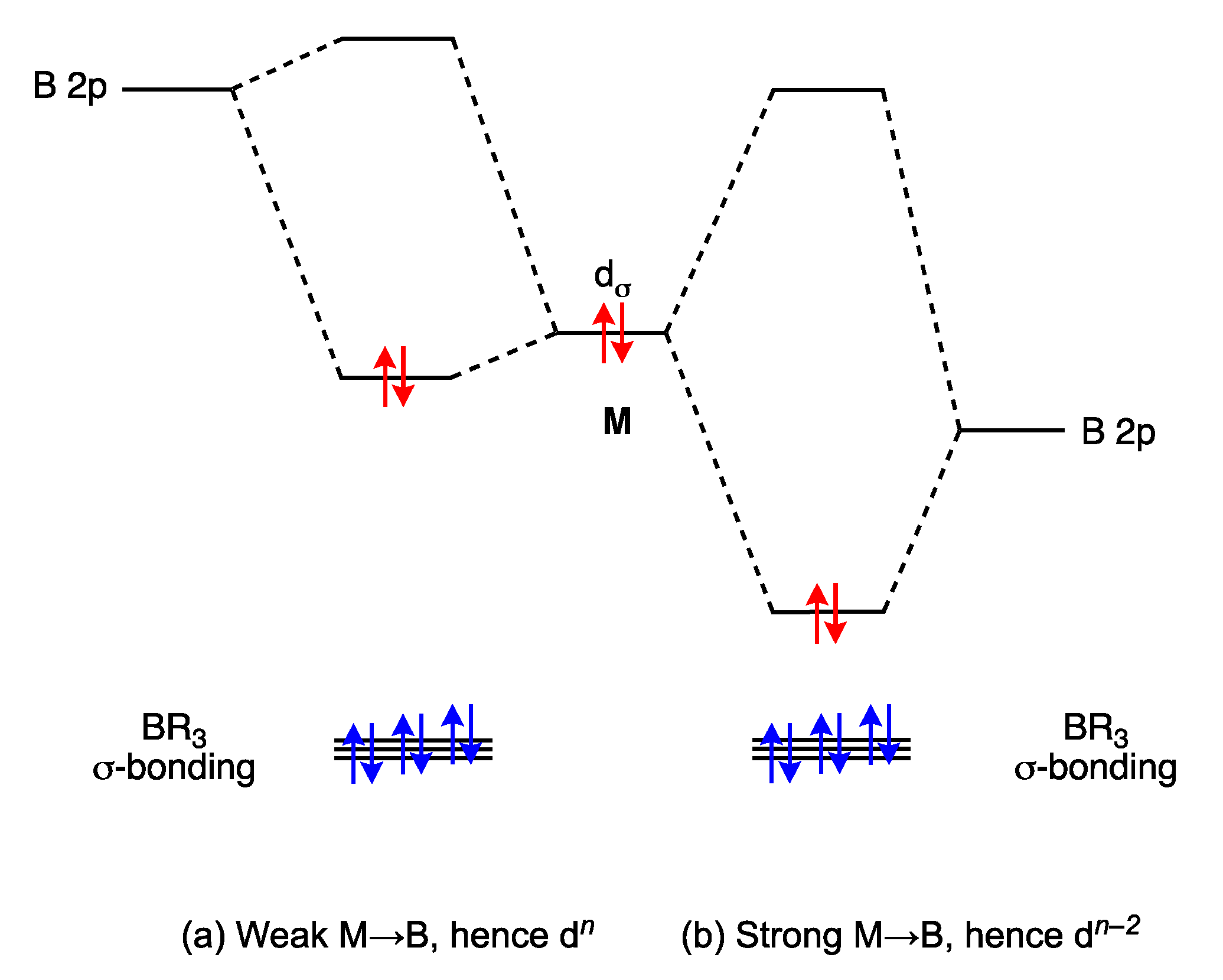
6. σ Acceptor (σ Z) Ligands: Boranes
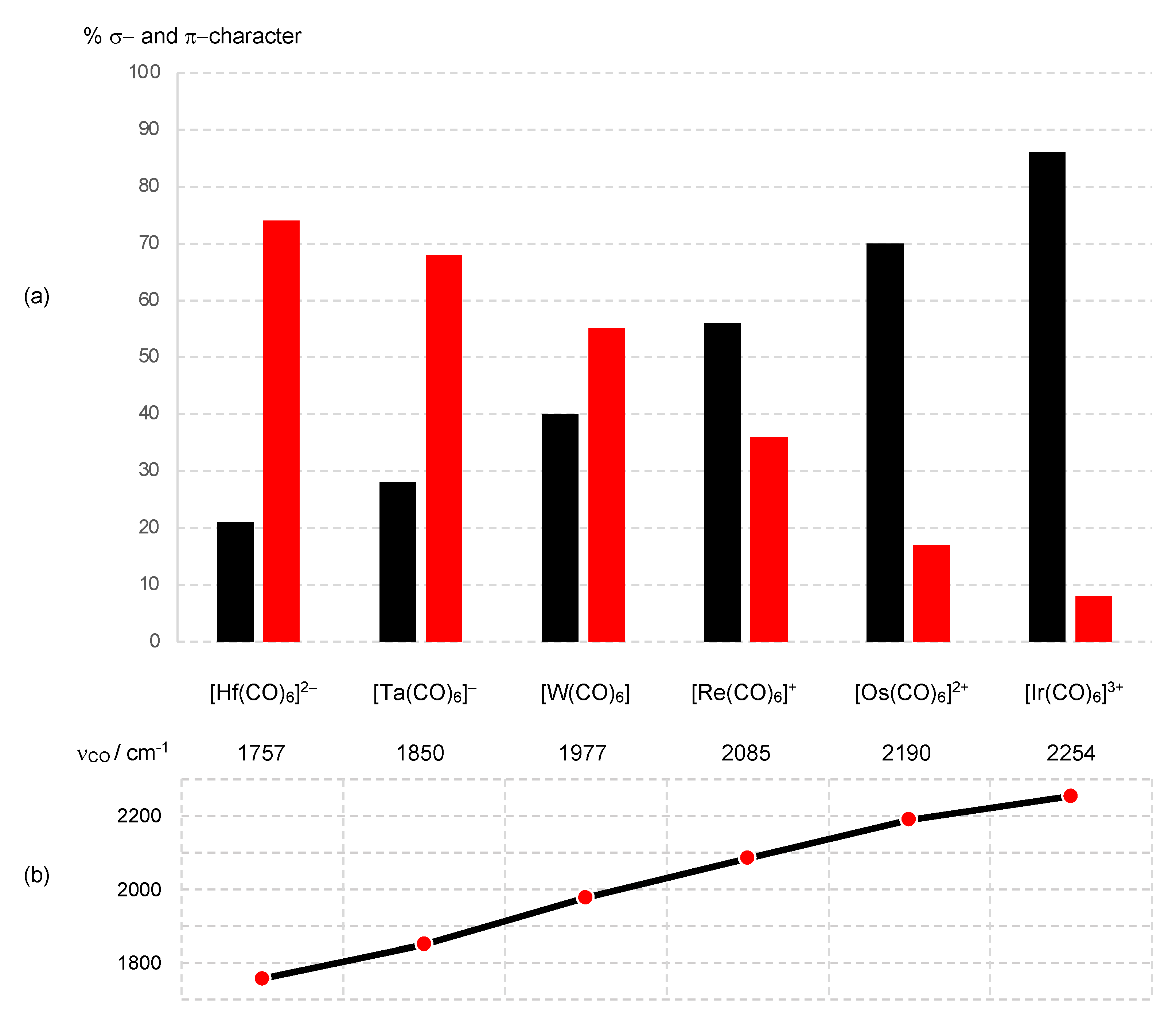
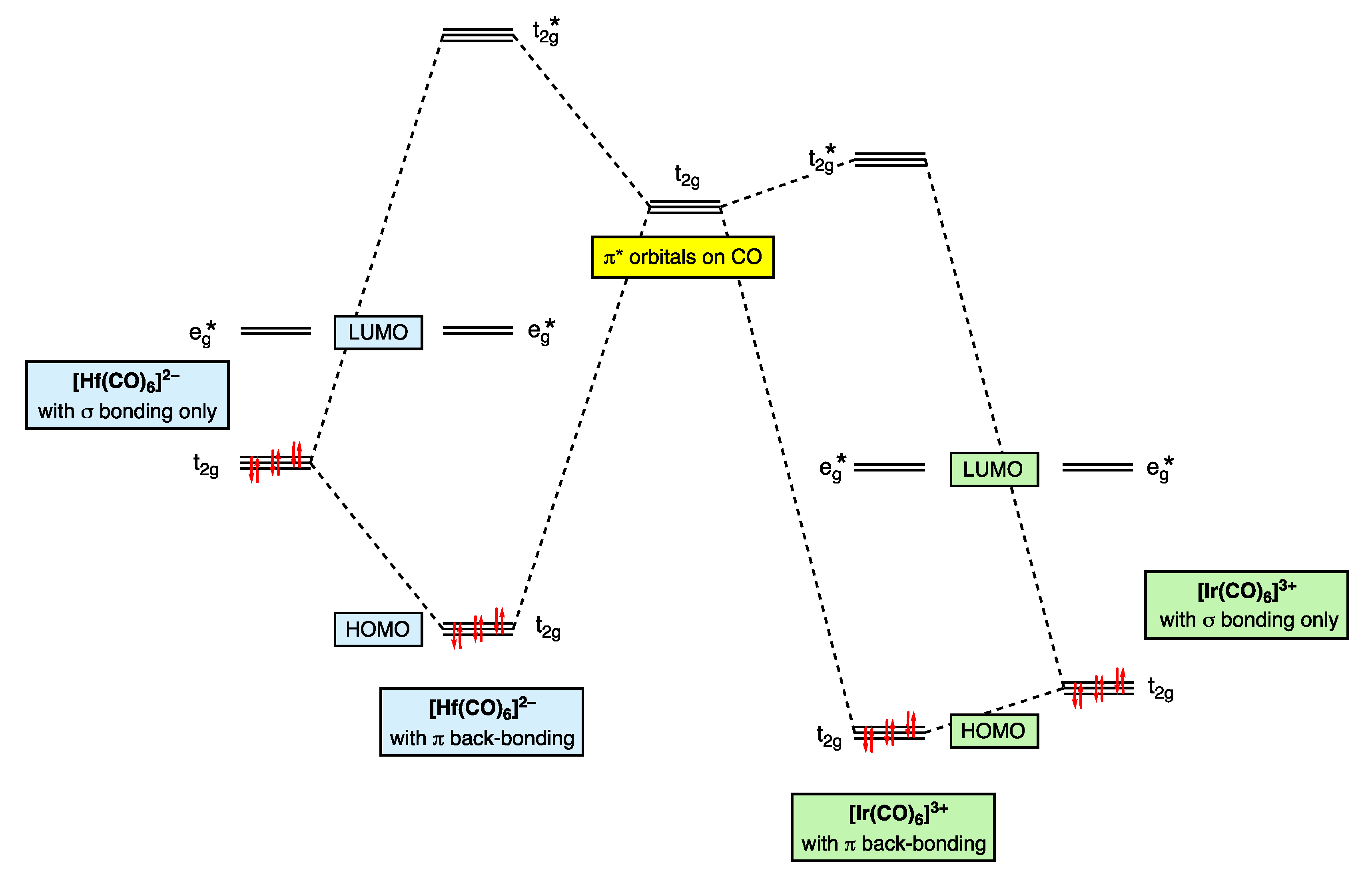
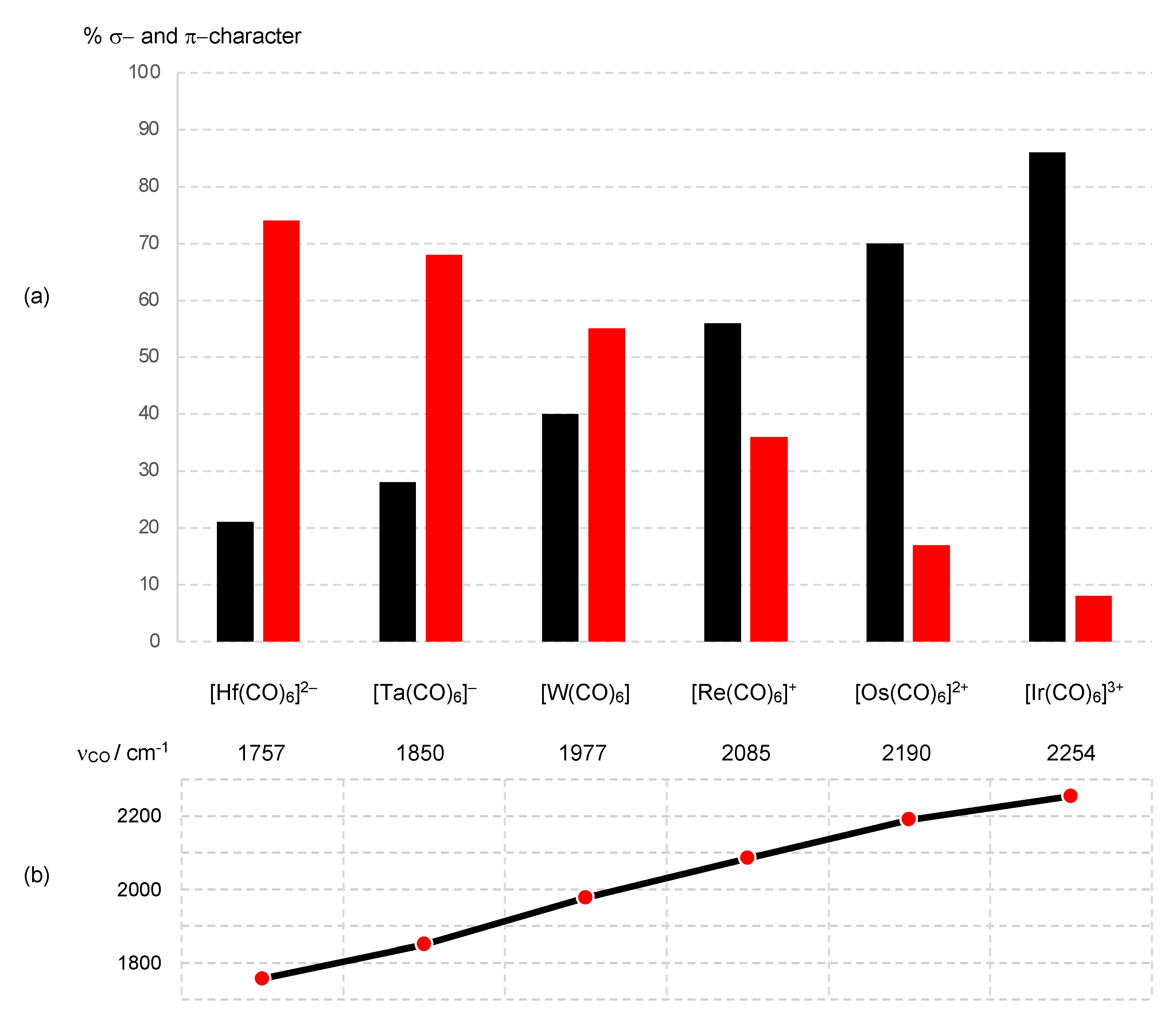
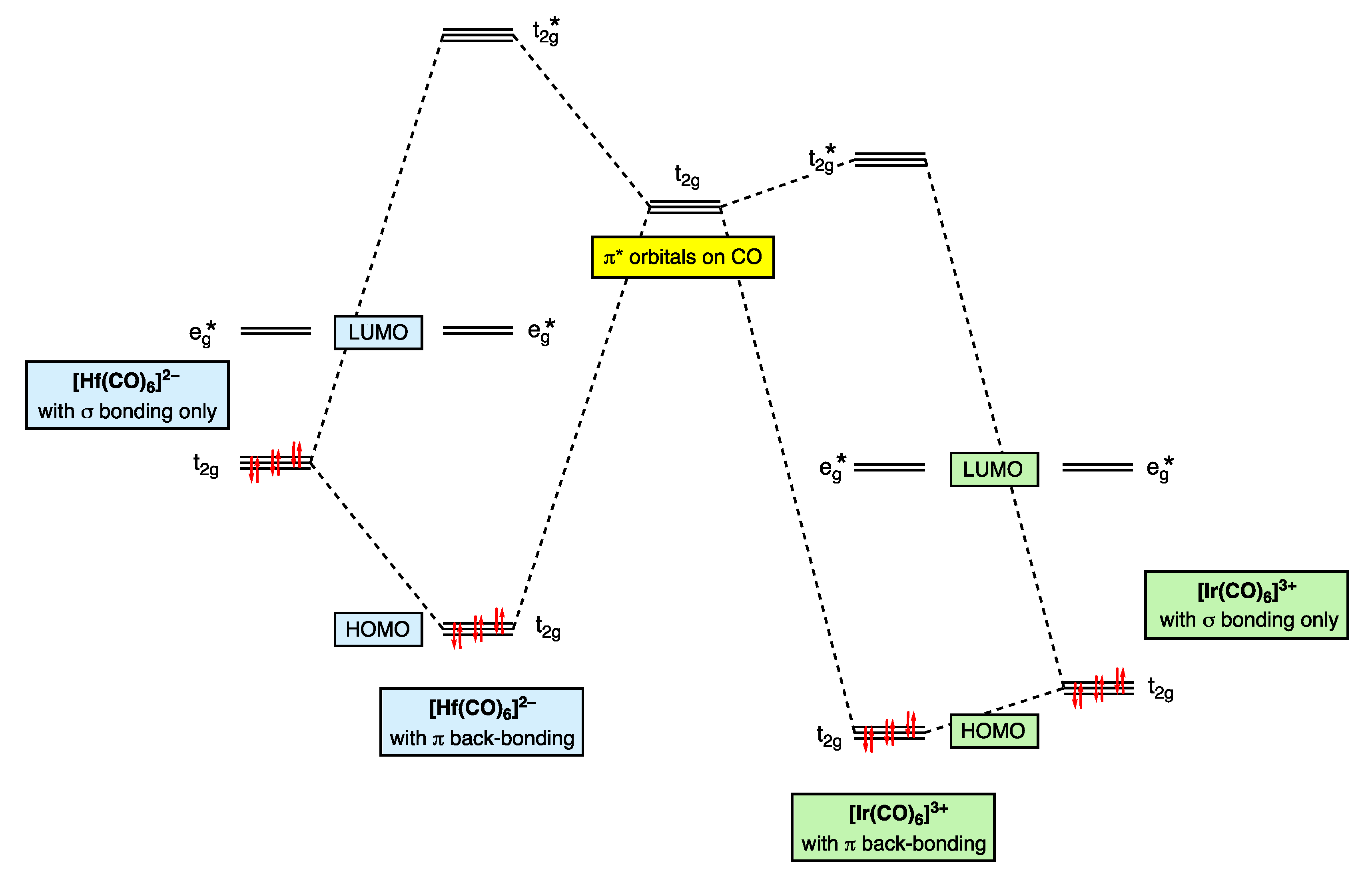
7. π Acceptor (π Z) Ligands: CO
Thus, the t2g orbitals transform from being classified as largely non-bonding in [Ir(CO)6]3+ to bonding in [Hf(CO)6]2−. This progression is necessarily accompanied by a decrease in the dn configuration using the definition that this quantity refers to electrons in d-based molecular orbitals that are not considered to be bonding.
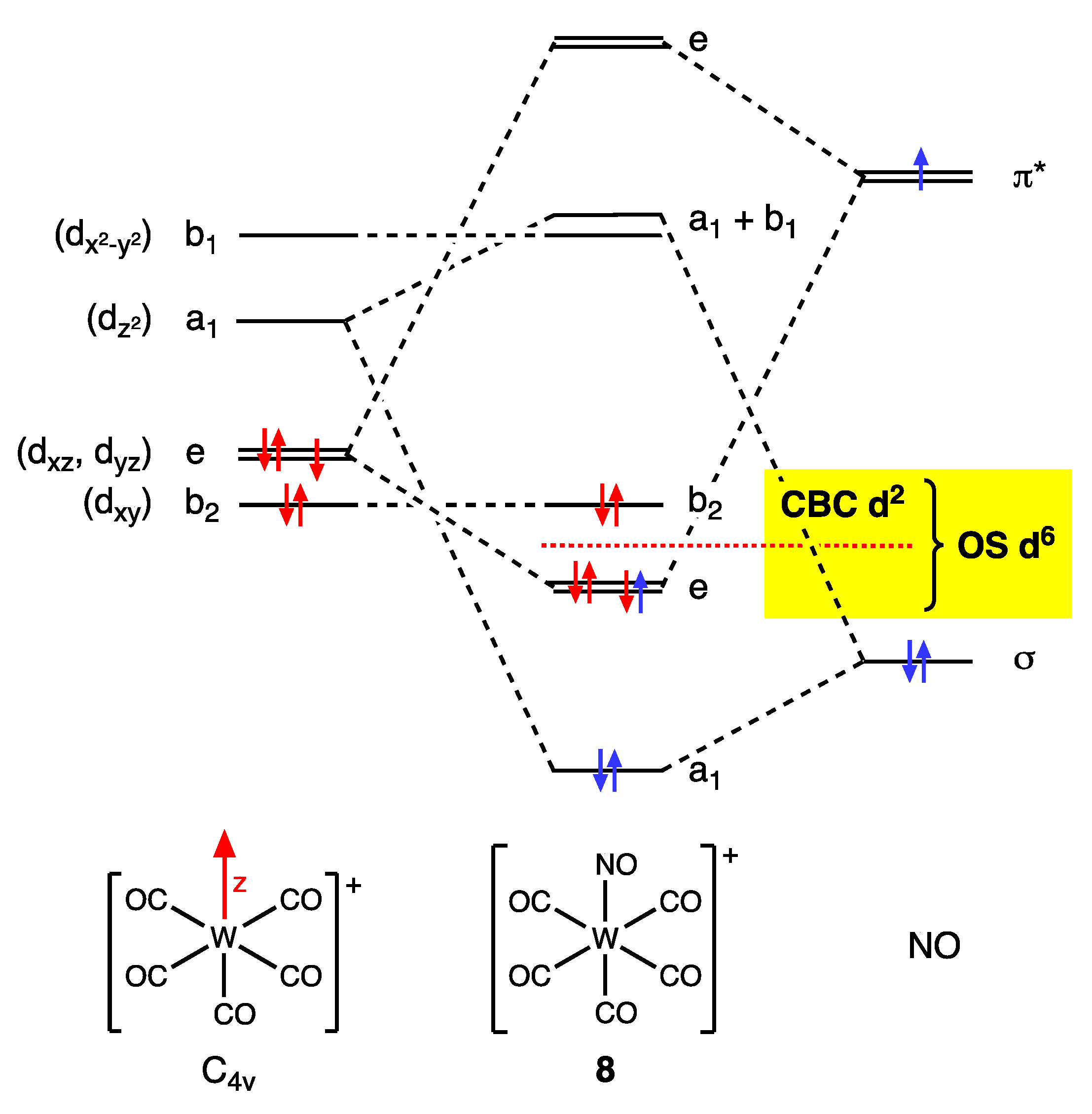
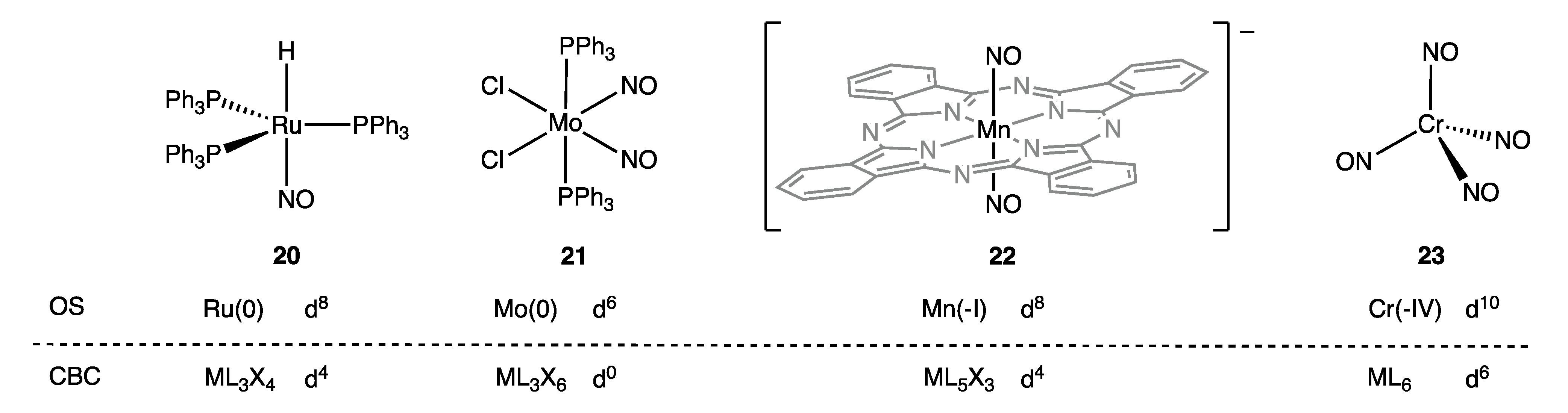
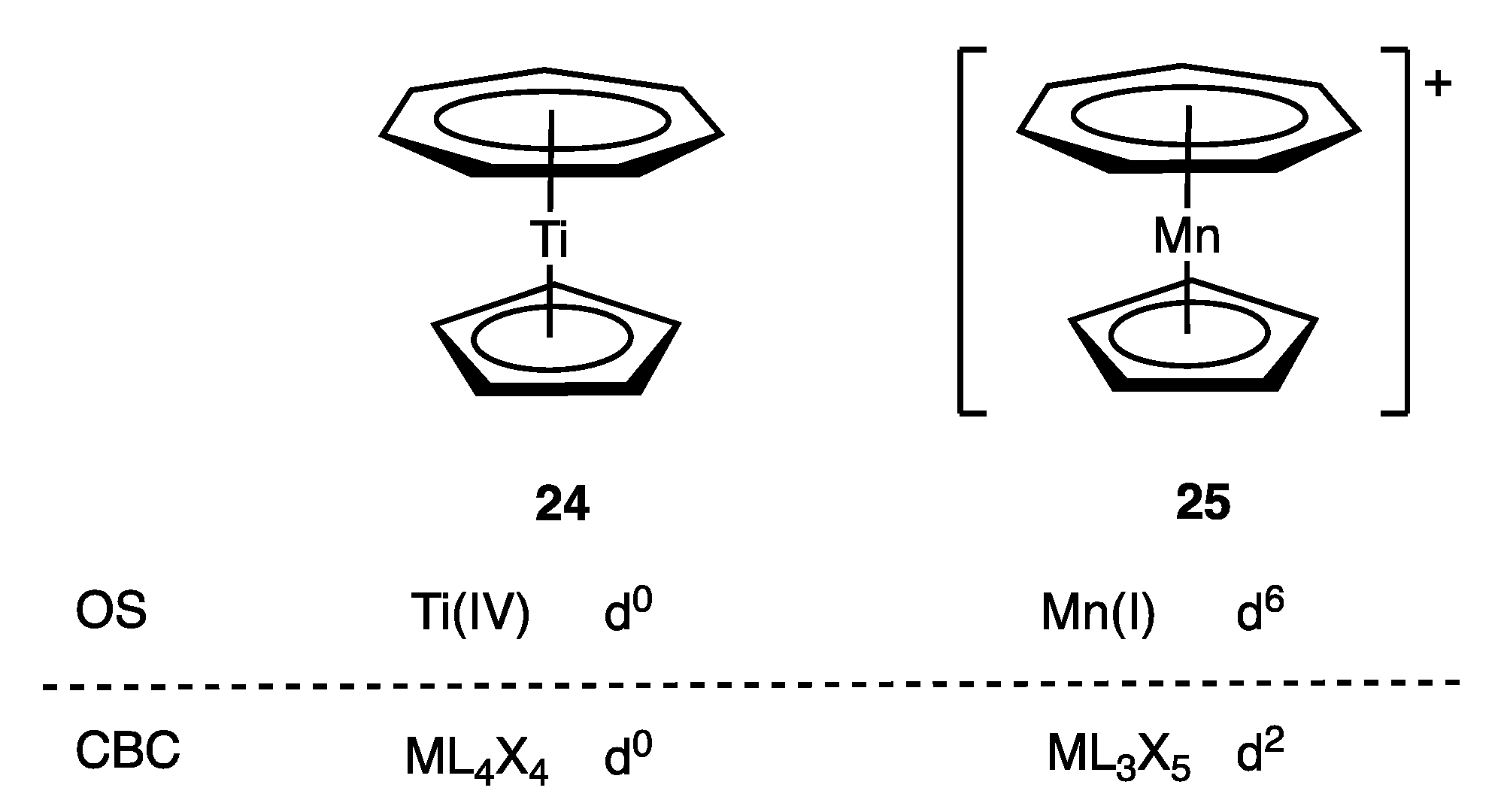
8. Cationic Ligands: Linear NO and Tropylium
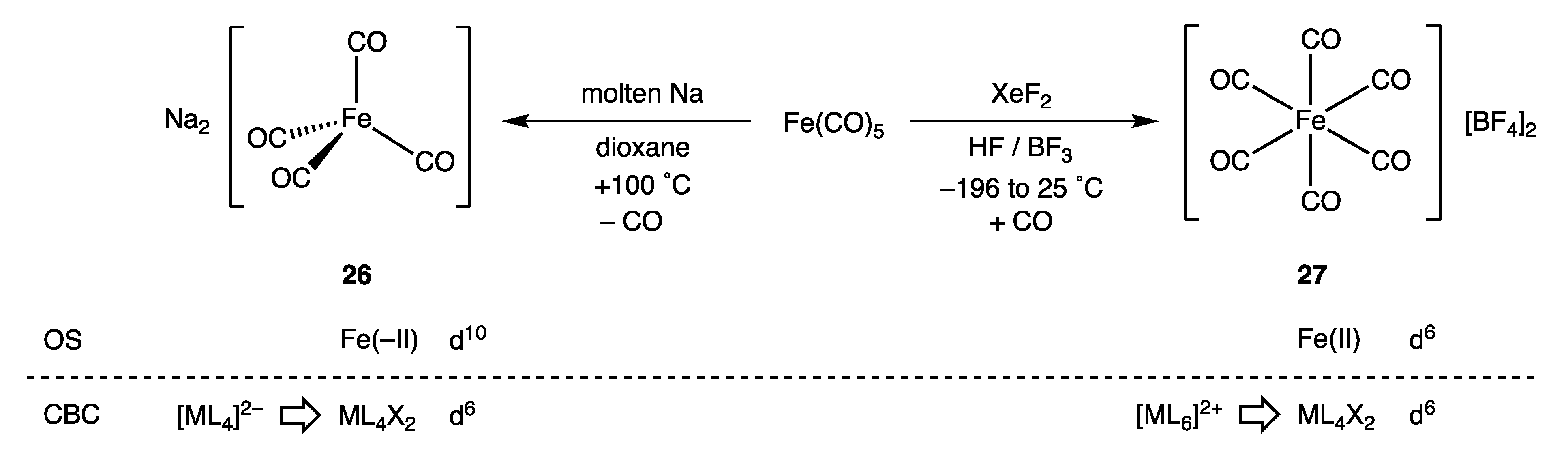
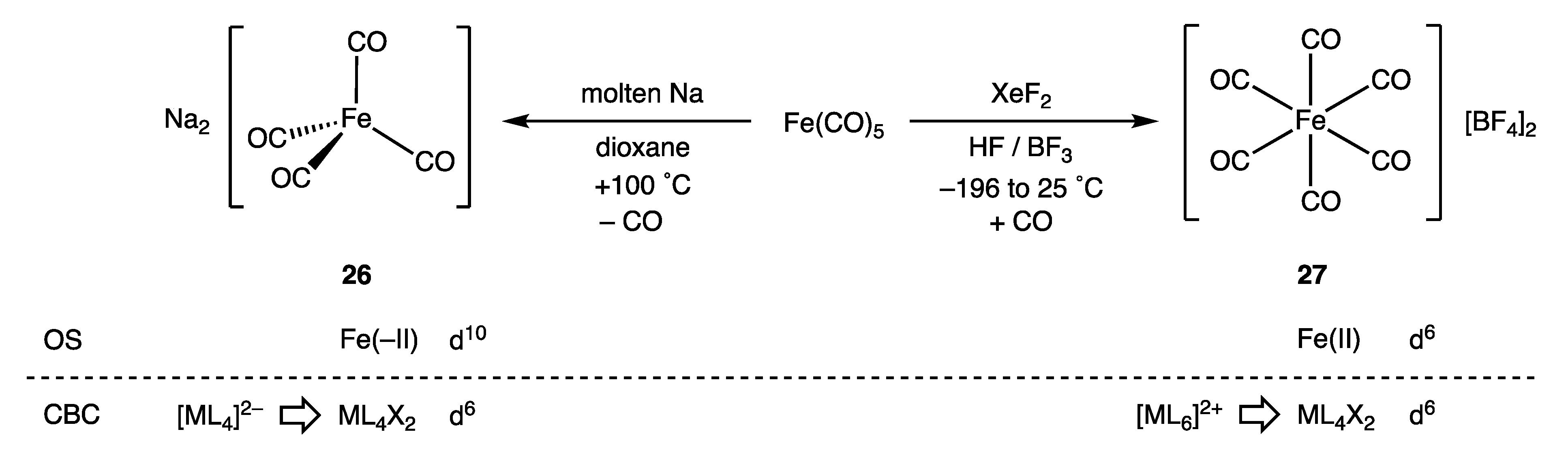
9. Negative Oxidation States
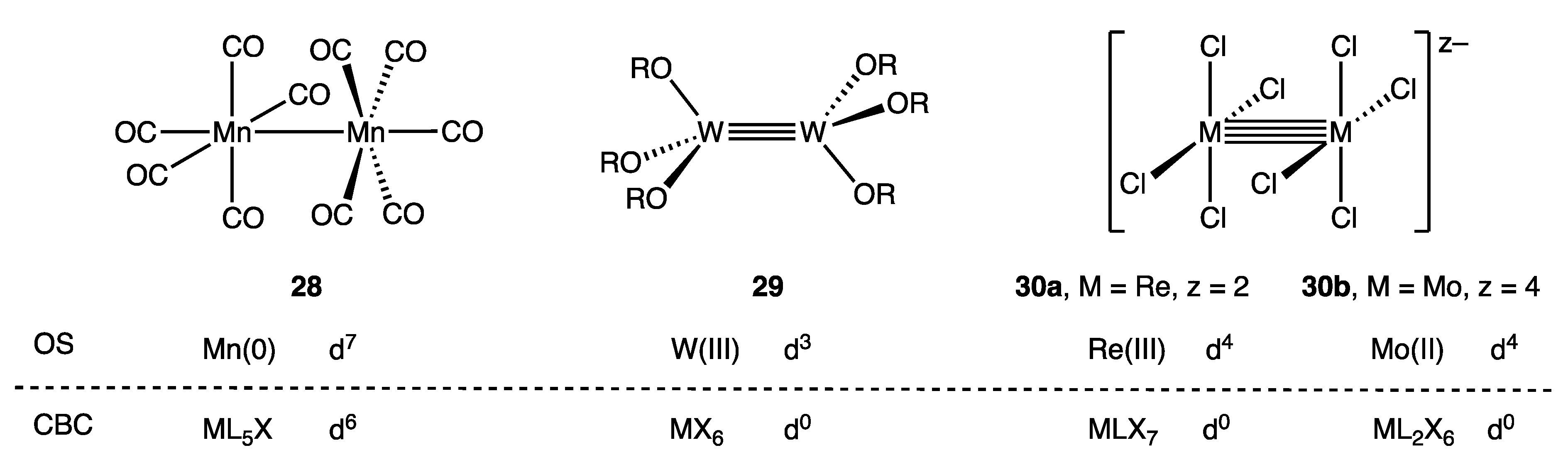
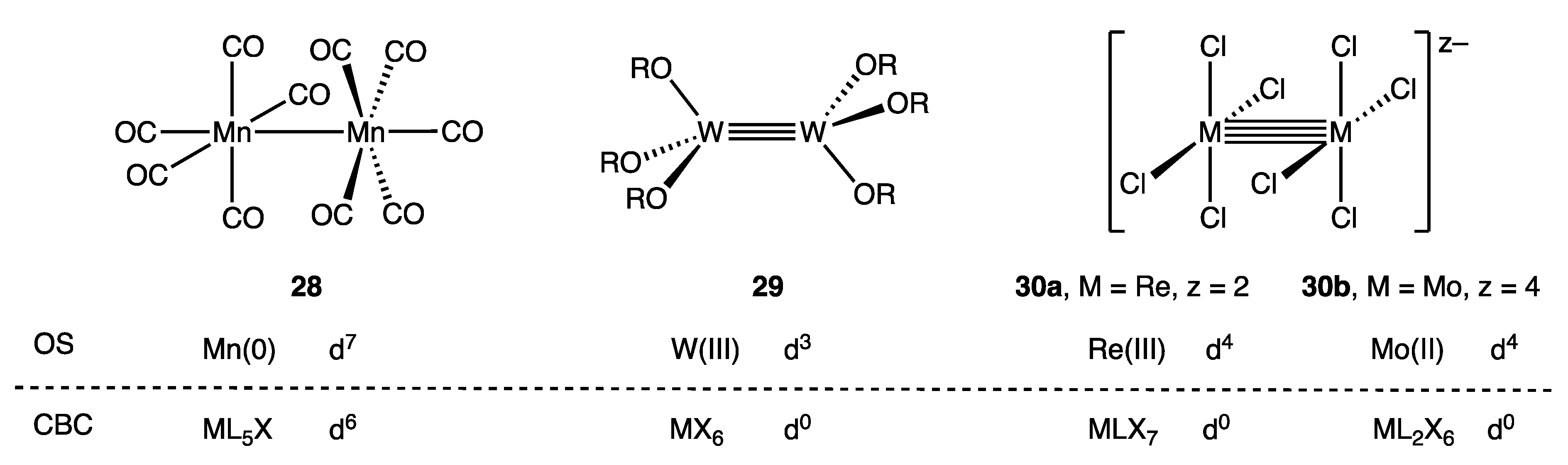
10. Compounds Containing Metal–Metal Bonds
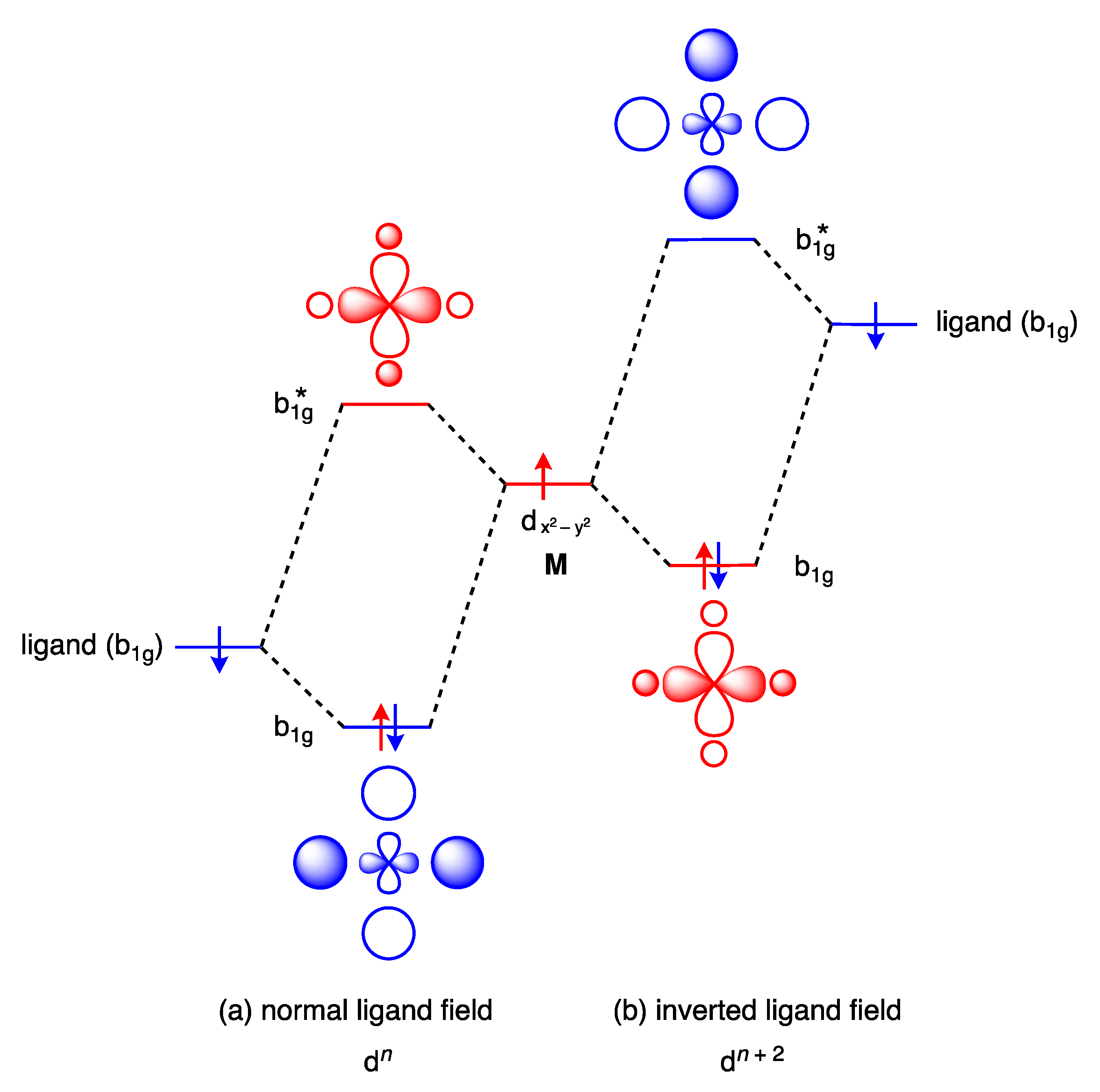
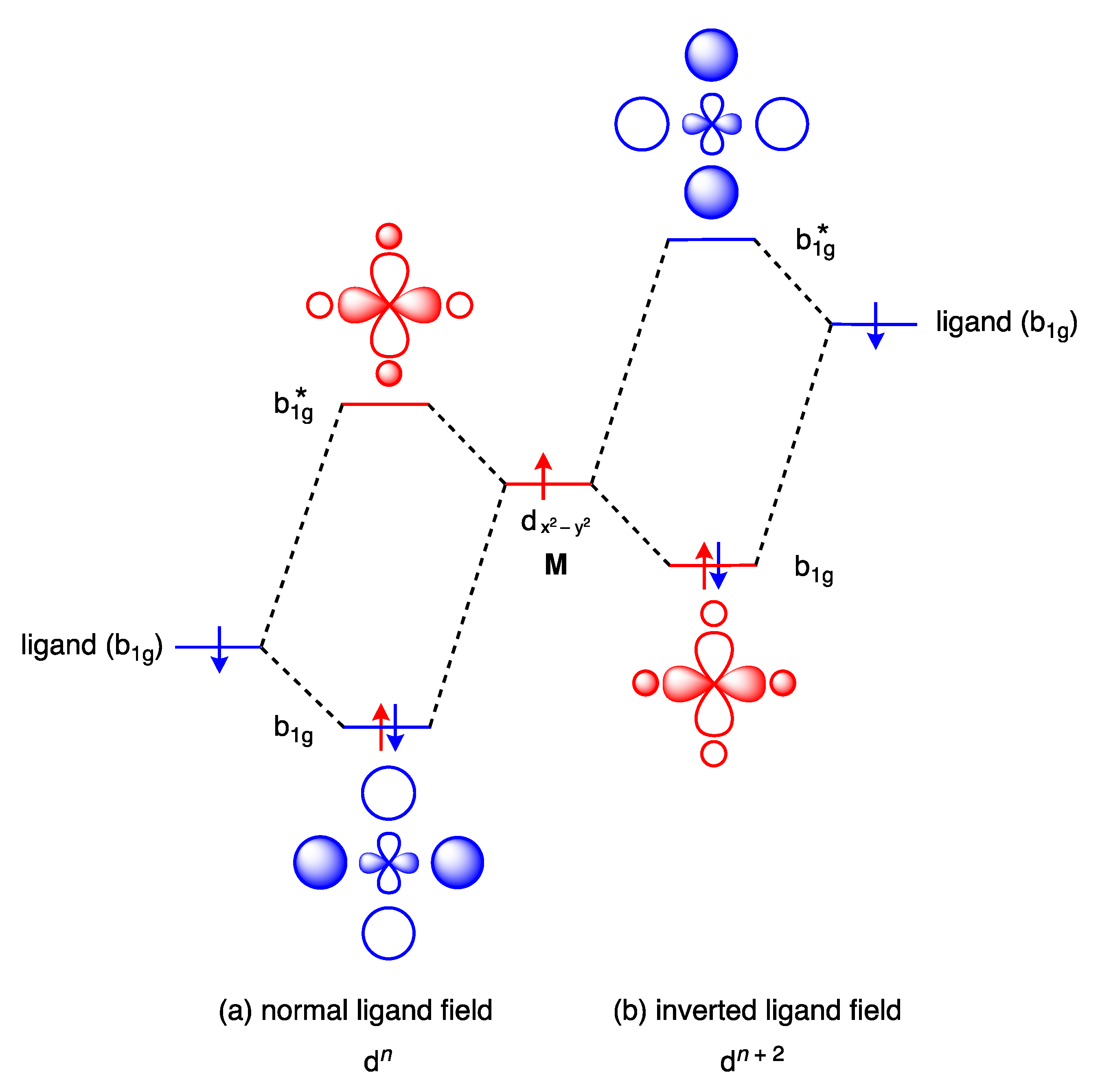
11. Inverted Ligand Fields
12. Concluding Discussion and a Modified Definition of dn
- (i)
- (ii)
- Complexes containing cationic ligands (Section 8).
- (iii)
- Complexes in which the metal is in a negative oxidation state (Section 9).
- (iv)
- Complexes with metal–metal bonds (Section 10).
However, whereas oxidation numbers depend on how one chooses to deconstruct the molecule, the dn configuration is a function of the molecule and must be independent of how one decides to deconstruct it. As such, a system of evaluating molecules that results in different dn configurations for the same molecule according to the preference of the author is unsatisfactory.[original emphasis]
The dn number of a transition metal complex is assigned based on the number of electrons that occupy the frontier orbitals, which have the same symmetry as metal d-orbitals.
- (1)
- It removes the issues around the subjective interpretation of the terms ‘primary’ or ‘primarily’ with regard to whether or not electrons reside in bonding orbitals (see Section 2).
- (2)
- It preserves the useful link between the value of dn and the structural and chemical properties listed in Section 2.
- (3)
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Notes
- The terms ‘transition elements’ and ‘d-block elements’ will be considered as fungible in this article without concern as to whether certain elements merit inclusion within those categories or not. Thus, arguments, for example, about whether the Group 12 elements are best described as transition elements or main group elements will be left to others.
- For more detail on how to assign an oxidation state, see ref. [14] and for an abbreviated summary, see ref. [15]. See also the IUPAC ‘Gold Book’ definitions of oxidation state [16] and the related term, oxidation number [17]. For the purposes of this article, the distinction between oxidation state and oxidation number is unimportant.
- Many discussions of whether a given compound will be high spin or low spin (where there is a distinction to be made) focus on the magnitude of Δ and the factors which affect it (the nature of the ligand, the row in the periodic table in which the metal resides and the metal oxidation state) and the pairing energy (P) associated with placing two electrons in the same orbital. With regard to the pairing energy, in ref. [5], the exchange energy associated with each of the different possible electron configurations is evaluated, which is a rather more rigorous approach.
- There are a few exceptions to this generalisation, such as certain square planar d10 Ag(I) complexes, which have been known for over 50 years. However, they are rare, and the planar geometry is normally associated either with a rigidity inherent in the chelating ligands or as a result of the geometry being crystallographically imposed. See, for example, ref. [18].
- In neutral gas-phase atoms, ns orbitals are populated in addition to (n − 1)d orbitals as reflected in the electronic configurations often presented in periodic tables; for example, the ground state electronic configuration of Fe is written as 4s2 3d6. In metal complexes, however, the (n − 1)d orbitals are generally lower in energy than the ns orbitals such that all the valence electrons are allocated to the d orbitals. Within the CFT (and LFT) models, Fe(0) is therefore designated as d8 and Fe(III), for example, as d5.
- This general point is captured for the t2g and eg orbitals in the following quote taken from ref. [5]: ‘Taken together, the 2eg and the 1t2g are described as the metal-based orbitals.’ [the numbers in front of eg and t2g reflect the numbering of the orbitals in the diagram presented in ref. [5] and are of no consequence for the argument presented here].
- For the purposes of this article, the terms carbene and alkylidene will be treated as synonymous, and the term carbene will be used throughout.
- For a more general article on this topic, see ref. [41].
- The gradation from M(η2-H2) to M(H)2 represents the simplest continuum in bonding from ML to MX2 (and hence a dn to dn−2 transition); see ref. [42] and refs. therein.
- Neutral ambiphilic ligands of the type considered here are multidentate ligands which contain a Z-type boron centre attached to, usually, one, two or three S- or P-donor sites, each one of which can be labelled as an L ligand. For a review of this general topic, see ref. [43].
- More generally, {MNO}n may be written as {M(NO)x}n to accommodate poly-nitrosyl complexes. As stated in the text, the values of n in the expressions {MNO}n (or {M(NO)x}n) and dn are the same if the NO ligand is treated as NO+. Alternatively, the value of n in the expression {MNO}n can be determined by adding the value of n in the dn number of the complex without the NO ligands to the number of electrons present in the NO π* orbitals; one per NO.
- For a computational study which considers the 3d element series, [Ti(CO)6]2− to [Fe(CO)6]2+, see ref. [55].
- It is worth noting that were the π Z interaction ignored in [Hf(CO)6]2−, an initial CBC assignment of [ML6]2− becomes, according to the transformation L− → LX, ML6X2 from which a d2 configuration would result based on Equation (3), but see ref. [9] for a more detailed discussion of how to consider this transformation in species where there is undoubtedly a significant π component.
- Versions of Figure 9 appear in most inorganic chemistry textbooks, but they do not generally take into account the following: (i) the interactions between the metal t2g orbitals and the filled π bonding orbitals on the CO, which also have t2g symmetry, (ii) the effect that the positive charge has on increasing the strength of the M–CO σ bonding and hence the increasing energy of the eg* orbitals and (iii) the mutual σ/π bond-strengthening known as the synergic effect. One text that does consider these matters in more detail is ref. [56], specifically, p. 862.
- An interesting approach referred to as Charge Distribution via Reporters (CDVR) can be found in ref. [57] which bears on some of the issues discussed in this section, although not specifically the dn number.
- For further recent examples of papers by Klüfers et al. that deal with the bonding in metal nitrosyls, see refs. [63,64] and for an interesting discussion about how to consider the NO ligand in the complex [TpMe2]NiNO (where [TpMe2] is the tris(3,5-dimethylpyrazolyl)hydroborato ligand), see refs. [65,66]. For a recent paper which considers how to estimate charge distribution in metal nitrosyl complexes, see ref. [67] and for a discussion of the factors which affect the linear vs. bent (or intermediate) geometries in the cationic species [MCl(NO)2(PPh3)2]+ (M = Ru, Os), see ref. [68].
- [Mn(CO)(NO)3] and [FeX(NO)3] (X = Cl, Br, I) are examples of M(NO)3 complexes which have C3v symmetry with a pseudo tetrahedral geometry at the metal centre. They are d10 complexes according to the OS method [Mn(-III) and Fe(-II), respectively]. We are not aware that an MLX assignment has been proposed for the M(NO)3 unit, but it is expected that the d6 configuration that the CBC method suggests for other [M(CO)n(NO)4–n] complexes also holds for [Mn(CO)(NO)3] which leads to an ML5X designation, and the classification of a M(NO)3 group in a tetrahedral geometry of L4X; [FeX(NO)3] will then be d6-ML4X2.
- The electronic structure of the paramagnetic complex 22 has been reported as d6 with, in addition, two electrons occupying a doubly degenerate HOMO, which is localised on the NO ligands, see ref. [69].
- NO and C7H7 are treated somewhat differently due to the inclusion of the Z component. Thus, NO is initially designated as LXZ, which, since LZ is equivalent to X2 [Scheme 1 (iii)], becomes X3. C7H7 is initially classified as L3XZ, which, again because LZ is equivalent to X2, becomes L2X3.
- The problem disappears if the equivalent neutral class formalism is abandoned for complexes in negative oxidation states whereby [Fe(CO)4]2− would be considered simply as [FeL4]2− in which the two additional electrons would convert d8-FeL4 to d10-[FeL4]2−.
- The addition of a haloalkane RX to 26 (Collman’s Reagent) to give the versatile intermediate species [RFe(CO)4]− is a pivotal reaction for the synthetic applications of [Fe(CO)4]2− and can be viewed as involving two electrons from the Fe centre in 26 to form the Fe–R bond in [RFe(CO)4]−; effectively, an SN2 reaction. In terms of OS, this process is readily understood as an oxidation of a d10-Fe(-II) to give a d8-Fe(0) species. By contrast, in terms of the CBC, this reaction involves a transformation of a d6-ML4X2 to a d8-ML5 species, which is less easily reconciled with electronic changes at the Fe centre.
- A computational study which contrasts the number of electrons actually associated with metal–metal bonding with a simplistic accounting based on the M–M bond order is reported in ref. [86].
- For a general article on PSEPT, see ref. [87].
- If the significant vs. insignificant π back-bonding judgment is to be evidenced by the position of the νCO bands in the vibrational spectra, then complications will arise with substituted metal carbonyls. For example, the νCO bands for cis-[Cr(CO)2(dmpe)2] (dmpe = Me2PCH2CH2PMe2) occur at 1856 and 1798 cm−1, which are in the vicinity of the νCO (T1u) bands for [V(CO)6]−. See ref. [96].
- In line with this suggestion, and following the point made in (Appendix A Note 31), it may be noted that metal–metal bonded binuclear compounds are sometimes written with a similar type of descriptor to indicate the metal–metal bond order, for example, [Mo2(CO)6(η-C5H5)2] (M–M) and [Mo2(CO)4(η-C5H5)2] (M≡M).
References
- Cotton, F.A.; Wilkinson, G.; Gaus, P.L. Basic Inorganic Chemistry, 3rd ed.; Wiley: Hoboken, NJ, USA, 1995. [Google Scholar]
- Cotton, F.A.; Wilkinson, G.; Murillo, C.A.; Bochmann, M. Advanced Inorganic Chemistry, 6th ed.; Wiley: Hoboken, NJ, USA, 1999. [Google Scholar]
- Housecroft, C.E.; Sharpe, A.G. Inorganic Chemistry, 5th ed.; Pearson: London, UK, 2018. [Google Scholar]
- Weller, M.T.; Overton, T.L.; Rourke, J.P.; Armstrong, F.A. Inorganic Chemistry, 7th ed.; Oxford University Press: Oxford, UK, 2018. [Google Scholar]
- Keeler, J.; Wothers, P. Chemical Structure and Reactivity: An Integrated Approach, 2nd ed.; Oxford University Press: Oxford, UK, 2014. [Google Scholar]
- Parkin, G. A Simple Description of the Bonding in Transition-Metal Borane Complexes. Organometallics 2006, 25, 4744–4747. [Google Scholar] [CrossRef]
- Jean, Y. Molecular Orbitals of Transition Metal Complexes; Oxford University Press: Oxford, UK, 2005. [Google Scholar]
- Hartwig, J. Organotransition Metal Chemistry: From Bonding to Catalysis; University Science Books: Sausalito, CA, USA, 2010. [Google Scholar]
- Parkin, G. Impact of the Coordination of Multiple Lewis Acid Functions on the Electronic Structure and vn Configuration of a Metal Centre. Dalton Trans. 2022, 51, 411–427. [Google Scholar] [CrossRef]
- Parkin, G. Valence, Oxidation Number, and Formal Charge: Three Related but Fundamentally Different Concepts. J. Chem. Ed. 2006, 83, 791–799. [Google Scholar] [CrossRef]
- Smith, D.W. Valence, Covalence, Hypervalence, Oxidation State and Coordination Number. J. Chem. Ed. 2005, 82, 1202–1204. [Google Scholar] [CrossRef]
- Norman, N.C.; Pringle, P.G. Hypervalence: A Useful Concept or One That Should Be Gracefully Retired? Chemistry 2022, 4, 1226–1249. [Google Scholar] [CrossRef]
- Norman, N.C. Periodicity and the s- and p-Block Elements, 2nd ed.; Oxford University Press: Oxford, UK, 2021. [Google Scholar]
- Karen, P.; McArdle, P.; Takats, J. Toward a Comprehensive Definition of Oxidation State (IUPAC Technical Report). Pure Appl. Chem. 2014, 86, 1017–1081. [Google Scholar] [CrossRef]
- Karen, P.; McArdle, P.; Takats, J. Comprehensive Definition of Oxidation State (IUPAC Recommendations 2016). Pure Appl. Chem. 2016, 88, 831–839. [Google Scholar] [CrossRef]
- McNaught, A.D.; Wilkinson, A. (Eds.) 1997. IUPAC. Compendium of Chemical Terminology, 2nd ed.; the “Gold Book”. Oxidation State. Available online: https://goldbook.iupac.org/terms/view/O04365 (accessed on 10 July 2023).
- McNaught, A.D.; Wilkinson, A. (Eds.) 1997. IUPAC. Compendium of Chemical Terminology, 2nd ed.; The “Gold Book”. Oxidation Number. Available online: https://goldbook.iupac.org/terms/view/O04363 (accessed on 10 July 2023).
- Young, A.G.; Hanton, L.R. Square Planar Silver(I) Complexes: A Rare but Increasingly Observed Stereochemistry for Silver(I). Coord. Chem. Rev. 2008, 252, 1346–1386. [Google Scholar] [CrossRef]
- Albright, T.A.; Burdett, J.K.; Whangbo, M.H. Orbital Interactions in Chemistry, 2nd ed.; Wiley: Hoboken, NJ, USA, 2013. [Google Scholar]
- Fekl, U. Covalent d-Block Organometallics: Teaching Lewis Structures and sd/sd2 Hybridisation Gives Students Additional Explanations and Powerful Predictive Tools. J. Chem. Ed. 2021, 98, 3189–3206. [Google Scholar] [CrossRef]
- Landis, C.R.; Weinhold, F. 18-Electron Rule and the 3c/4e Hyperbonding Saturation Limit. J. Comput. Chem. 2016, 37, 237–241. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R.; Glendening, E.D. What is NBO Analysis and How is it Useful? Int. Rev. Phys. Chem. 2016, 35, 399–440. [Google Scholar] [CrossRef]
- Crabtree, R.H. The Organometallic Chemistry of the Transition Metals, 7th ed.; Wiley: Hoboken, NJ, USA, 2019. [Google Scholar]
- Green, M.L.H. A New Approach to the Formal Classification of Covalent Compounds and Elements. J. Organomet. Chem. 1995, 500, 127–148. [Google Scholar] [CrossRef]
- Green, M.L.H.; Parkin, G. Application of the Covalent Bond Classification Method for the Teaching of Inorganic Chemistry. J. Chem. Ed. 2014, 91, 807–816. [Google Scholar] [CrossRef]
- Parkin, G. Classification of Organometallic Compounds. Comprehensive Organometallic Chemistry III; Elsevier: Amsterdam, The Netherlands, 2007; Volume 1, pp. 1–57. [Google Scholar]
- Bohnenberger, J.; Feuerstein, W.; Himmel, D.; Daub, M.; Breher, F.; Krossing, I. Stable Salts of the Hexacarbonyl Chromium(I) Cation and its Pentacarbonyl-Nitrosyl Chromium(I) Analogue. Nat. Commun. 2019, 10, 624. [Google Scholar] [CrossRef]
- Grubbs, R.H.; Trnk, T.M.; Sandford, M.S. Fundamentals of Molecular Catalysis; Kurosawa, H., Yamamoto, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Montgomery, C.D. Fischer and Schrock Carbene Complexes: A Molecular Modeling Exercise. J. Chem. Ed. 2015, 92, 1653–1660. [Google Scholar] [CrossRef]
- Esteruelas, M.A.; González, A.I.; López, A.M.; Oñate, E. An Osmium-Carbene Complex with Fischer–Schrock Ambivalent Behavior. Organometallics 2003, 22, 414–425. [Google Scholar] [CrossRef]
- Casey, C.P.; Vosejpka, P.C.; Askham, F.R. Amphiphilic Carbene Complexes: Both Electrophiles and Nucleophiles Attack the Carbene Carbon of C5H5(CO)2Re:CHR. J. Am. Chem. Soc. 1990, 112, 3713–3715. [Google Scholar] [CrossRef]
- Santamaría, J.; Aguilar, E. Beyond Fischer and Schrock Carbenes: Non-Heteroatom-Stabilized Group 6 Metal Carbene Complexes—A General Overview. Org. Chem. Front. 2016, 3, 1561–1588. [Google Scholar] [CrossRef]
- Hopkinson, M.N.; Richter, C.; Schedler, M.; Glorius, F. An Overview of N-Heterocyclic Carbenes. Nature 2014, 510, 485–496. [Google Scholar] [CrossRef]
- Soleilhavoup, M.; Bertrand, G. Cyclic (Alkyl)(Amino)Carbenes (CAACs): Stable Carbenes on the Rise. Acc. Chem. Res. 2015, 48, 256–266. [Google Scholar] [CrossRef]
- Occhipinti, G.; Jensen, V.R. Nature of the Transition Metal Carbene Bond in Grubbs Olefin Metathesis Catalysts. Organometallics 2011, 30, 3522–3529. [Google Scholar] [CrossRef]
- Clark, G.R.; Hoskins, S.V.; Jones, T.C.; Roper, W.R. Oxidation State Control of the Reactivity of a Transition Metal-Carbon Double Bond. Synthesis, X-Ray Crystal Structure, and Reactions of the Zerovalent Difluorocarbene Complex [Ru(=CF2)(CO)2(PPh3)2]. J. Chem. Soc. Chem. Commun. 1983, 13, 719–721. [Google Scholar] [CrossRef]
- Clark, G.R.; Hoskins, S.V.; Roper, W.R. Difluorocarbene Complexes of Ruthenium Derived from Trifluoromethyl Compounds. RuCl2(CF2)(CO)(PPh3)2, RuCl2(CFNMe2)(CO)(PPh3)2 and the Structure of Ru(CF3)(HgCF3)(CO)2(PPh3)2. J. Organomet. Chem. 1982, 234, C9–C12. [Google Scholar] [CrossRef]
- Fu, X.-P.; Xue, X.-S.; Zhang, X.-Y.; Xiao, Y.-L.; Zhang, S.; Guo, Y.-L.; Leng, X.; Houk, K.N.; Zhang, X. Controllable Catalytic Difluorocarbene Transfer Enables Access to Diversified Fluoroalkylated Arenes. Nat. Chem. 2019, 11, 948–956. [Google Scholar] [CrossRef]
- Zhou, W.; Pan, W.-J.; Chen, J.; Zhang, M.; Lin, J.-H.; Cao, W.; Xiao, J.-C. Transition-Metal Difluorocarbene Complexes. Chem. Commun. 2021, 57, 9316–9329. [Google Scholar] [CrossRef]
- Cohen, S.A.; Auburn, P.R.; Bercaw, J.E. Structure and Reactivity of Bis(pentamethylcyclopentadienyl)(ethylene)titanium(II), a Simple Olefin Adduct of Titanium. J. Am. Chem. Soc. 1983, 105, 1136–1143. [Google Scholar] [CrossRef]
- Jerabek, P.; Schwerdtfeger, P.; Frenking, G. Dative and Electron-Sharing Bonding in Transition Metal Compounds. J. Comput. Chem. 2019, 40, 247–264. [Google Scholar] [CrossRef]
- Crabtree, R.H. Dihydrogen Complexation. Chem. Rev. 2016, 116, 8750–8769. [Google Scholar] [CrossRef]
- Bouhadirab, G.; Bourissou, D. Complexes of Ambiphilic Ligands: Reactivity and Catalytic Applications. Chem. Soc. Rev. 2016, 45, 1065–1079. [Google Scholar] [CrossRef]
- Sircoglou, M.; Bontemps, S.; Mercy, M.; Saffon, N.; Takahashi, M.; Bouhadir, G.; Maron, L.; Bourissou, D. Transition-Metal Complexes Featuring Z-Type Ligands: Agreement or Discrepancy between Geometry and dn Configuration? Angew. Chem. Int. Ed. 2007, 46, 8583–8586. [Google Scholar] [CrossRef]
- Kameo, H.; Tanaka, Y.; Shimoyama, Y.; Izumi, D.; Matsuzaka, H.; Nakajima, Y.; Lavedan, P.; Le Gac, A.; Bourissou, D. Square-Planar Anionic Pt0 Complexes. Angew. Chem. Int. Ed. 2023, 62, e202301509. [Google Scholar] [CrossRef]
- Bontemps, S.; Bouhadir, G.; Miqueu, K.; Bourissou, D. On the Versatile and Unusual Coordination Behavior of Ambiphilic Ligands o-R2P(Ph)BR’2. J. Am. Chem. Soc. 2006, 128, 12056–12057. [Google Scholar] [CrossRef]
- Hill, A.F. An Unambiguous Electron-Counting Notation for Metallaboratranes. Organometallics 2006, 25, 4741–4743. [Google Scholar] [CrossRef]
- Enemark, J.H.; Feltham, R.D. Stereochemical Control of Valence, III. The {CoNO}8 Group in Ligand Fields C4v, C2v, and Cs Symmetry. J. Am. Chem. Soc. 1974, 96, 5004–5005. [Google Scholar] [CrossRef]
- Enemark, J.H.; Feltham, R.D. Principles and Structure, Bonding and Reactivity for Metal Nitrosyl Complexes. Coord. Chem. Rev. 1974, 13, 339–406. [Google Scholar] [CrossRef]
- Schild, D.J.; Nurdin, L.; Moret, M.-E.; Oyala, P.H.; Peters, J.C. Characterisation of a Proposed Terminal Iron(III) Nitride Intermediate of Nitrogen Fixation Stabilized by a Trisphosphine-Borane Ligand. Angew. Chem. Int. Ed. 2022, 61, e202209655. [Google Scholar] [CrossRef]
- Frenking, G.; Fernández, I.; Holzmann, N.; Pan, S.; Krossing, I.; Zhou, M. Metal–CO Bonding in Mononuclear Transition Metal Carbonyl Complexes. JACS Au 2021, 1, 623–645. [Google Scholar] [CrossRef]
- Frenking, G. Understanding the Nature of the Bonding in Transition Metal Complexes: From Dewar’s Molecular Orbital Model to an Energy Partitioning Analysis of the Metal-Ligand Bond. J. Organomet. Chem. 2001, 635, 9–23. [Google Scholar] [CrossRef]
- Bach, C.; Willner, H.; Aubke, F.; Wang, C.; Rettig, S.J.; Trotter, J. Cationic Iridium(III) Carbonyl Complexes: [Ir(CO)6]3+ and [Ir(CO)5Cl]2+. Angew. Chem. Int. Ed. Engl. 1996, 35, 1974–1976. [Google Scholar] [CrossRef]
- Ellis, J.E.; Chi, K.-M. Synthesis, Isolation, and Characterisation of [K(cryptand 2.2.2)]2[Hf(CO)6], the First Substance to Contain Hafnium in a Negative Oxidation State. Structural Characterisation of [K(cryptand 2.2.2)]2[M(CO)6].Pyridine (M = Ti, Zr, and Hf). J. Am. Chem. Soc. 1990, 112, 6022–6025. [Google Scholar] [CrossRef]
- van der Lubbe, S.C.C.; Vermeeren, P.; Fonseca Guerra, C.; Bickelhaupt, F.M. The Nature of Nonclassical Carbonyl Ligands Explained by Kohn-Sham Molecular Orbital Theory. Chem. Eur. J. 2020, 26, 15690–15699. [Google Scholar] [CrossRef]
- Purcell, K.F.; Kotz, J.C. Inorganic Chemistry; W. B. Saunders Company Ltd.: London, UK, 1977. [Google Scholar]
- Wolczanski, P.T. Flipping the Oxidation State Formalism: Charge Distribution in Organometallic Complexes as Reported by Carbon Monoxide. Organometallics 2017, 36, 622–631. [Google Scholar] [CrossRef]
- Stepanenko, I.; Zalibera, M.; Schaniel, D.; Telser, J.; Arion, V.B. Ruthenium-Nitrosyl Complexes as NO-Releasing Molecules, Potential Anticancer Drugs, and Photoswitches Based on Linkage Isomerism. Dalton Trans. 2022, 51, 5367–5393. [Google Scholar] [CrossRef]
- Gouveia Júnior, F.S.; de Moraes Silveira, J.A.; Holanda, T.M.; Marinho, A.D.; Ridnour, L.A.; Wink, D.A.; Bezerra de Siqueira, R.J.; Azul Monteiro, H.S.; Silva de Sousa, E.H.; de França Lopes, L.G. New Nitrosyl Ruthenium Complexes with Combined Activities for Multiple Cardiovascular Disorders. Dalton Trans. 2023, 52, 5176–5191. [Google Scholar] [CrossRef]
- Norman, N.C.; Pringle, P.G. In Defence of Oxidation States. Dalton Trans. 2022, 51, 400–410. [Google Scholar] [CrossRef]
- Ampßler, T.; Monsch, G.; Popp, J.; Riggenmann, T.; Salvador, P.; Schröder, D.; Klüfers, P. Not Guilty on Every Count: The “Non-Innocent” Nitrosyl Ligand in the Framework of IUPAC’s Oxidation-State Formalism. Angew. Chem. Int. Ed. 2020, 59, 12381–12386. [Google Scholar] [CrossRef]
- Popp, J.; Riggenmann, T.; Schröder, D.; Ampßler, T.; Salvador, P.; Klüfers, P. Bent and Linear {CoNO}8 Entities: Structure and Bonding in a Prototypic Class of Nitrosyls. Inorg. Chem. 2021, 60, 15980–15996. [Google Scholar] [CrossRef]
- Popp, J.; Klüfers, P. Bond Strength of a Diatomic Acceptor Ligand: A Reliable Measure of its Antibond Occupation and its Charge? Eur. J. Inorg. Chem. 2022, 32, e202200374. [Google Scholar] [CrossRef]
- Schröder, D.; Klüfers, P. Bonding in PdCl(NO) and Related Nitrosylmetal Species of the Enemark-Feltham {MNO}10 Type. Z. Anorg. Allg. Chem. 2023, 649, e202200338. [Google Scholar] [CrossRef]
- Landry, V.K.; Pang, K.; Quan, S.M.; Parkin, G. Tetrahedral Nickel Nitrosyl Complexes with Tripodal [N3] and [Se3] Donor Ancillary Ligands: Structural and Computational Evidence that a Linear Nitrosyl is a Trivalent Ligand. Dalton Trans. 2007, 8, 820–824. [Google Scholar] [CrossRef]
- Tomson, N.C.; Crimmin, M.R.; Petrenko, T.; Rosebrugh, L.E.; Sproules, S.; Boyd, W.C.; Bergman, R.G.; DeBeer, S.; Toste, F.D.; Wieghardt, K. A Step Beyond the Feltham-Enemark Notation: Spectroscopic and Correlated ab Initio Computational Support for an Antiferromagnetically Coupled M(II)-(NO)− Description of Tp*M(NO) (M = Co, Ni). J. Am. Chem. Soc. 2011, 133, 18785–18801. [Google Scholar] [CrossRef]
- D’Arpino, A.A.; Cundari, T.R.; Wolczanski, P.T.; MacMillan, S.N. Transition Metal Nitrosyls: Statistics, Charge Estimates via CDVR, and Studies of Tetradentate Chelate Diamides (pddi)M and (pddi)MNO (M = Cr, Fe, and Co). Organometallics 2023, 42, 2747–2761. [Google Scholar] [CrossRef]
- Mingos, D.M.P. A Theoretical Analysis of Ambivalent and Ambiphilic Lewis Acids/Bases with Symmetry Signatures. Coord. Chem. Rev. 2015, 293–294, 2–18. [Google Scholar] [CrossRef]
- Tangen, E.; Conradie, J.; Svadberg, A.; Ghosh, A. Understanding the Unexpected Linearity of the trans-{Mn(NO)2}8 Unit in a Phthalocyanine Complex: Some Thoughts on Dinitrosylheme Intermediates in Biology. J. Inorg. Biochem. 2005, 99, 55–59. [Google Scholar] [CrossRef]
- Menconi, G.; Kaltsoyannis, N. Nature of the Transition Metal-Cycloheptatrienyl Bond. Computational Studies of the Electronic Structure of [M(h7-C7H7)(h5-C5H5)] (M = Groups 4-6). Organometallics 2005, 24, 1189–1197. [Google Scholar] [CrossRef]
- Basse, R.; Vanicek, S.; Höfer, T.; Kopacka, H.; Wurst, K.; Muller, T.; Schwartz, H.A.; Olthof, S.; Casper, L.A.; Nau, M.; et al. Cationic Cycloheptatrienyl Cyclopentadienyl Manganese Sandwich Complexes: Tromancenium Explored with High-Power LED Photosynthesis. Organometallics 2021, 40, 2736–2749. [Google Scholar] [CrossRef]
- Goedken, V.L.; Vallarino, L.M.; Quagliano, J.V. Cationic Ligands. Coordination of the 1,1,1 -Trimethylhydrazinium Cation to Nickel(II). Inorg. Chem. 1971, 10, 2682–2685. [Google Scholar] [CrossRef]
- Feil, C.M.; Hettich, T.D.; Beyer, K.; Sondermann, C.; Schlindwein, S.H.; Nieger, M.; Gudat, D. Comparing the Ligand Behavior of N-Heterocyclic Phosphenium and Nitrosyl Units in Iron and Chromium Complexes. Inorg. Chem. 2019, 58, 6517–6528. [Google Scholar] [CrossRef]
- Malchau, C.; Loose, F.; Mees, Y.; Duppe, J.; Sun, Y.; Niedner-Schatteburg, G.; Thiel, W.R. Electronic Properties of a Cationic Triphenylphosphine Ligand Decorated with a (η5-C5H5)Fe Group in Late-Transition-Metal Complexes. Organometallics 2020, 39, 3335–3343. [Google Scholar] [CrossRef]
- Schroers, J.P.; Kliemann, M.N.; Kollath, J.M.A.; Tauchert, M.E. How Cationic Metalloligands Affect the Coordination of Lewis Basic Ligands in RhI Complexes. Organometallics 2021, 40, 3893–3906. [Google Scholar] [CrossRef]
- Maser, L.; Vogt, M.; Langer, R. Cationic Ligands Between σ-Donation and Hydrogen-Bridge-Bond-Stabilisation of Ancillary Ligands in Coinage Metal Complexes with Protonated Carbodiphosphoranes. Dalton Trans. 2022, 51, 17397–17404. [Google Scholar] [CrossRef]
- Finze, M.; Bernhardt, E.; Willner, H.; Lehmann, C.W.; Aubke, F. Homoleptic, σ-Bonded Octahedral Superelectrophilic Metal Carbonyl Cations of Iron(II), Ruthenium(II), and Osmium(II). Part 2: Syntheses and Characterizations of [M(CO)6][BF4]2 (M = Fe, Ru, Os). Inorg. Chem. 2005, 44, 4206–4214. [Google Scholar] [CrossRef]
- Collman, J.P. Disodium Tetracarbonylferrate—A Transition Metal Analog of a Grignard Reagent. Acc. Chem. Res. 1975, 8, 342–347. [Google Scholar] [CrossRef]
- Cotton, F.A.; Murillo, C.A.; Walton, R.A. (Eds.) Multiple Bonds between Metal Atoms, 3rd ed.; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- Falvello, L.R.; Foxman, B.M.; Murillo, C.A. Fitting the Pieces of the Puzzle: The d bond. Inorg. Chem. 2014, 53, 9441–9456. [Google Scholar] [CrossRef]
- Krapp, A.; Lein, M.; Frenking, G. The Strength of the s-, p-, and d-bonds in Re2Cl82–. Theor. Chem. Acc. 2008, 120, 313–320. [Google Scholar] [CrossRef]
- Carlin, R.T.; Osteryoung, R.A. Electrochemistry of Molybdenum Chloride Dimers in a Basic Ambient-Temperature Molten Salt. Inorg. Chem. 1988, 27, 1482–1488. [Google Scholar] [CrossRef]
- Nuzzo, R.G.; Simon, H.J.; San Filippo, J., Jr. Selective Reduction of Sulfoxides. J. Org. Chem. 1977, 42, 568–569. [Google Scholar] [CrossRef]
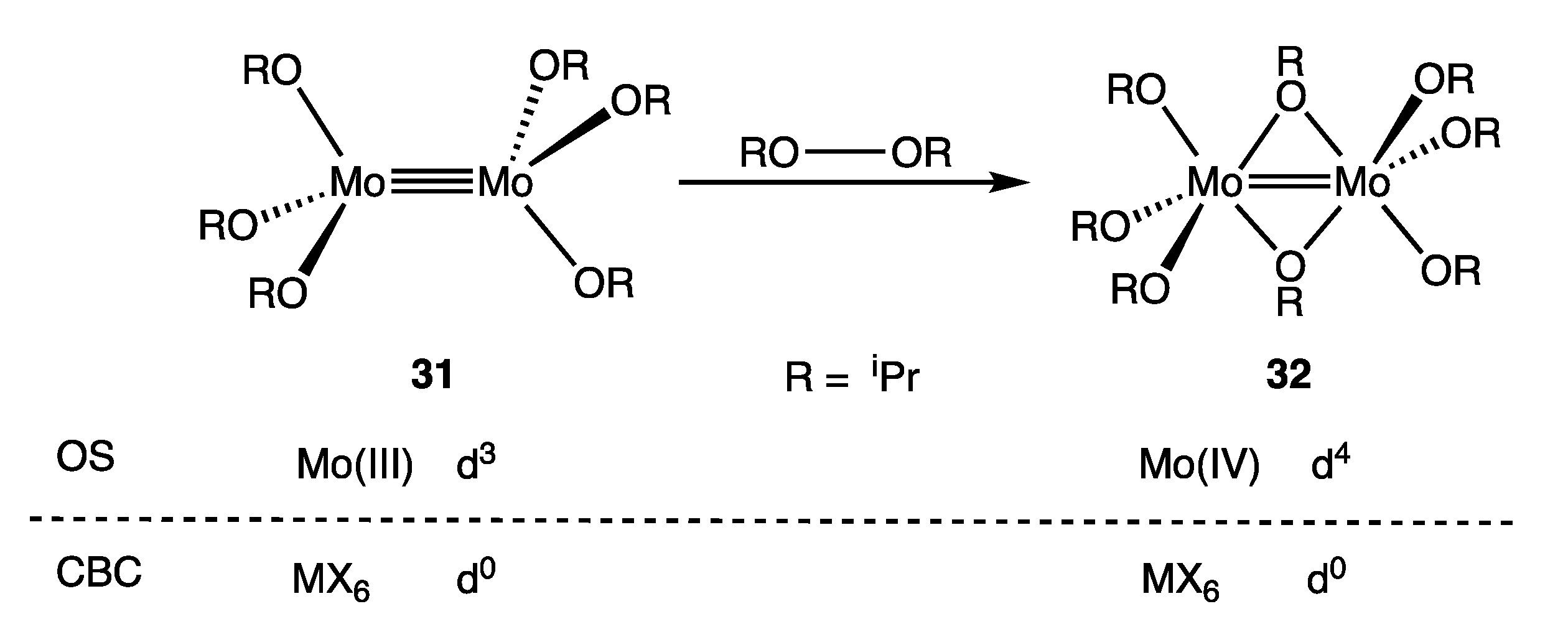
- Chisholm, M.H.; Kirkpatrick, C.C.; Huffman, J.C. Reactions of Metal–Metal Multiple Bonds. 7. Addition of the Halogens Chlorine, Bromine and Iodine and Diisopropyl Peroxide to Hexaisopropoxydimolybdenum (M≡M). Dinuclear Oxidative-Addition Reactions Accompanied by Metal–Metal Bond-Order Changes from 3 to 2 to 1. Inorg. Chem. 1981, 20, 871–876. [Google Scholar]
- Norman, N.C.; Pringle, P.G. Reply to the ‘Comment on “In Defence of Oxidation States”’ by J. C. Green. Dalton Trans. 2022, 51, 748–749. [Google Scholar] [CrossRef]
- Gatti, C.; Lasi, D. Source Function Description of Metal–Metal Bonding in d-Block Organometallic Compounds. Faraday Discuss. 2007, 135, 55–78. [Google Scholar] [CrossRef]
- Mingos, D.M.P. Polyhedral Skeletal Electron Pair Approach. Acc. Chem. Res. 1984, 17, 311–319. [Google Scholar] [CrossRef]
- Hoffmann, R.; Alvarez, S.; Mealli, C.; Falceto, A.; Cahill III, T.J.; Zeng, T.; Manca, G. From Widely Accepted Concepts in Coordination Chemistry to Inverted Ligand Fields. Chem. Rev. 2016, 116, 8173–8192. [Google Scholar] [CrossRef]
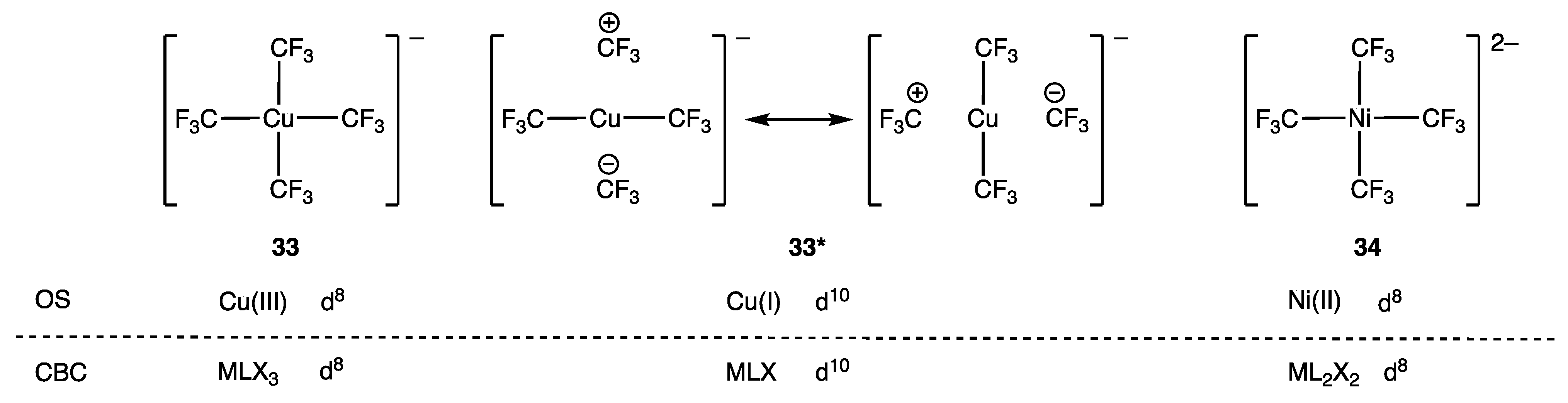
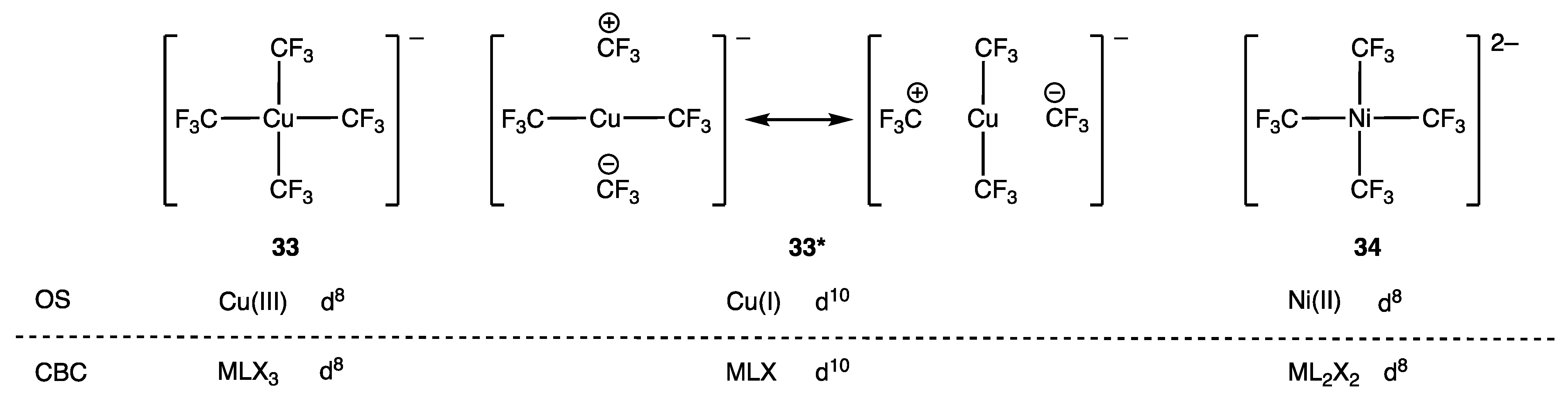
- Walroth, R.C.; Lukens, J.T.; MacMillan, S.N.; Finkelstein, K.D.; Lancaster, K.M. Spectroscopic Evidence for a 3d10 Ground State Electronic Configuration and Ligand Field Inversion in [Cu(CF3)4]1−. J. Am. Chem. Soc. 2016, 138, 1922–1931. [Google Scholar] [CrossRef]
- Leach, I.F.; Havenith, R.W.A.; Klein, J.E.M.N. Revisiting Formal Copper(III) Complexes: Bridging Perspectives with Quasi-d10 Configurations. Eur. J. Inorg. Chem. 2022, 27, e202200247. [Google Scholar] [CrossRef]
- Demonti, L.; Tabikh, H.; Saffon-Merceron, N.; Nebra, N. Safe Entry to Ag(I)/Ag(III) Mixed Valence Salts Containing the Homoleptic Ag(III) Anion [Ag(CF3)4]−. Eur. J. Inorg. Chem. 2023, 26, e202300042. [Google Scholar] [CrossRef]
- Shreiber, S.T.; DiMucci, I.M.; Khrizanforov, M.N.; Titus, C.J.; Nordlund, D.; Dudkina, Y.; Cramer, R.E.; Budnikova, Y.; Lancaster, K.M.; Vicic, D.A. [(MeCN)Ni(CF3)3]− and [Ni(CF3)4]2−: Foundations Toward the Development of Trifluoromethylations at Unsupported Nickel. Inorg. Chem. 2020, 59, 9143–9151. [Google Scholar] [CrossRef]
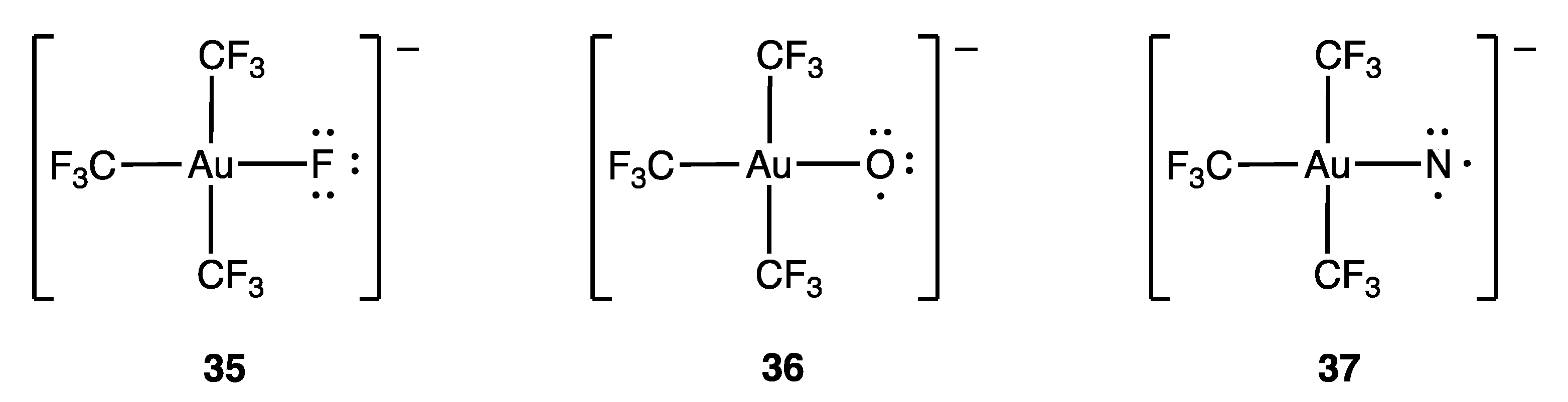
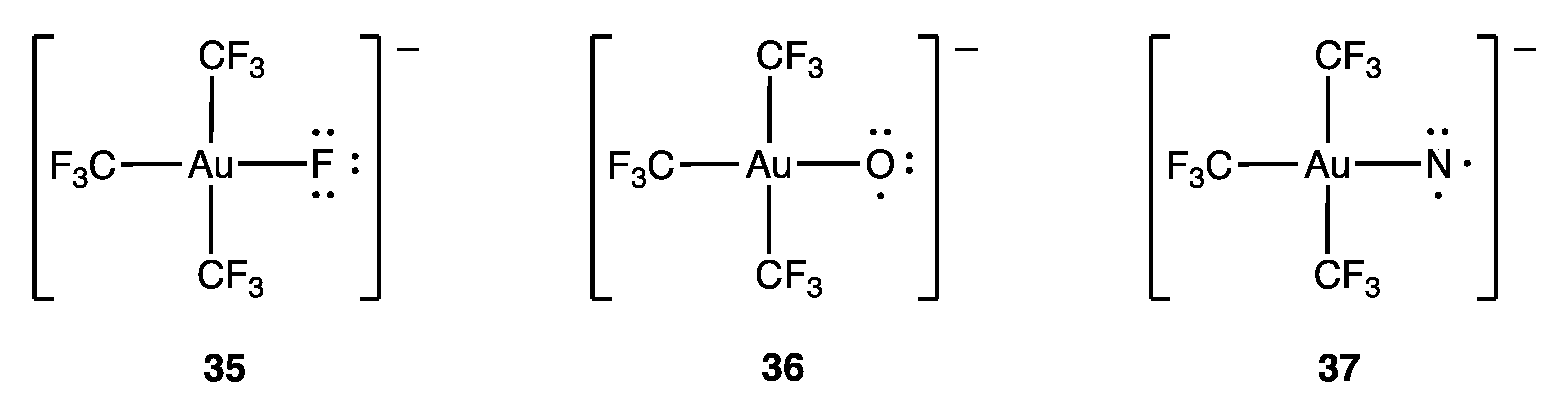
- Pérez-Bitrián, A.; Alvarez, S.; Baya, M.; Echeverría, J.; Martín, A.; Orduna, J.; Menjón, B. Terminal Au–N and Au–O Units in Organometallic Frames. Chem. Eur. J. 2023, 29, e202203181. [Google Scholar] [CrossRef]
- Ellis, J.E. Adventures with Substances Containing Metals in Negative Oxidation States. Inorg. Chem. 2006, 45, 3167–3186. [Google Scholar] [CrossRef]
- Ellis, J.E. Metal Carbonyl Anions: From [Fe(CO)4]2− to [Hf(CO)6]2− and Beyond. Organometallics 2003, 22, 3322–3338. [Google Scholar] [CrossRef]
- Connor, J.A.; McEwen, G.K.; Rix, C.J. Ditertiary Alkylphosphine Derivatives of the Group VI Metal Carbonyls and their Oxidation with Iodine. J. Chem. Soc. Dalton Trans. 1974, 6, 589–595. [Google Scholar] [CrossRef]
- Sidgwick, N.V. The Electronic Theory of Valence; Oxford University Press: Oxford, UK, 1927. [Google Scholar]
- Zhao, L.; Pan, S.; Holzmann, N.; Schwerdtfeger, P.; Frenking, G. Chemical Bonding and Bonding Models in Main-Group Chemistry. Chem. Rev. 2019, 119, 8781–8845. [Google Scholar] [CrossRef]
- Gimferrer, M.; Aldossary, A.; Salvador, P.; Head-Gordon, M. Oxidation State Localised Orbitals: A Method for Assigning Oxidation States Using Optimally Fragment-Localised Orbitals and a Fragment Orbital Localisation Index. J. Chem. Theory Comput. 2022, 18, 309–322. [Google Scholar] [CrossRef]
- Rupf, S.M.; Pan, S.; Moshtaha, A.L.; Frenking, G.; Malischewski, M. Structural Characterization and Bonding Analysis of [Hg{Fe(CO)5}2]2+[SbF6]−2. J. Am. Chem. Soc. 2023, 145, 15353–15359. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
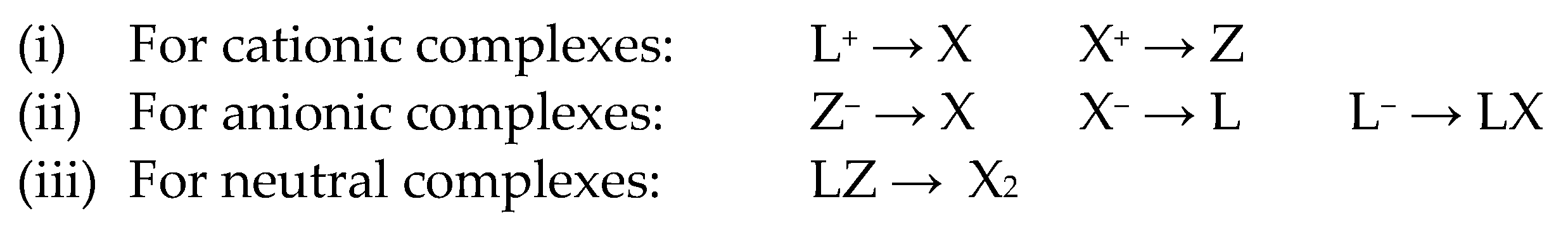{kind=link}





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Norman, N.C.; Pringle, P.G. The dn Number in Transition Metal Chemistry: Its Utility and Limitations. Chemistry 2023, 5, 2630-2656. https://doi.org/10.3390/chemistry5040170
Norman NC, Pringle PG. The dn Number in Transition Metal Chemistry: Its Utility and Limitations. Chemistry. 2023; 5(4):2630-2656. https://doi.org/10.3390/chemistry5040170
Chicago/Turabian StyleNorman, Nicholas C., and Paul G. Pringle. 2023. "The dn Number in Transition Metal Chemistry: Its Utility and Limitations" Chemistry 5, no. 4: 2630-2656. https://doi.org/10.3390/chemistry5040170
APA StyleNorman, N. C., & Pringle, P. G. (2023). The dn Number in Transition Metal Chemistry: Its Utility and Limitations. Chemistry, 5(4), 2630-2656. https://doi.org/10.3390/chemistry5040170