1. Introduction
The application of high-energy materials (explosives, fuels, and propellants) in the industry is largely limited by their sensitivities toward detonation [
1,
2,
3,
4,
5,
6]. Multiple factors affect the sensitivity of high-energy materials towards detonation, like free space in the crystal lattice, the heat of detonation, positive values of electrostatic potential above the central regions of the molecular surface, and the presence of hydrogen bonds [
7,
8,
9,
10,
11,
12]. Modification of these factors is a promising strategy for the fine-tuning of the detonation characteristics of explosives and the design of new types of HEMs with lower sensitivity towards detonation [
13,
14].
Although modifying the sensitivity of conventional explosives towards detonation is possible, numerous obstacles make it difficult to design conventional explosives with moderate sensitivity and satisfactory efficiency. One of the key problems related to the design of new explosives with improved properties lies in the fact that highly efficient explosives are usually very sensitive to detonation [
15,
16,
17,
18]. Strategies for the modification of the properties of explosives rely on the adjustment of molecular factors that determine the sensitivity of explosives toward detonation [
13,
15]. As already pointed out, one of the main molecular factors that affect the sensitivity of explosives towards detonation is the positive value of electrostatic potential over the central region of the molecular surface. Although the exact role of positive electrostatic potential in defining the detonation characteristics of explosives is not completely revealed, it is believed that positive values of electrostatic potentials in the central regions of molecules are an indicator of the withdrawal of the negative charge by substituents like NO
2 groups from the center to the periphery of the molecules [
16]. This type of charge distribution results in weaker C-N bonds and higher sensitivity towards detonation due to the easier initiation of the bond-breaking process. It is important to note that dissociation of the C-NO
2 bond is the first step in the process of decomposition of the nitroaromatic explosives [
19]. Nevertheless, targeted modification of the electrostatic potential that would lead to the design of explosives with desired detonation properties is still a challenge for material design scientists.
A promising approach for the modification of the electrostatic potential values and detonation properties of classical explosives is co-crystallization. It is shown that the co-crystallisation of the TNT molecules with CL-20 molecules affects both the sensitivity and performance of these explosives [
13]. However, the design of high-energy co-crystals with improved properties is mainly based on a trial-and-error approach since the properties of high-energy co-crystals are often unpredictable.
Another approach for the design of HEM molecules with desired properties is the introduction of additional structural features in the HEM molecule that may provide new tools for the modification of the highly energetic properties of these compounds. A good example in this respect represents the class of high-energy transition metal complexes [
2,
20,
21,
22,
23,
24,
25,
26,
27,
28,
29]. Examples of simple Werner-type transition metal complexes that can act as explosives when exposed to adequate external stimuli have been known for more than 50 years [
26]. The structures of transition metal complexes allow the combining of ligands and counterions with reductive and oxidative characteristics, making them candidates for high-energy compounds. There are well-known cases of highly energetic complexes like nickel hydrazinium nitrate and nickel hydrazinium perchlorate in which hydrazine ligands were used as reducing agents while nitrate or perchlorate counterions were used as oxidizing agents [
30]. This approach was also used in the study conducted by Myers and co-workers, in which stable Fe(II) complexes containing tetrazine ligands and perchlorate counterions were synthesized and initiated using low-energy lasers [
2].
However, combining the oxidizing and reducing agents in coordination compounds is not the only strategy to modify their detonation characteristics. The coordination of high-energy organic molecules may lead to significant changes in their electrostatic potential values on their surfaces. Furthermore, the quantum chemical study performed on a series of chelate complexes showed that a careful combination of transition metal ions and ligands in different acetylacetonato complexes can lead to a variety of different electrostatic potential values on the molecular surface, including surprising cases of negative values of the electrostatic potential above the metal atoms in chelate complexes [
31]. Since the high values of positive electrostatic potentials in the center of the molecular surface are related to the high impact sensitivity, modification of electrostatic potential values can lead to the development of new types of high-energy transition metal explosives.
Our recent Density Functional Theory study showed that nitro-bis(acetylacetonato) complexes may act as high-energy molecules with moderate sensitivity toward detonation [
22]. In this study, two important indicators of the sensitivity of HEMs were analyzed: positive values of electrostatic potential and Bond Dissociation Energies of the weakest chemical bond in the molecule (the C-NO
2 bond). Analysis of electrostatic potential maps of the nitro-bis(acetylacetonato) complex of copper (Cu(AcAc-NO
2)
2 complex) showed that the value of the positive potential in the central regions of this complex (25.25 kcal/mol) is similar to the value of the electrostatic potential in the central regions of classical explosives like 2,4,6-trinitrotoluene—TNT (23.76 kcal/mol). Also, the calculated BDE value for the C-NO
2 bond of the Cu(AcAc-NO
2)
2 complex (59.50 kcal/mol) is very similar to the BDE value calculated for the C-NO
2 bond of the 2,4,6-trinitrotoluene (58.90 kcal/mol) [
19,
22]. However, the Cu(AcAc-NO
2)
2 complex forms coordination polymers in which monomer units are linked through nitro-groups, and this type of structure may prevent the dissociation of C-NO
2 bonds [
32].
Here we studied indicators of sensitivity towards detonation of tris(3-nitropentane-2,4-dionato-κ2 O,O′) complexes (nitro-tris(acetylacetonato) complexes) of Cr(III), Mn(III), Fe(III), and Co(III) by DFT calculations and analysis of experimental data. Previous studies showed that DFT calculations are an excellent tool for determining different properties of materials and their structural features, from highly energetic properties to the isomeric stability of fullerenes and the nature of methyl rotation barriers [
2,
7,
8,
9,
10,
33,
34,
35,
36,
37]. The influence of the presence of different transition metals and additional chelate rings on the molecular electrostatic potentials and BDE values of the C-NO
2 bonds was analyzed. Changes in the BDE values of coordinated and non-coordinated nitroacetylacetone were also evaluated.
2. Materials and Methods
All geometry optimizations, vibrational frequency calculations, wave function file calculations, and BDE calculations were performed using the Gaussian09 program package [
38]. Geometry optimizations, vibrational frequency calculations, and electrostatic potential map calculations were performed using the M06 functional and cc-PVDZ basis sets since this level of theory showed satisfactory results when applied to similar systems [
22]. BDE values were calculated using the B3LYP/6-311++G** level of theory, following the previously proposed methodology [
19]. Calculated infrared spectra and three-dimensional coordinates of optimized molecules are given in the
Supplementary Materials (
Figure S1–S5 and
Tables S1–S5). The obtained wave function files were used for calculations of the Molecular Electrostatic Potentials of the studied complexes. Electrostatic potential maps were calculated and visualized using the Wave Function Analysis—Surface Analysis Suite (WFA-SAS) software [
39]. Visualization of the optimized geometries and analysis of crystal structure were performed using Mercury software, while visualization of infrared spectra was performed using the Avogadro program [
40,
41]. The crystal structures of the tris-(3-nitroacetylacetonato) complexes were extracted from the Cambridge Structural Database [
42].
Nitro-tris(acetylacetonato)cobalt(III) complex was synthesized according to the general procedure proposed by Collman et al. [
43]. To a 100 cm
3 volume of acetic anhydride, 0.02 moles of copper (II) nitrate trihydrate were added. The resultant mixture was stirred for 15 min at 0 °C. Subsequently, 0.007 moles of cobalt (III) acetylacetonate were added to the aforementioned mixture. The mixture was stirred for 2 h at 0 °C, followed by an additional hour at room temperature. The resultant slurry was mixed with 300 mL of water, 300 g of ice, and 7.5 g of sodium acetate. From this mixture, a green precipitate consisting of Co(AcAc-NO
2)
3 was collected. The infrared spectrum was recorded in the 4000–450 cm
−1 region using the Thermo Scientific Nicolet Summit FT-IR Spectrometer (Massachusetts, MA, USA), while UV/VIS spectra were recorded in the 190–1000 nm region using chloroform as a solvent. The heat of combustion (higher calorific value) was measured using an IKA 200 calorimeter. The experiment was performed in an oxygen atmosphere using 0.2 g of the Co(AcAc-NO
2)
3 sample.
3. Results
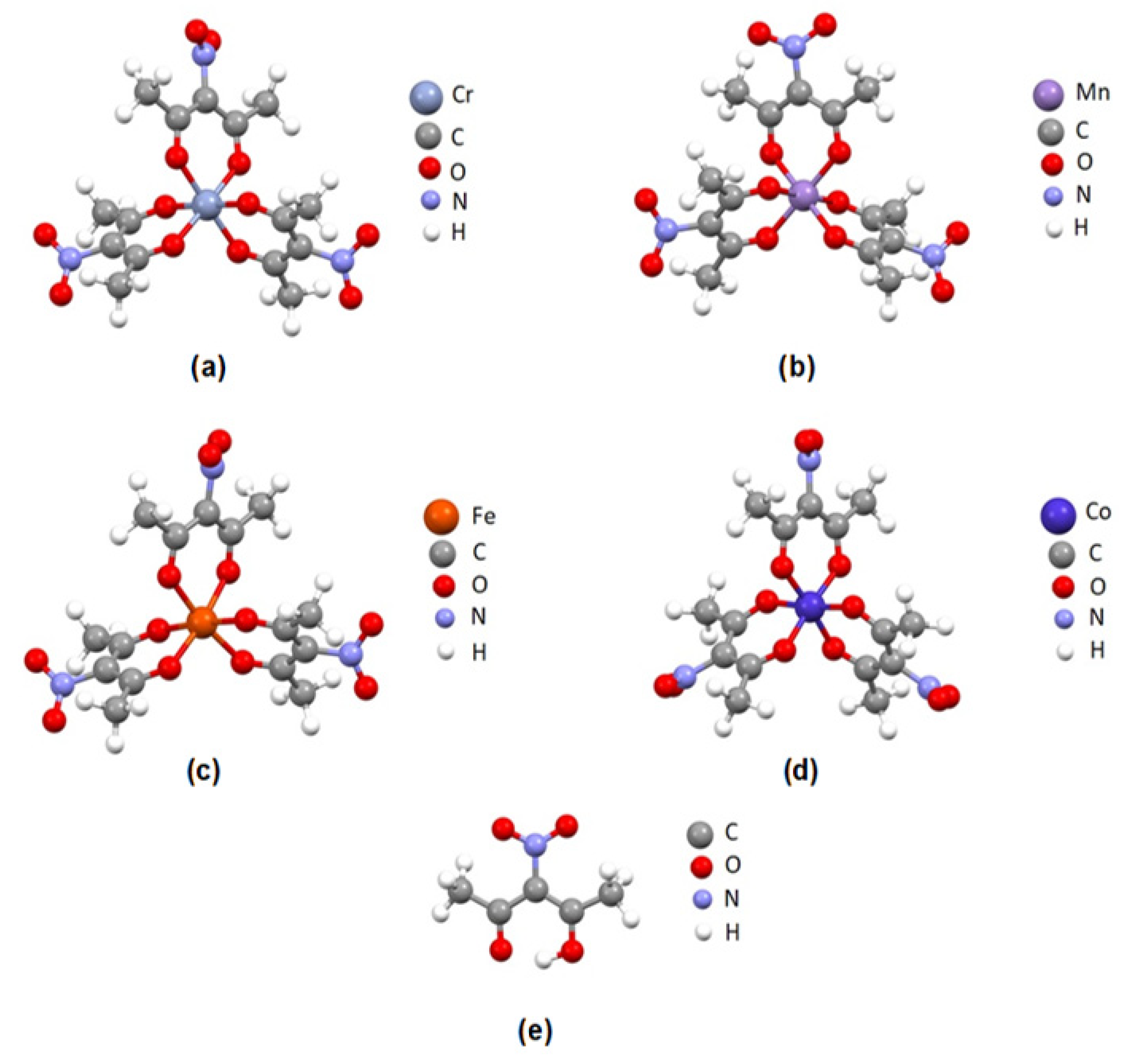
3.1. Quantum Chemical Calculations
The geometries of four nitro-tris(acetylacetonato) complexes (nitro-tris(acetylacetonato) chromium(III), nitro-tris(acetylacetonato) manganese(III), nitro-tris(acetylacetonato) iron(III) and nitro-tris(acetylacetonato) cobalt(III) complexes) and nitroacetylacetone ligand were optimized using M06/cc-PVDZ level of theory. Optimized geometries of transition metal complexes were used to calculate electrostatic potential maps, while optimized geometries of all five molecules were used for bond dissociation energy calculations of the C-NO
2 bonds. Optimized geometries of studied molecules are given in
Figure 1.
After the geometry optimizations, the structures of all four nitro-tris(acetylacetonato) complexes adopted octahedral geometry. Possible hydrogen bonds between oxygen atoms from NO2 groups and hydrogen atoms from neighboring methyl groups were identified in optimized structures.
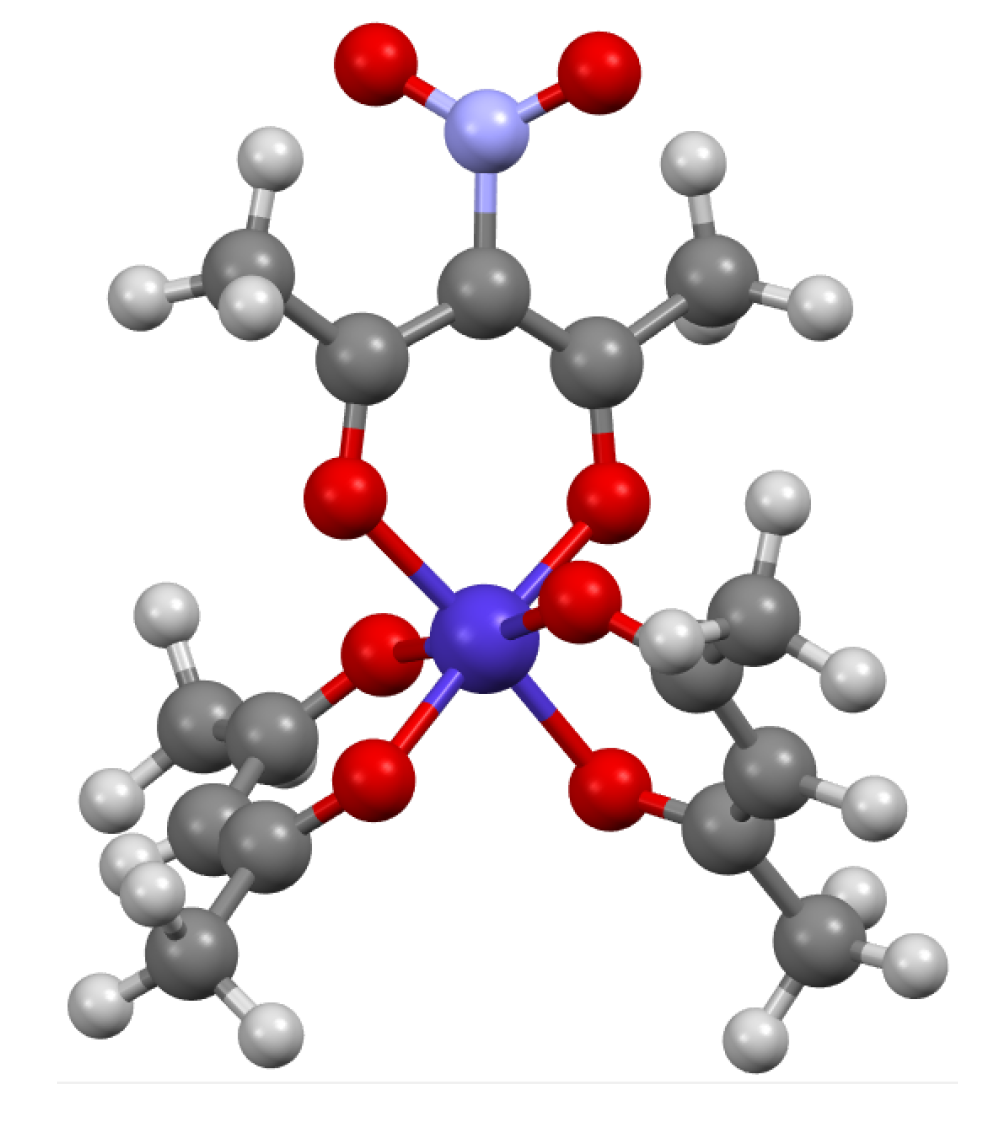
To compare computationally optimized geometries with experimental data, the Cambridge Structural Database was searched for all structures containing nitro-tris(acetylacetonato) transition metal complexes. Only crystal structures of nitro-tris(acetylacetonato) chromium(III) and nitro-tris(acetylacetonato) cobalt(III) were found and extracted (refcodes: MALBOT and LAMWUT, respectively). The geometries of both nitro-tris(acetylacetonato) complexes found in crystal structures were octahedral, which was in agreement with the results of the geometry optimization calculations. Moreover, in both crystal structures, short contacts between oxygen atoms from NO
2 groups and hydrogen atoms from neighboring CH
3 groups were identified (N-O
…H-C distances: 2.431 Å and 2.443 Å in the MALBOT and LAMWUT crystal structures, respectively). The nitro-acetylacetonato fragment was also noticed in the bis(acetylacetonato)-(nitro-acetylacetonato) cobalt(III) complex extracted from the KUCCAM crystal structure (
Figure 2).
In this structure, a short contact between the oxygen atom from the NO2 group and the hydrogen atom from the CH3 group was detected, too (N-O…H-C distance: 2.095 Å).
The Hirshfeld surface was mapped with d
norm and analyzed (
Figure S6). Analysis showed that oxygen atoms could interact with hydrogen atoms in the CH
3 group. A filtered 2-D fingerprint plot (
Figure S6) showed that O
…H interactions represent 17.7% of all noncovalent interactions involving this complex.
Electrostatic potential maps were calculated for all nitro-tris(acetylacetonato) complexes, and the results were analyzed to identify regions of strong positive electrostatic potential that may indicate the highly energetic properties of these molecules. Calculated electrostatic potential maps are given in
Figure 3.
In all four cases, an area of positive electrostatic potential (yellow and red) was identified in the central regions of chelate rings. This indicates possible highly energetic characteristics of the studied complexes. Also, electrostatic potentials in the area of C-NO2 bonds are positive (yellow and red), which indicates that these chemical bonds are prone to rupture and that the bond-breaking process is likely to occur. This is the consequence of the presence of the electron-withdrawing NO2 substituents on the chelate rings.
To further examine the tendency of the C-NO
2 bonds to undergo the bond-breaking process, BDE values for C-NO
2 bonds in all four studied complexes were calculated and analyzed. Calculated BDE values for C-NO
2 bonds in these complexes are given in
Table 1.
Both non-corrected and ZPE-corrected BDE values calculated for the C-NO
2 bonds in nitro-tris(acetylacetonato) complexes are similar to the BDE values of the C-NO
2 bonds present in thermally stable and insensitive conventional nitroaromatic explosives (the previously calculated BDE value for the C-NO
2 bond in a 2,4,6-triamino-1,3,5-trinitrobenzene (TATB) molecule is 69.4 kcal/mol) [
19]. The strongest BDE value was calculated for the C-NO
2 bond from the Co(AcAc-NO
2)
3 complex (68.42 kcal/mol), while the weakest BDE value was calculated for the C-NO
2 bond from the Mn(AcAc-NO
2)
3 complex (64.91 kcal/mol). These results indicate that the Co(AcAc-NO
2)
3 complex may have the lowest sensitivity towards detonation compared to other studied complexes. It is important to note that the calculated BDE value of the C-NO
2 bond from the nitroacetylacetone (AcAc-NO
2) (71.39 kcal/mol) molecule is significantly larger compared to the BDE values of the C-NO
2 bonds present in the coordinated nitroacetylacetone fragments. The decrease in these BDE values upon the formation of the transition metal complex indicates that coordination, in principle, can be used as a tool for the adjustment of the detonation properties of high-energy materials.
3.2. Synthesis and Characterization of the Nitro-Tris(acetylacetonato) Cobalt(III) Complex
Since the results of the DFT calculations indicated that the Co(AcAc-NO
2)
3 complex may act as a high-energy material with a lower sensitivity towards detonation compared to the other studied complexes, this complex was synthesized according to the previously established procedure [
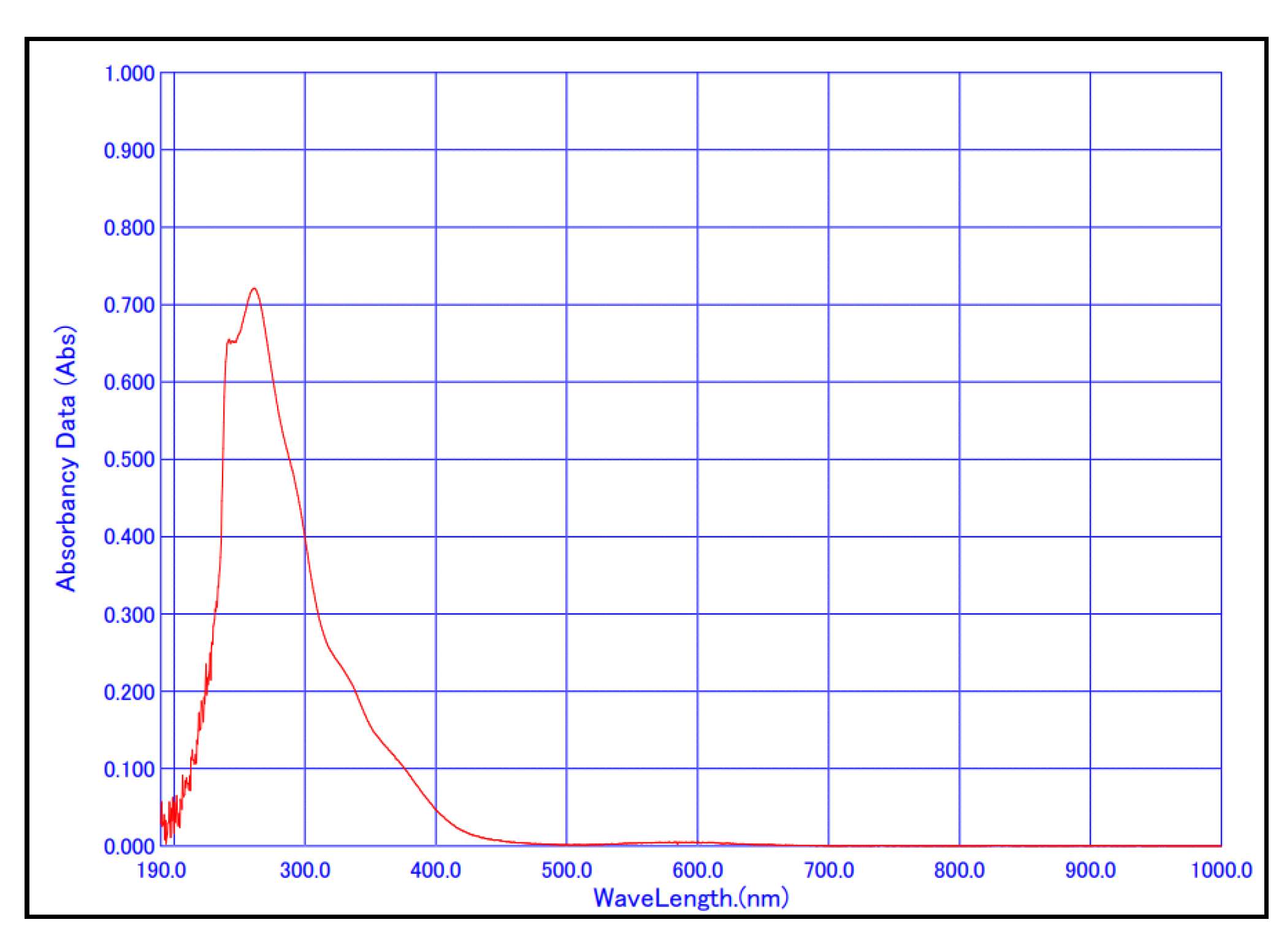
43]. Green crystals were collected and characterized by UV/Vis and FT-IR spectroscopy experiments. The UV/Vis spectrum of the chloroform solution of the prepared Co(AcAc-NO
2)
3 complex is given in
Figure 4.
The absorption maximum in the UV region appeared at λ
max = 261.0 nm, while the absorption maximum in the visible region appeared at λ
max = 583.0 nm. These λ
max values are in good agreement with the previously obtained results for this complex (λ
max (UV) = 258.0 nm and λ
max (Vis) = 588.0 nm) [
44].
The synthesized Co(AcAc-NO
2)
3 complex was also characterized by an FT-IR spectroscopy experiment, and spectral data were analyzed in the context of the possible bond-breaking process of the C-NO
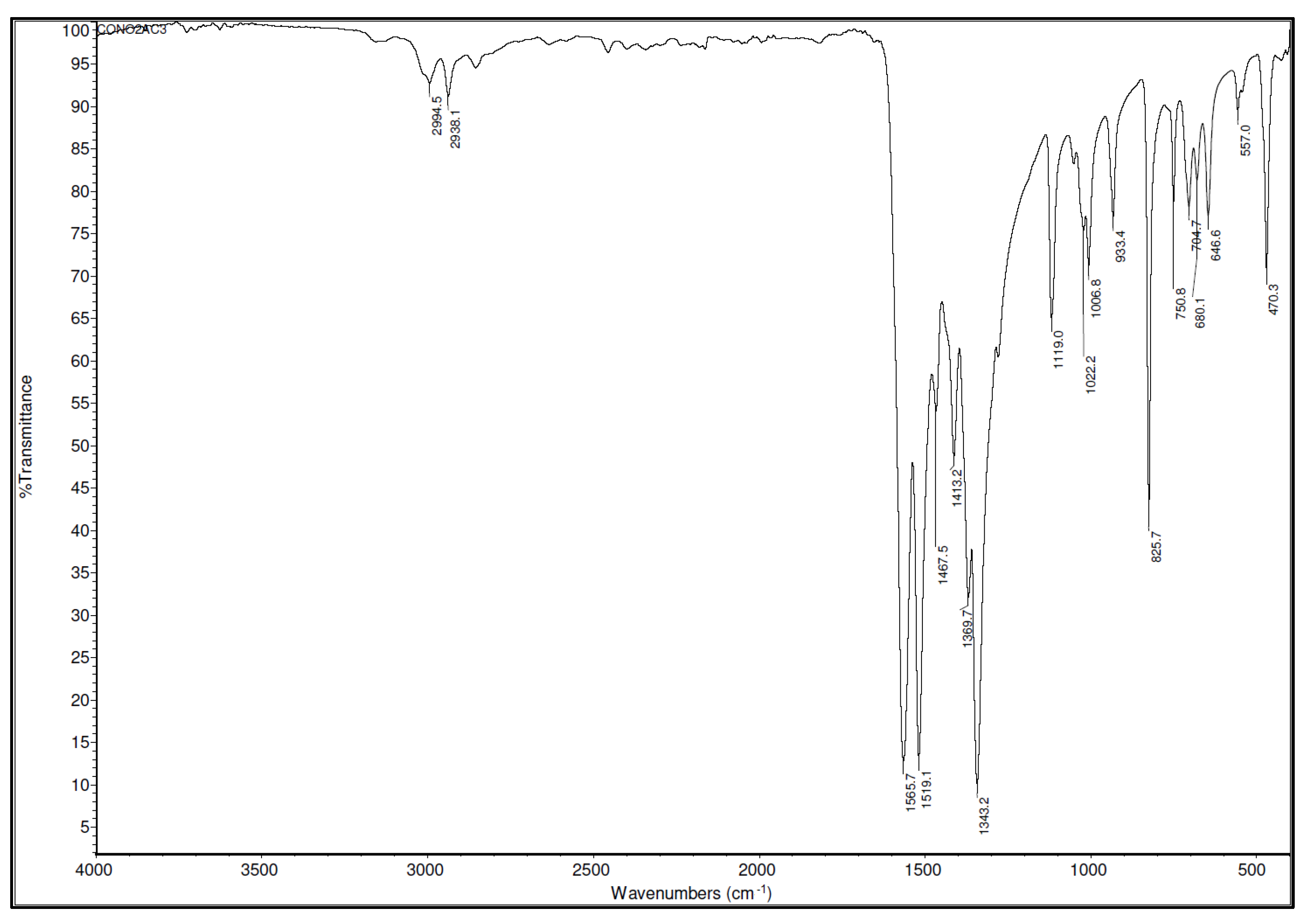
2 bond. The IR spectrum for the Co(AcAc-NO
2)
3 complex is given in
Figure 5. Since there is a general relation between bond strength and the position of the corresponding bands in vibrational spectra, analysis of IR spectral data was used not only for the identification of the product but also for a rough assessment of the characteristics of C-N bonds in terms of their strength.
The strong band that can be observed at 1565.7 cm−1 is the consequence of the mixed C=C and C=O vibrations of the chelate ring. The second strong band (1519.1 cm−1) corresponds to the asymmetric stretching vibrations of the NO2 group mixed with C=C and C=O vibrations of the chelate ring. The bands that appear at 1369.7, 1413.2, and 1467.5 cm−1 are the consequence of asymmetric NO2 stretching vibrations and CH3 fragment deformations.
The peak at 1343.2 cm−1 is assigned to the C-N and N-O stretching vibrations. The band at 1119.0 cm−1 is assigned to C-C=C and stretching N-O vibrations. Bands detected at 1006.8 and 1022.2 cm−1 are the consequence of the CH3 rocking vibrations. A band at 933.4 cm−1 is assigned to the C-CH3 vibrations. A sharp band at 825.7 cm−1 is the consequence of the NO2 scissoring vibrations coupled with C-C=C bending vibrations. Bands in the region of 500–700 cm−1 are mainly the consequence of Co-O stretching vibrations.
Since the values of IR frequencies are related to the bond strength (frequency increases as bond strength increases), analysis of bands assigned to C-N vibrations may be used to roughly predict the bond-breaking tendency of the C-N bond from the Co(AcAc-NO
2)
3 complex. The frequency of the C-N vibration in the Co(AcAc-NO
2)
3 complex (1343.2 cm
−1) is very similar to the frequency of the C-N vibration of the TNT (1324 cm
−1) [
32]. This indicates that the tendency of the C-N bond from Co(AcAc-NO
2)
3 to undergo a bond-breaking process is comparable with the tendency of some well-known classical explosives like TNT.
To test the high-energy properties of synthesized Co(AcAc-NO
2)
3, we initiated a decomposition reaction by exposing a small amount of the dry product to an open flame (
Figure 6).
Upon initiation by an open flame, the sample rapidly decomposed, releasing a large amount of heat and gaseous products in the process (Video S1).
To further assess the high-energy properties of synthesized Co(AcAc-NO2)3, we performed a calorimetry experiment and measured the heat of combustion. Results showed that the heat of combustion of Co(AcAc-NO2)3 is 14,133 J/g, which indicates that this compound is a high-energy material.
4. Discussion
Analysis of the molecular electrostatic potentials calculated for the nitro-tris(acetylacetonato) complexes of Cr(III), Mn(III), Fe(III), and Co(III) showed that in all studied compounds, positive values of electrostatic potential were identified in the central regions of chelate rings. Since it is known that positive values of electrostatic potential in the central regions of HEM molecules are related to their high sensitivity towards detonation, these results indicate that all four studied complexes can act as high-energy materials. Furthermore, positive values of electrostatic potentials were observed in the regions of the C-NO
2 bonds. Positive values of electrostatic potentials above the C-NO
2 bonds in nitroaromatic explosives are also an indicator of high sensitivity towards detonation. This is in agreement with previous observations that nitro-tris(acetylacetonato) complexes of Al(III) and Ga(III) spontaneously ignite in the air upon heating [
45]. It is also in agreement with the results of the recent study demonstrating that nitro-acetylacetonato complexes of iridium, gallium, and zinc can act as fuel in the processing of certain types of thin films [
46]. However, since positive electrostatic potential over the molecular center is only one indicator of the sensitivity of HEM molecules, bond dissociation energies were calculated for C-NO
2 bonds of studied complexes, and results were analyzed. To assess the influence of coordination on the strength of C-NO
2 bonds from ligands, BDE values were also calculated for the C-NO
2 bond present in the non-coordinated nitroacetylacetone.
In general, BDE values calculated for the C-NO
2 bonds present in nitro-tris(acetylacetonato) complexes of Cr(III), Mn(III), Fe(III), and Co(III) are comparable with the BDE values of C-NO
2 bonds present in conventional nitroaromatic explosives. The lowest BDE was calculated for the C-NO
2 bond present in the Mn(AcAc-NO
2)
3 complex (64.91 kcal/mol), and this value is similar to the BDE value previously calculated for the C-NO
2 bond present in the classical nitroaromatic explosive 1,3,5-trinitrobenzene (64.0 kcal/mol) [
19]. The highest BDE value was calculated for the C-NO
2 bond present in the Co(AcAc-NO
2)
3 complex (68.42 kcal/mol). A relatively high BDE value calculated for the C-NO
2 bond from the Co(AcAc-NO
2)
3 complex indicates that this complex has low sensitivity towards detonation.
These values are higher than previously obtained values for nitro-bis(acetylacetonato) complexes of Ni(II), Co(II), Cu(II), and Zn(II) (58.09–60.48 kcal/mol) [
22]. Analysis based on the comparison of the BDE values for C-NO
2 bonds present in nitro-bis(acetylacetonato) complexes and nitro-tris(acetylacetonato) complexes indicates that nitro-tris(acetylacetonato) complexes have lower sensitivity towards detonation. This result also indicates that additional chelate rings affect the BDE of C-NO
2 bonds and that the addition of chelate rings may be used as a new tool for controlling the sensitivity towards detonation of high-energy coordination compounds.
It is also important to note that the BDE value of C-NO2 bonds in non-coordinated nitroacetylacetone (71.39 kcal/mol) is significantly higher compared to that of coordinated nitroacetylacetone (64.91–68.42 kcal/mol). This result demonstrates that coordination can be used as a tool for fine-tuning the properties of high-energy molecules.
The difference in the BDE values calculated for C-NO
2 bonds of nitro-tris(acetylacetonato) complexes containing different transition metals shows that it is possible to fine-tune the sensitivity toward detonation of these HEMs by careful selection of transition metal atoms. In some cases, these differences are quite significant: the difference between the calculated ZPE-corrected BDE value for the C-NO
2 bond of the nitro-tris(acetylacetonato) Mn(III) complex and the nitro-tris(acetylacetonato) Co(III) complex is 3.51 kcal/mol. For example, the difference in the BDE values calculated for the C-NO
2 bond of picric acid (60.1 kcal/mol) and some well-known stable explosive molecules like TNT (58.9 kcal/mol) is only 1.20 kcal/mol [
19].
Short distances between hydrogen atoms from CH
3 groups and oxygen atoms from NO
2 groups were detected in both optimized geometries of studied compounds and crystal structures of nitro-acetylacetonato complexes, indicating the existence of weak hydrogen bonds. Possible weak hydrogen bonds between oxygen atoms from NO
2 groups and neighboring hydrogen atoms may affect the BDE values of C-NO
2 bonds and their sensitivities toward detonation by making the bond-breaking process less likely to occur. That is not uncommon since some of the most stable nitroaromatic explosives owe their stability to the intramolecular hydrogen bonds between NO
2 groups and hydrogen atoms from neighboring groups. An obvious example is the TATB, which is one of the best-known nitroaromatic explosives with moderate sensitivity towards detonation. In the structure of TATB, a network of intramolecular N-H
…O-N hydrogen bonds between the NO
2 and NH
2 groups was recognized [
47].
Since the results of the BDE calculations indicated that the tris-nitro(acetylacetonato) complex of Co(III) has the lowest sensitivity towards detonation, this complex was synthesized and characterized by UV and IC spectroscopy experiments. Analysis of the obtained UV/Vis spectrum was used to confirm that the synthesized compound was indeed a Co(AcAc-NO
2)
3 complex. The chloroform solution of this complex showed an absorption maximum in the UV region at λmax = 261.0 nm and an absorption maximum in the visible region at λmax = 583.0 nm. This is in agreement with previously obtained results (258.0 nm in the UV and 588.0 nm in the visible region) for this complex [
44]. This is also similar to the values of absorption maxima in the UV region previously collected for tris-nitro(acetylacetonato) complexes of Cr(III), Mn(III), and Fe(III), which are found to be at 272.0 nm, 273.0 nm, and 270.0 nm, respectively [
44]. In the case of absorption maxima in the visible region, the value collected for the Co(AcAc-NO
2)
3 complex is similar to the values previously collected for Cr(III) and Mn(III) complexes (576.0 nm and 548.0 nm, respectively), while absorption maxima in the visible region for the Fe(III) complex were found to be at 435.0 nm [
44]. Analysis of IR spectra showed that frequencies corresponding to the vibrations of C-N bonds from the Co(AcAc-NO
2)
3 complex (1343.2 cm
−1) have similar values as frequencies of vibrations of C-N bonds from conventional explosive TNT (1324 cm
−1) [
32]. Since the values of the vibrational frequencies of C-N bonds are directly related to the bond strength, this result demonstrates that C-N bonds in both the Co(AcAc-NO
2)
3 complex and TNT molecules have a similar tendency to undergo a bond-breaking process. This is especially important since the rupture of the C-NO
2 bond is the first step in the reaction of decomposition of the vast majority of nitroaromatic explosives. The vigorous reaction that occurred when the Co(AcAc-NO
2)
3 complex was initiated by an open flame confirms that this complex has highly energetic characteristics. Results of the calorimetry experiment showed that the heat of combustion of the nitro-tris(acetylacetonato) cobalt(III) complex in oxygen is 14,133 J/g, which is very similar to the previously measured heat of combustion of TNT in oxygen (14,958 J/g) [
48].
The present study has successfully demonstrated proof-of-concept for the potential utilization of nitro-tris(acetylacetonato) complexes as high-energy materials possessing a degree of sensitivity that is within acceptable limits. Further studies of the high-energy properties of nitro-tris(acetylacetonato) complexes are needed. This includes Thermogravimetric Analysis (TGA) and Differential Scanning Calorimetry (DSC) experiments, which would provide a complete quantitative description of thermal properties and the mechanism of decomposition of tris-nitro(acetylacetonato) complexes. However, we believe that results showing that the high-energy properties of nitro-tris(acetylacetonato) complexes could be tuned by careful selection of metal atoms and the addition of chelate rings are of great significance for researchers in the field of the design of high-energy materials. This study offers general guidelines for the design of not only tris-nitro(acetylacetonato) HEMs but also other chelate compounds with possible highly energetic properties and moderate sensitivity towards detonation.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}