
Optimization of Enzymatic Synthesis of D-Glucose-Based Surfactants Using Supported Aspergillus niger Lipase as Biocatalyst




Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Solubility Test
2.3. Enzymatic Synthesis
- 0.125 g, 34.6% isolated yield for initial synthesis conditions in 2M2B/DMSO.
- Rf 0.18 (AcOEt/petroleum ether 4:1 v/v).
- FTIR–ATR (vmax/cm−1): 3300 (OH), 2852–2920 (aliphatic CH), 1730 (ester C=O), 1010 (–O), 952 (OH deformation), 722 ((CH2)n, n ≥ 4).
- 1H NMR (DMSO-d6): δ ppm 5.08 (m, 1H), 4.90 (d, J = 4.6 Hz, 1H), 4.28 (dd, J = 13.5 Hz, 2.3 Hz, 1H), 3.97 (dd, J = 11.6 Hz, 6.3 Hz, 2H), 3.76 (m, 1H), 3.43 (m, 1H), 3.14 (m, 1H), 3.04 (t, J = 8,7 Hz, 1H), 2.27 (t, J = 7.2 Hz, 2H), 1.50 (m, 2H), 1.24 (m, 16H, alkyl chain), 0.85 (t, J = 6.7 Hz, 3H, CH3).
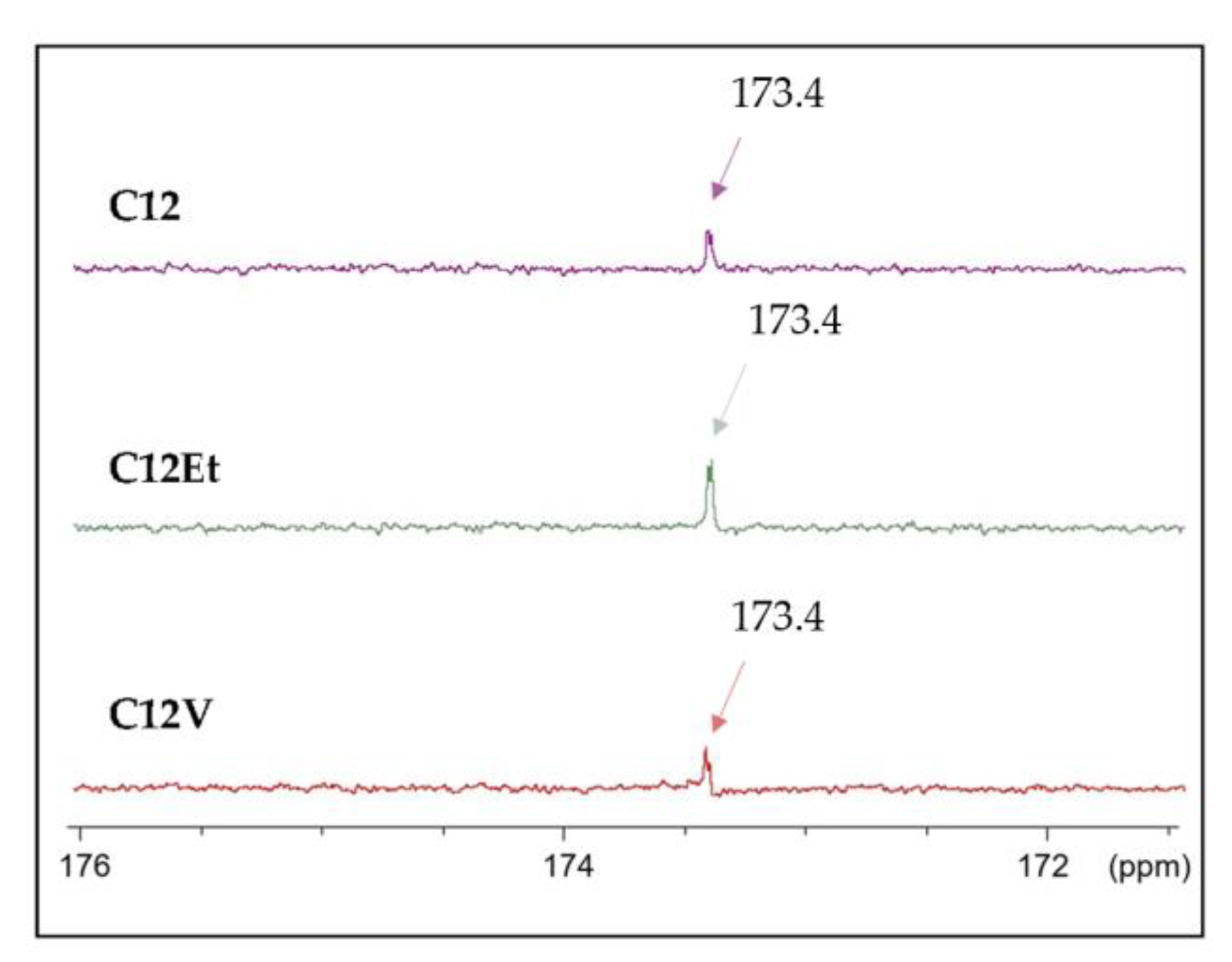
- 13C NMR (DMSO-d6): δ ppm 97.35–92.76 (1C, α/β monoester), 173.4 (C=O), 76.58 (1C), 74.86 (1C), 73.02 (1C), 72.35 (1C), 64.35 (1C), 33.88–22.51 (10C, alkyl chain), 14.40 (1C, CH3).
- DEPT analysis δ ppm 173.4, 64.35. The alkyl chain was identified from 33.88 to 22.5.
- MS(EI) = m/z 384.7 [M + Na]+.
2.4. Analytical Methods
2.5. Statistical Analyses
3. Results and Discussion
- reaction volume of 20 mL
- 1 g of 3 Å molecular sieve
- ratio of 1 molar equivalent (eq) of glucose (180 mg) to 3 eq of fatty acid
- 1% lipase (w/v)
- 56 °C, 240 rpm, 72 h.
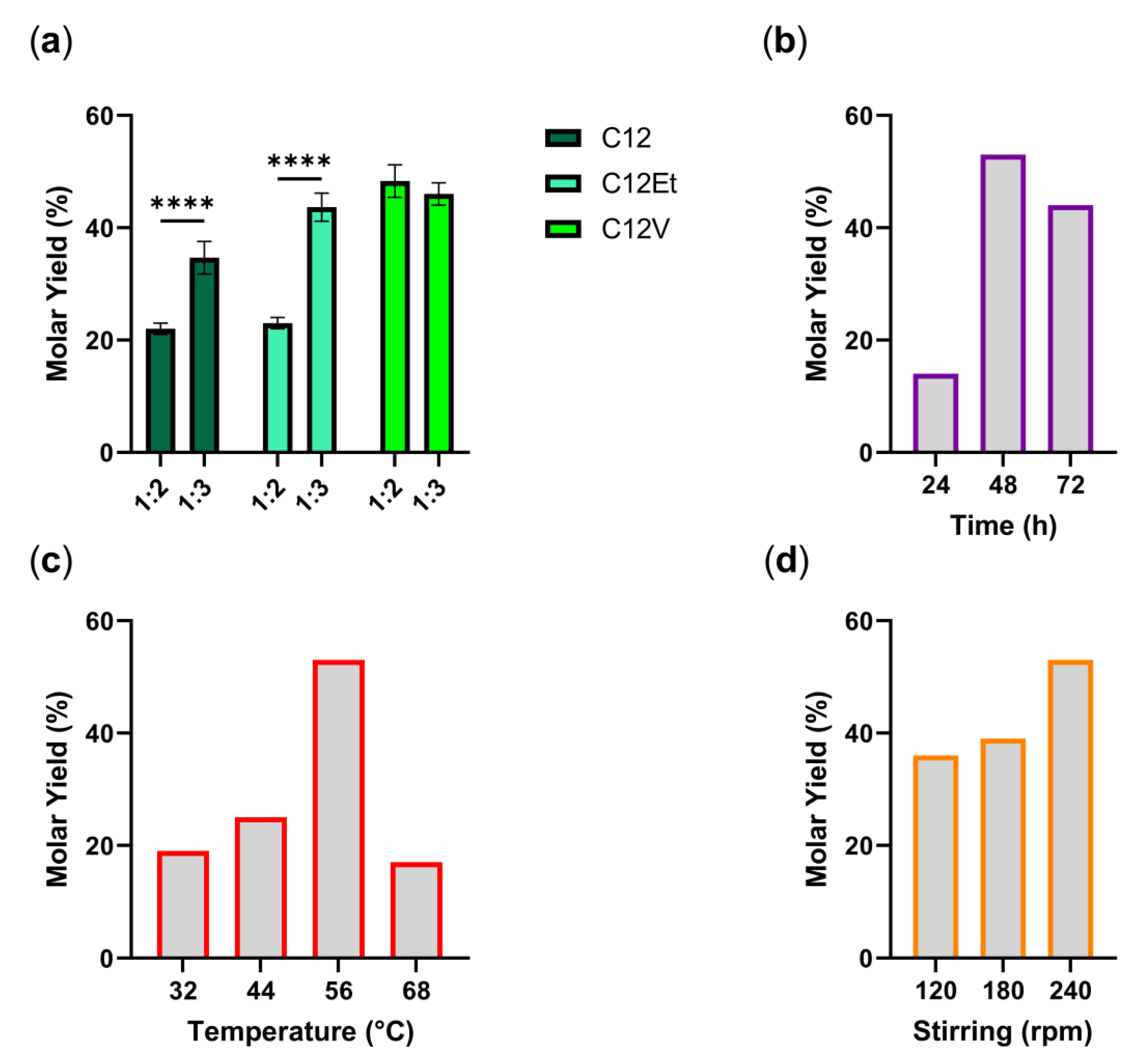
3.1. Optimization of Key Parameters
- types of substrates
- ratio between the different reagents
- reaction time
- temperature
- stirring speed.
- 1% Aspergillus niger lipase (w/v)
- co-solvent reaction medium, 2M2B/DMSO 20% (v/v).
- ratio of 1 molar equivalent of glucose to 3 equivalents of ethyl laurate
- 48 h, 56 °C, 240 rpm, 1 g 3 Å sieve.
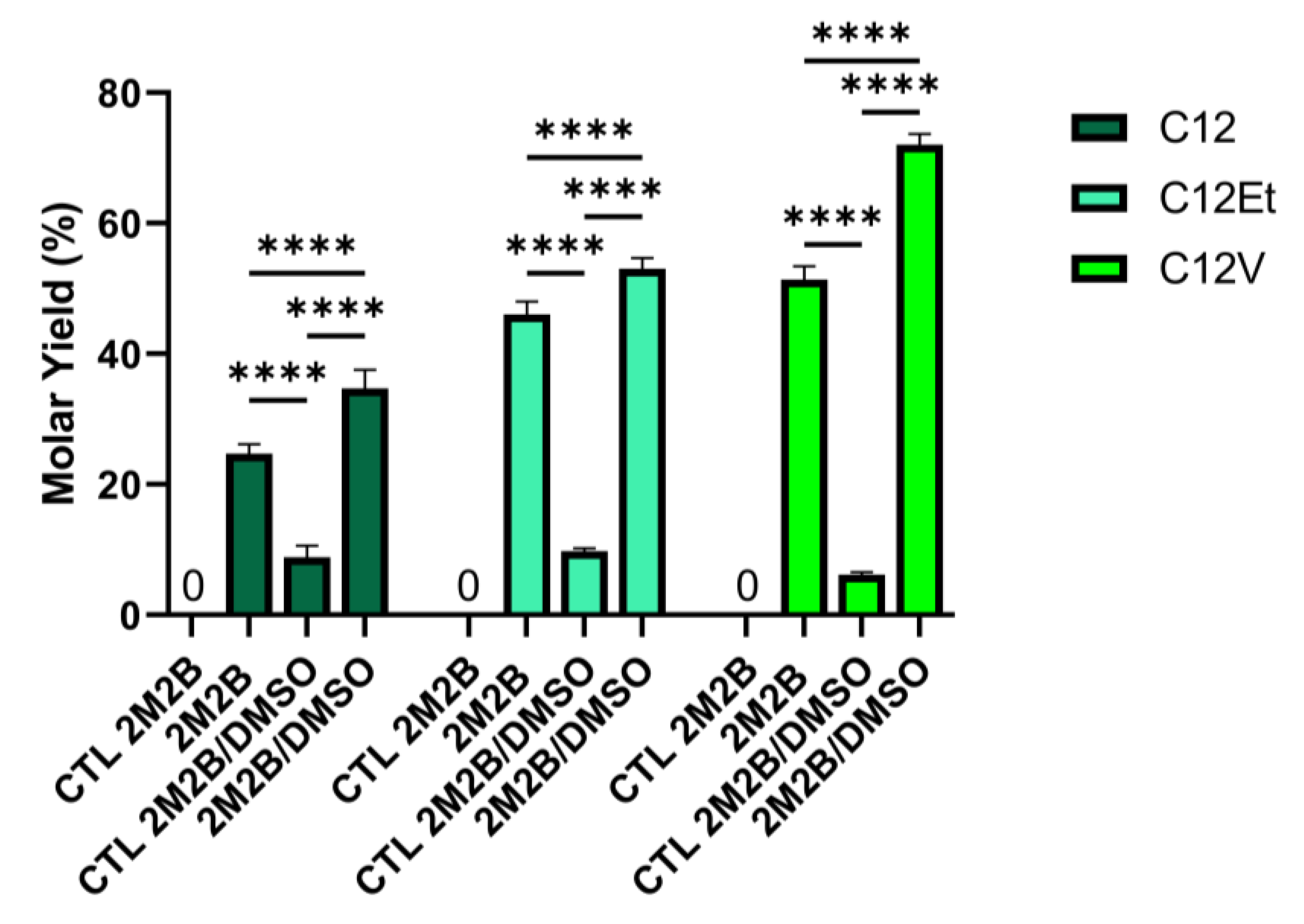
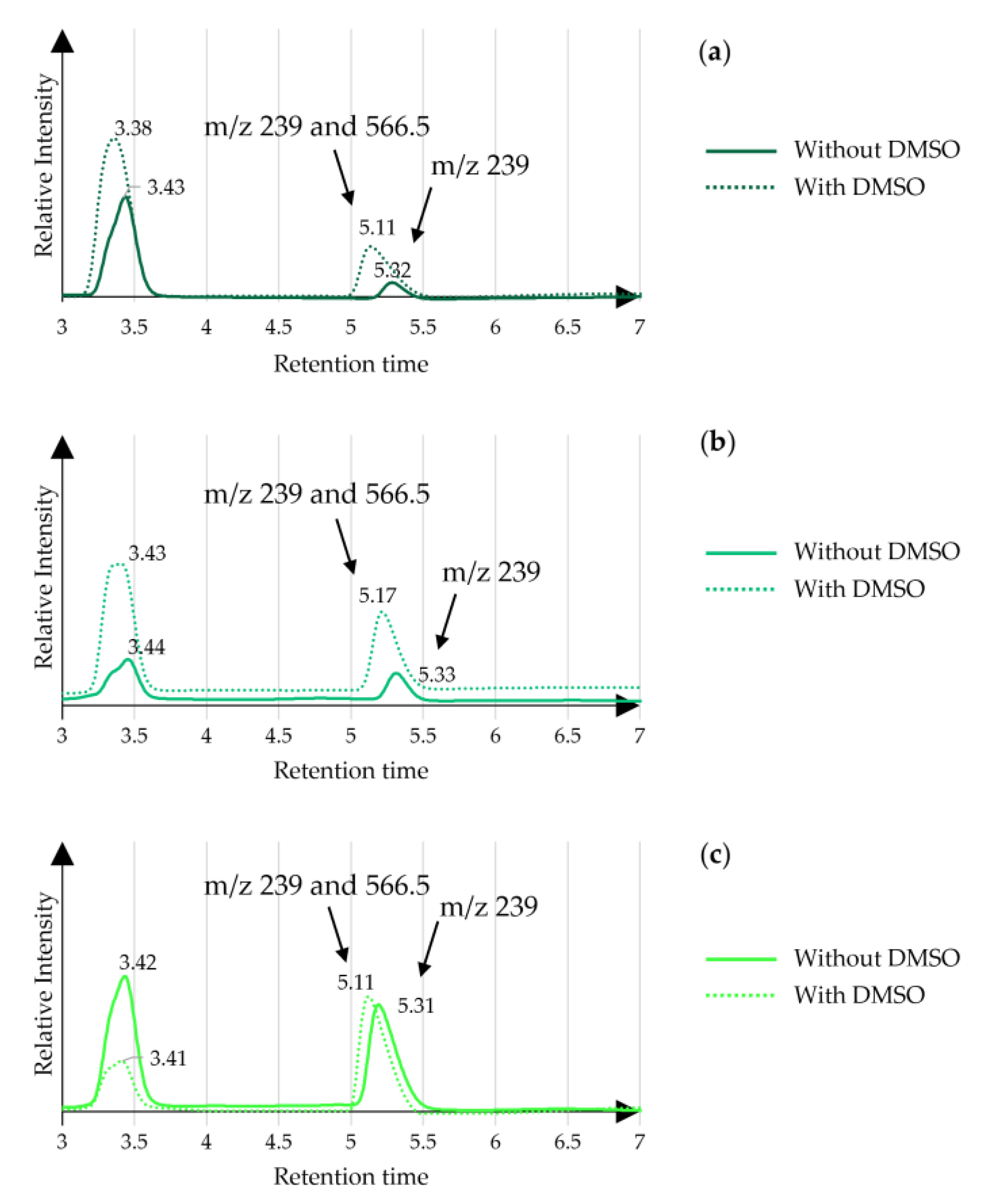
3.2. Enzyme Selectivity
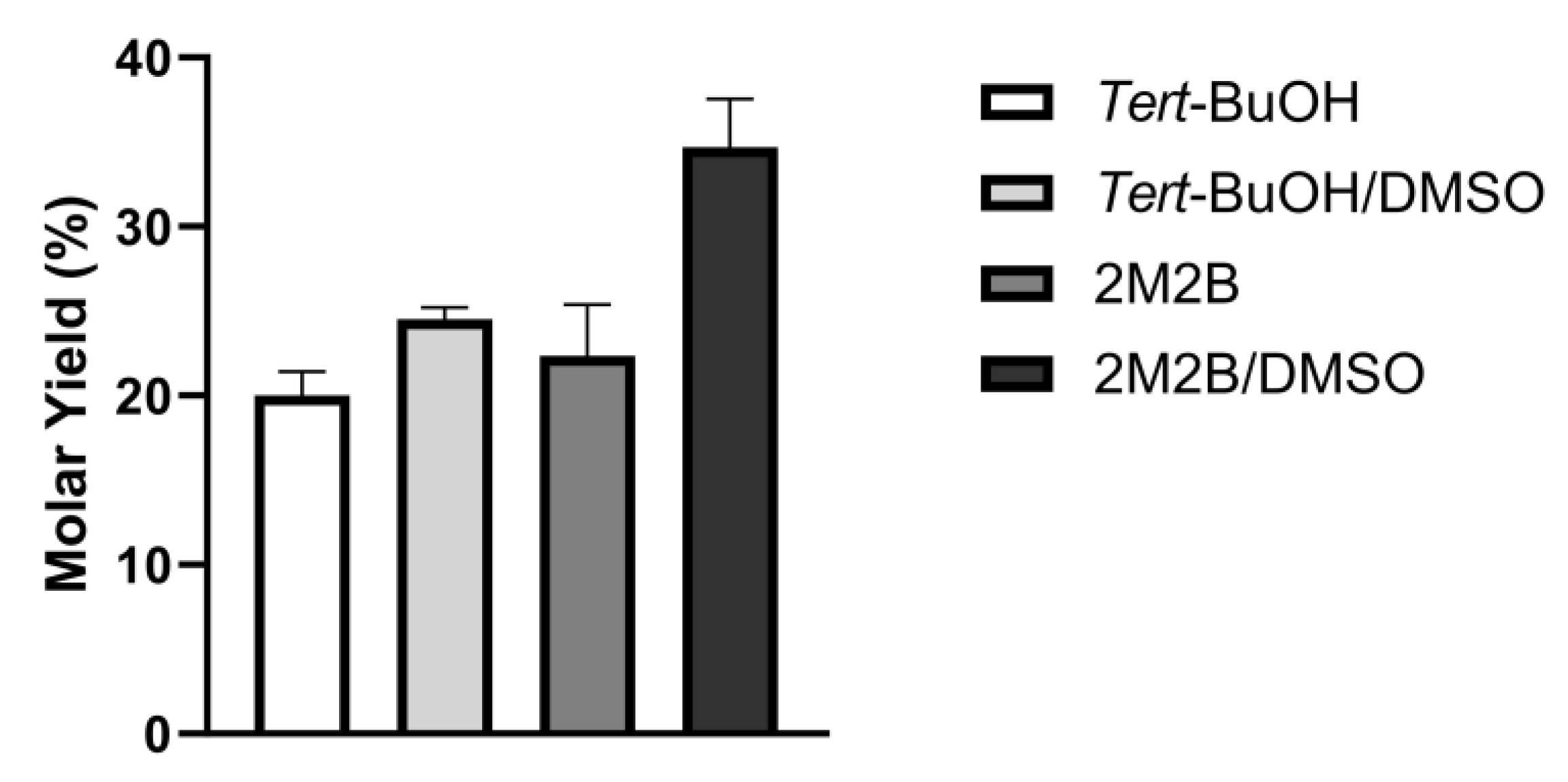
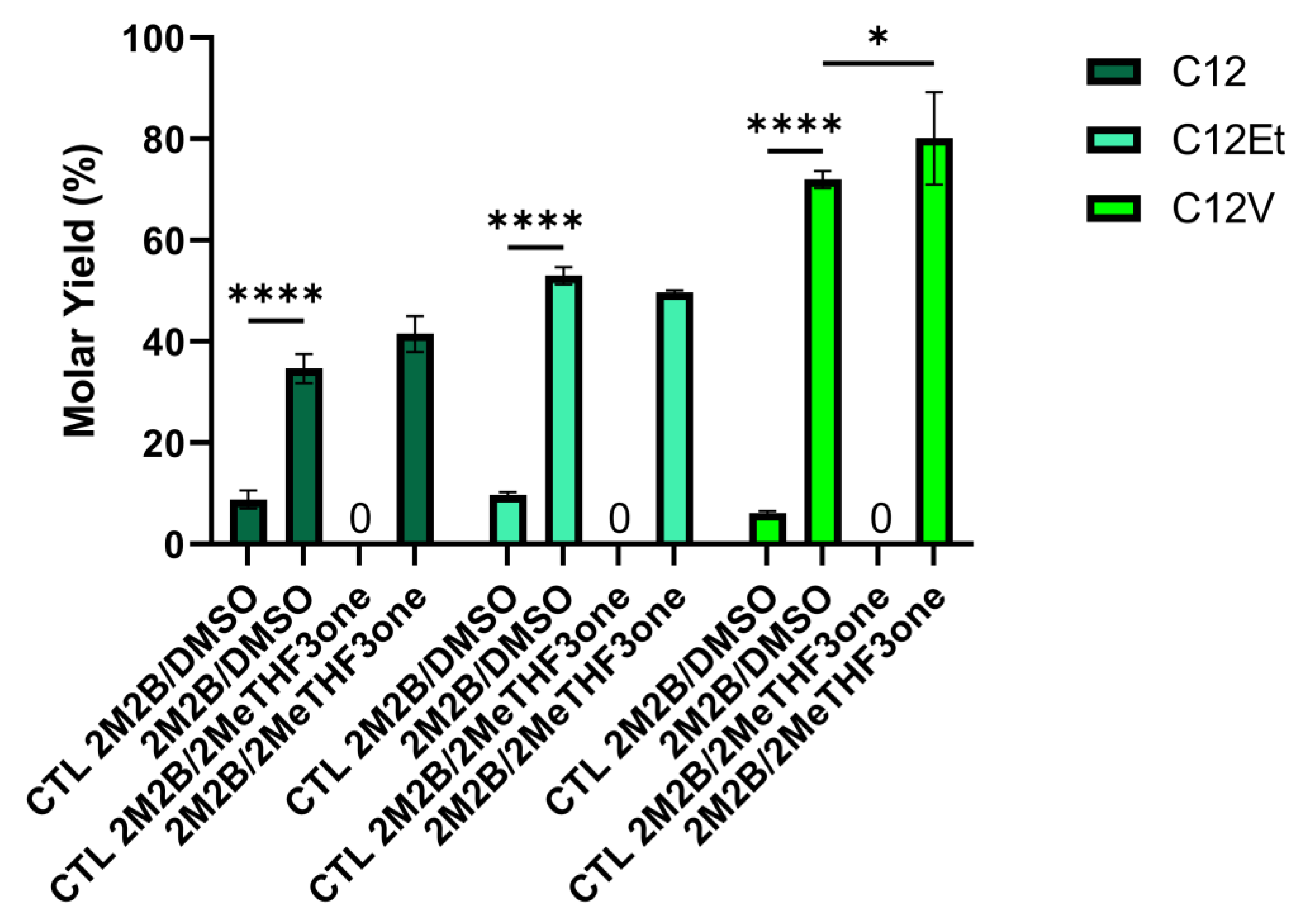
3.3. Greener Co-Solvents
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Deleu, M.; Paquot, M. From Renewable Vegetables Resources to Microorganisms: New Trends in Surfactants. Comptes Rendus Chim. 2004, 7, 641–646. [Google Scholar] [CrossRef]
- Farias, C.B.B.; Almeida, F.C.G.; Silva, I.A.; Souza, T.C.; Meira, H.M.; Soares da Silva, R.D.C.F.; Luna, J.M.; Santos, V.A.; Converti, A.; Banat, I.M.; et al. Production of Green Surfactants: Market Prospects. Electron. J. Biotechnol. 2021, 51, 28–39. [Google Scholar] [CrossRef]
- Puterka, G.J.; Farone, W.; Palmer, T.; Barrington, A. Structure-Function Relationships Affecting the Insecticidal and Miticidal Activity of Sugar Esters. J. Econ. Entomol. 2003, 96, 636–644. [Google Scholar] [CrossRef]
- Ferrer, M.; Soliveri, J.; Plou, F.J.; López-Cortés, N.; Reyes-Duarte, D.; Christensen, M.; Copa-Patiño, J.L.; Ballesteros, A. Synthesis of Sugar Esters in Solvent Mixtures by Lipases from Thermomyces Lanuginosus and Candida Antarctica B, and Their Antimicrobial Properties. Enzym. Microb. Technol. 2005, 36, 391–398. [Google Scholar] [CrossRef]
- Nott, K.; Richard, G.; Laurent, P.; Jérôme, C.; Blecker, C.; Wathelet, J.-P.; Paquot, M.; Deleu, M. Enzymatic Synthesis and Surface Properties of Novel Rhamnolipids. Process Biochem. 2013, 48, 133–143. [Google Scholar] [CrossRef]
- Gloster, T.M. Exploitation of Carbohydrate Processing Enzymes in Biocatalysis. Curr. Opin. Chem. Biol. 2020, 55, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Pyser, J.B.; Chakrabarty, S.; Romero, E.O.; Narayan, A.R.H. State-of-the-Art Biocatalysis. ACS Cent. Sci. 2021, 7, 1105–1116. [Google Scholar] [CrossRef]
- Wu, S.; Snajdrova, R.; Moore, J.C.; Baldenius, K.; Bornscheuer, U.T. Biocatalysis: Enzymatic Synthesis for Industrial Applications. Angew. Chem. Int. Ed. 2021, 60, 88–119. [Google Scholar] [CrossRef]
- Nikulin, M.; Švedas, V. Prospects of Using Biocatalysis for the Synthesis and Modification of Polymers. Molecules 2021, 26, 2750. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Brady, D. Green Chemistry, Biocatalysis, and the Chemical Industry of the Future. ChemSusChem 2022, 15, e202102628. [Google Scholar] [CrossRef]
- Perugino, G. Oligosaccharide Synthesis by Glycosynthases. Trends Biotechnol. 2004, 22, 31–37. [Google Scholar] [CrossRef]
- Chandra, P.; Enespa; Singh, R.; Arora, P.K. Microbial Lipases and Their Industrial Applications: A Comprehensive Review. Microb. Cell Factories 2020, 19, 169. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Perez, S.; Turati, D.F.M.; Borges, J.P.; Luna, P.; Señorans, F.J.; Guisan, J.M.; Fernandez-Lorente, G. Critical Role of Different Immobilized Biocatalysts of a Given Lipase in the Selective Ethanolysis of Sardine Oil. J. Agric. Food Chem. 2017, 65, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, D.P.; Cameotra, S.S. Biosurfactants in Agriculture. Appl. Microbiol. Biotechnol. 2013, 97, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- Stergiou, P.-Y.; Foukis, A.; Filippou, M.; Koukouritaki, M.; Parapouli, M.; Theodorou, L.G.; Hatziloukas, E.; Afendra, A.; Pandey, A.; Papamichael, E.M. Advances in Lipase-Catalyzed Esterification Reactions. Biotechnol. Adv. 2013, 31, 1846–1859. [Google Scholar] [CrossRef]
- Okumura, S.; Iwai, M.; Tominaga, Y. Synthesis of Ester Oligomer by Aspergillus Niger Lipase. Agric. Biol. Chem. 1984, 48, 2805–2808. [Google Scholar] [CrossRef]
- Romero, C.M.; Pera, L.M.; Loto, F.; Vallejos, C.; Castro, G.; Baigori, M.D. Purification of an Organic Solvent-Tolerant Lipase from Aspergillus Niger MYA 135 and Its Application in Ester Synthesis. Biocatal. Agric. Biotechnol. 2012, 1, 25–31. [Google Scholar] [CrossRef]
- Lin, X.-S.; Zhao, K.-H.; Zhou, Q.-L.; Xie, K.-Q.; Halling, P.J.; Yang, Z. Aspergillus Oryzae Lipase-Catalyzed Synthesis of Glucose Laurate with Excellent Productivity. Bioresour. Bioprocess. 2016, 3, 2. [Google Scholar] [CrossRef]
- Ren, K.; Lamsal, B.P. Synthesis of Some Glucose-Fatty Acid Esters by Lipase from Candida Antarctica and Their Emulsion Functions. Food Chem. 2017, 214, 556–563. [Google Scholar] [CrossRef]
- Bezbradica, D.; Mijin, D.; Šiler-Marinković, S.; Knežević, Z. The Effect of Substrate Polarity on the Lipase-Catalyzed Synthesis of Aroma Esters in Solvent-Free Systems. J. Mol. Catal. B Enzym. 2007, 45, 97–101. [Google Scholar] [CrossRef]
- Chamouleau, F.; Coulon, D.; Girardin, M.; Ghoul, M. Influence of Water Activity and Water Content on Sugar Esters Lipase-Catalyzed Synthesis in Organic Media. J. Mol. Catal. B Enzym. 2001, 11, 949–954. [Google Scholar] [CrossRef]
- An, D.; Zhang, X.; Liang, F.; Xian, M.; Feng, D.; Ye, Z. Synthesis, Surface Properties of Glucosyl Esters from Renewable Materials for Use as Biosurfactants. Colloids Surf. A Physicochem. Eng. Asp. 2019, 577, 257–264. [Google Scholar] [CrossRef]
- Zago, E.; Joly, N.; Chaveriat, L.; Lequart, V.; Martin, P. Enzymatic Synthesis of Amphiphilic Carbohydrate Esters: Influence of Physicochemical and Biochemical Parameters. Biotechnol. Rep. 2021, 30, e00631. [Google Scholar] [CrossRef]
- AlFindee, M.N.; Zhang, Q.; Subedi, Y.P.; Shrestha, J.P.; Kawasaki, Y.; Grilley, M.; Takemoto, J.Y.; Chang, C.-W.T. One-Step Synthesis of Carbohydrate Esters as Antibacterial and Antifungal Agents. Bioorganic Med. Chem. 2018, 26, 765–774. [Google Scholar] [CrossRef]
- Jia, C.; Zhao, J.; Feng, B.; Zhang, X.; Xia, W. A Simple Approach for the Selective Enzymatic Synthesis of Dilauroyl Maltose in Organic Media. J. Mol. Catal. B Enzym. 2010, 62, 265–269. [Google Scholar] [CrossRef]
- Ji, Q.; Wang, B.; Tan, J.; Zhu, L.; Li, L. Immobilized Multienzymatic Systems for Catalysis of Cascade Reactions. Process Biochem. 2016, 51, 1193–1203. [Google Scholar] [CrossRef]
- Degn, P.; Pedersen, L.H.; Duus, J.Ø.; Zimmermann, W. Lipase-Catalysed Synthesis of Glucose Fatty Acid Esters in Tert-Butanol. Biotechnol. Lett. 1999, 21, 275–280. [Google Scholar] [CrossRef]
- Ljunger, G.; Adlercreutz, P.; Mattiasson, B. Lipase Catalyzed Acylation of Glucose. Biotechnol. Lett. 1994, 16, 1167–1172. [Google Scholar] [CrossRef]
- Pappalardo, V.M.; Boeriu, C.G.; Zaccheria, F.; Ravasio, N. Synthesis and Characterization of Arabinose-Palmitic Acid Esters by Enzymatic Esterification. Mol. Catal. 2017, 433, 383–390. [Google Scholar] [CrossRef]
- Villeneuve, P. Lipases in Lipophilization Reactions. Biotechnol. Adv. 2007, 25, 515–536. [Google Scholar] [CrossRef]
- Reyes-Duarte, D.; Polaina, J.; López-Cortés, N.; Alcalde, M.; Plou, F.J.; Elborough, K.; Ballesteros, A.; Timmis, K.N.; Golyshin, P.N.; Ferrer, M. Conversion of a Carboxylesterase into a Triacylglycerol Lipase by a Random Mutation. Angew. Chem. (Int. Ed. Engl.) 2005, 44, 7553–7557. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Chu, T.; Chu, J.; Gao, B.; He, B. A Versatile Approach for Enzyme Immobilization Using Chemically Modified 3D-Printed Scaffolds. ACS Sustain. Chem. Eng. 2019, 7, 18048–18054. [Google Scholar] [CrossRef]
- Vuillemin, M.E.; Husson, E.; Laclef, S.; Jamali, A.; Lambertyn, V.; Pilard, S.; Cailleu, D.; Sarazin, C. Improving the Environmental Compatibility of Enzymatic Synthesis of Sugar-Based Surfactants Using Green Reaction Media. Process Biochem. 2022, 117, 30–41. [Google Scholar] [CrossRef]
- Shin, D.W.; Mai, N.L.; Bae, S.-W.; Koo, Y.-M. Enhanced Lipase-Catalyzed Synthesis of Sugar Fatty Acid Esters Using Supersaturated Sugar Solution in Ionic Liquids. Enzym. Microb. Technol. 2019, 126, 18–23. [Google Scholar] [CrossRef]
- Van den Broek, L.A.M.; Boeriu, C.G. Enzymatic Synthesis of Oligo- and Polysaccharide Fatty Acid Esters. Carbohydr. Polym. 2013, 93, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Delavault, A.; Grüninger, J.; Kapp, D.; Hollenbach, R.; Rudat, J.; Ochsenreither, K.; Syldatk, C. Enzymatic Synthesis of Alkyl Glucosides by β-Glucosidases in a 2-in-1 Deep Eutectic Solvent System. Chem. Ing. Tech. 2022, 94, 417–426. [Google Scholar] [CrossRef]
- Giorgi, V.; Botto, E.; Fontana, C.; Della Mea, L.; Vaz, S.; Menéndez, P.; Rodríguez, P. Enzymatic Production of Lauroyl and Stearoyl Monoesters of D-Xylose, l-Arabinose, and d-Glucose as Potential Lignocellulosic-Derived Products, and Their Evaluation as Antimicrobial Agents. Catalysts 2022, 12, 610. [Google Scholar] [CrossRef]
- Campana, R.; Merli, A.; Verboni, M.; Biondo, F.; Favi, G.; Duranti, A.; Lucarini, S. Synthesis and Evaluation of Saccharide-Based Aliphatic and Aromatic Esters as Antimicrobial and Antibiofilm Agents. Pharmaceuticals 2019, 12, 186. [Google Scholar] [CrossRef]
- Platel, R.; Chaveriat, L.; Le Guenic, S.; Pipeleers, R.; Magnin-Robert, M.; Randoux, B.; Trapet, P.; Lequart, V.; Joly, N.; Halama, P.; et al. Importance of the C12 Carbon Chain in the Biological Activity of Rhamnolipids Conferring Protection in Wheat against Zymoseptoria tritici. Molecules 2020, 26, 40. [Google Scholar] [CrossRef]
- Gonçalves, M.C.P.; Amaral, J.C.; Fernandez-Lafuente, R.; de Sousa Junior, R.; Tardioli, P.W. Lipozyme 435-Mediated Synthesis of Xylose Oleate in Methyl Ethyl Ketone. Molecules 2021, 26, 3317. [Google Scholar] [CrossRef]
- Wang, H.; Hu, Y.; Pan, J.; Yu, D. Arabidopsis VQ Motif-Containing Proteins VQ12 and VQ29 Negatively Modulate Basal Defense against Botrytis Cinerea. Sci. Rep. 2015, 5, 14185. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C.; Leo, A.; Hoekman, D.H. Exploring QSAR: Hydrophobic, Electronic, and Steric Constants; American Chemical Society: Washington, DC, USA, 1995; Volume 2, ISBN 978-0-8412-2991-4. [Google Scholar]
- Hsieh, S.-W.; Lee, M.-R.; Tsai, C.-W.; Lai, L.-Y.; Yeh, T.; Hsieh, C.-W.; Yang, T.-J.; Chang, S.-W. Enzymatic Synthesis, Purification and Identification of Bioactive Trehalose Ester Derivatives for Health Applications. Food Bioprod. Process. 2015, 95, 163–172. [Google Scholar] [CrossRef]
- Méline, T.; Muzard, M.; Deleu, M.; Rakotoarivonina, H.; Plantier-Royon, R.; Rémond, C. D-Xylose and L-Arabinose Laurate Esters: Enzymatic Synthesis, Characterization and Physico-Chemical Properties. Enzym. Microb. Technol. 2018, 112, 14–21. [Google Scholar] [CrossRef]
- Borges, M.R.; dos Santos, J.A.; Vieira, M.; Balaban, R. Polymerization of a Water Soluble Glucose Vinyl Ester Monomer with Tensoactive Properties Synthesized by Enzymatic Catalyst. Mater. Sci. Eng. C 2009, 29, 519–523. [Google Scholar] [CrossRef]
- Arcens, D.; Grau, E.; Grelier, S.; Cramail, H.; Peruch, F. 6-O-Glucose Palmitate Synthesis with Lipase: Investigation of Some Key Parameters. Mol. Catal. 2018, 460, 63–68. [Google Scholar] [CrossRef]
- Plat, T.; Linhardt, R.J. Syntheses and Applications of Sucrose-Based Esters. J. Surfactants Deterg. 2001, 4, 415–421. [Google Scholar] [CrossRef]
- Çetinkaya, S.; Yenidünya, A.F.; Başoğlu, F.; Polat, Z.A.; Savaş, S. Esterification of Fructose-Oleic Acid by Tert-Butanol/Dimethyl Sulfoxide and by 2-Methyl-2-Butanol/Dimethyl Sulfoxide. J. Oleo Sci. 2020, 69, 1281–1285. [Google Scholar] [CrossRef]
- Çetinkaya, S.; Yenidünya, A.F.; Başoğlu, F.; Saraç, K. D-Glucose-Fatty Acid Ester Synthesis with or without a Biocatalyst in the Same Organic Media. J. Oleo Sci. 2020, 69, 737–742. [Google Scholar] [CrossRef]
- Maurya, D.S.; Adamson, J.; Bensabeh, N.; Lligadas, G.; Percec, V. Catalytic Effect of DMSO in Metal-Catalyzed Radical Polymerization Mediated by Disproportionation Facilitates Living and Immortal Radical Polymerizations. J. Polym. Sci. 2023, 61, 959–978. [Google Scholar] [CrossRef]
- Giumanini, A.G.; Savoia, D. Disproportionation of 2-Iodothiophene in Dimethyl Sulfoxide. Available online: https://pubs.acs.org/doi/pdf/10.1021/jo00968a049 (accessed on 17 August 2023).
- Lei, P.; Wang, Y.; Mu, Y.; Wang, Y.; Ma, Z.; Feng, J.; Liu, X.; Szostak, M. Green-Solvent Selection for Acyl Buchwald–Hartwig Cross-Coupling of Amides (Transamidation). ACS Sustain. Chem. Eng. 2021, 9, 14937–14945. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DMSO 20% (v/v) | ||
|---|---|---|
| − | + | |
| tert-BuOH | 1.8 | 5.73 |
| 2M2B | 2.1 | 6.56 |
| Fatty Reagent | DMSO * | Yield before Treatment % | Yield after Treatment % |
|---|---|---|---|
| Lauric Acid C12 | − | 24 | 23 |
| + | 54 | 33 | |
| Ethyl Laurate C12Et | − | 47 | 44 |
| + | 66 | 52 | |
| Vinyl Laurate C12V | − | 49 | 49 |
| + | 93 | 71 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spalletta, A.; Joly, N.; Martin, P. Optimization of Enzymatic Synthesis of D-Glucose-Based Surfactants Using Supported Aspergillus niger Lipase as Biocatalyst. Chemistry 2023, 5, 1855-1869. https://doi.org/10.3390/chemistry5030127
Spalletta A, Joly N, Martin P. Optimization of Enzymatic Synthesis of D-Glucose-Based Surfactants Using Supported Aspergillus niger Lipase as Biocatalyst. Chemistry. 2023; 5(3):1855-1869. https://doi.org/10.3390/chemistry5030127
Chicago/Turabian StyleSpalletta, Alexis, Nicolas Joly, and Patrick Martin. 2023. "Optimization of Enzymatic Synthesis of D-Glucose-Based Surfactants Using Supported Aspergillus niger Lipase as Biocatalyst" Chemistry 5, no. 3: 1855-1869. https://doi.org/10.3390/chemistry5030127
APA StyleSpalletta, A., Joly, N., & Martin, P. (2023). Optimization of Enzymatic Synthesis of D-Glucose-Based Surfactants Using Supported Aspergillus niger Lipase as Biocatalyst. Chemistry, 5(3), 1855-1869. https://doi.org/10.3390/chemistry5030127