
Aptamer-Based Immune Drug Systems (AptIDCs) Potentiating Cancer Immunotherapy




Abstract
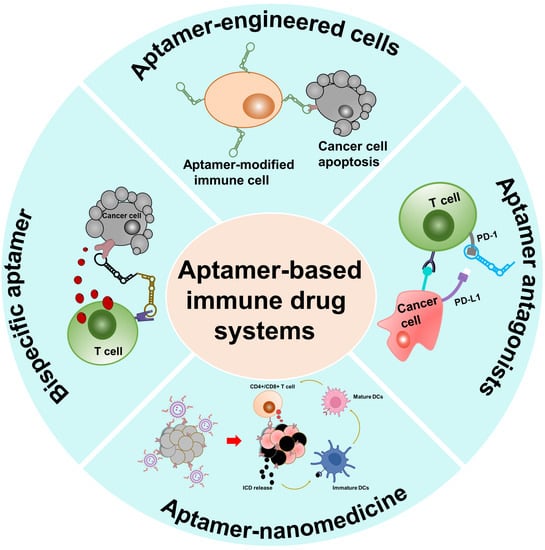
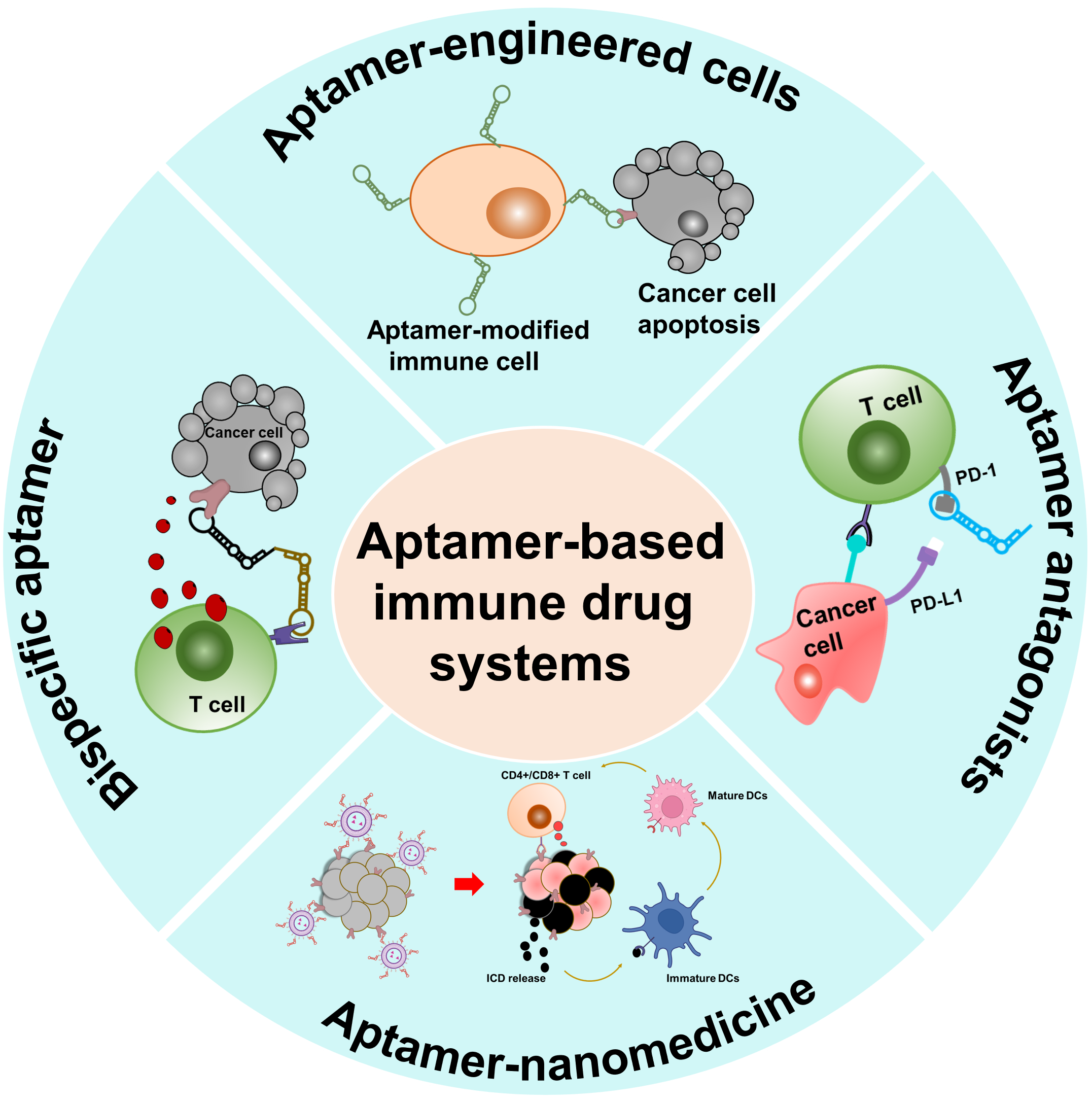
1. Introduction
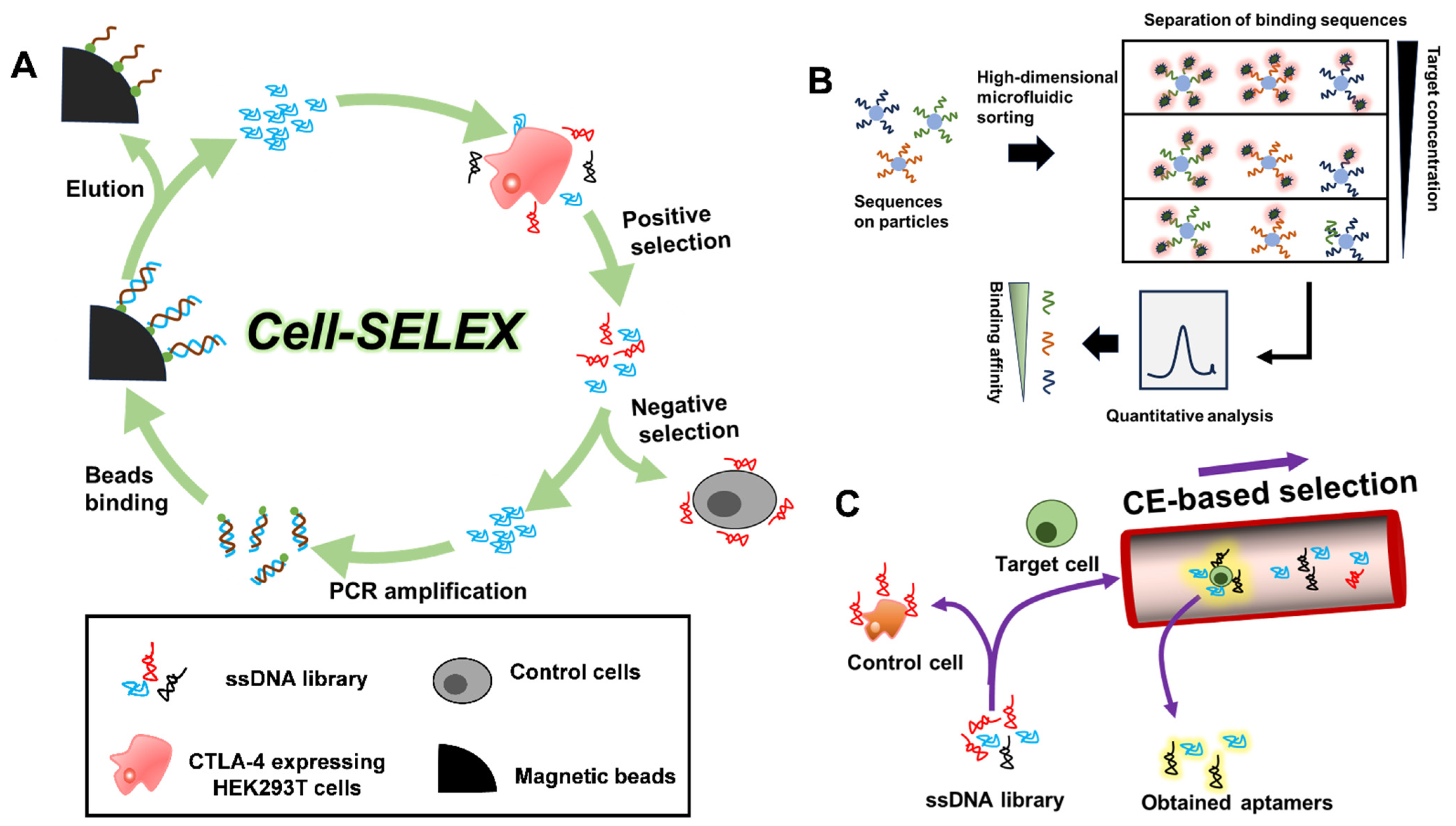
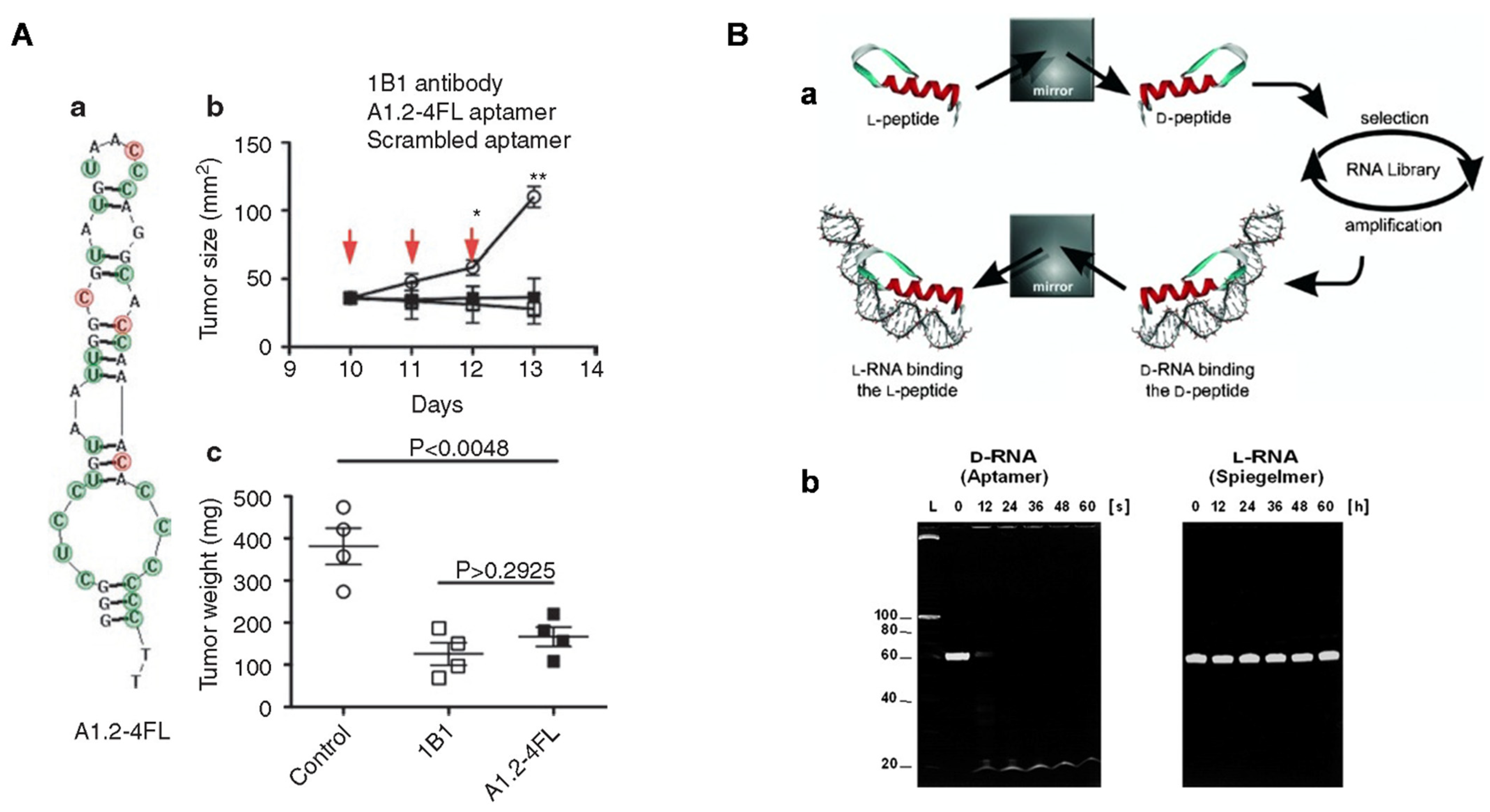
2. Aptamer Selection

3. AptIDCs for Enhanced Cancer Immunotherapy
3.1. Aptamer Agonists/Antagonists
3.1.1. Immune Checkpoints
3.1.2. Costimulatory Molecules
3.1.3. Immune Cytokines
3.2. Multivalent Bispecific Aptamers
3.2.1. The [1+1] bsApts

3.2.2. The [1+2] bsApts
3.2.3. The [2+2] bsApts
3.3. Aptamer–Nanoparticle Conjugates (AptNCs)
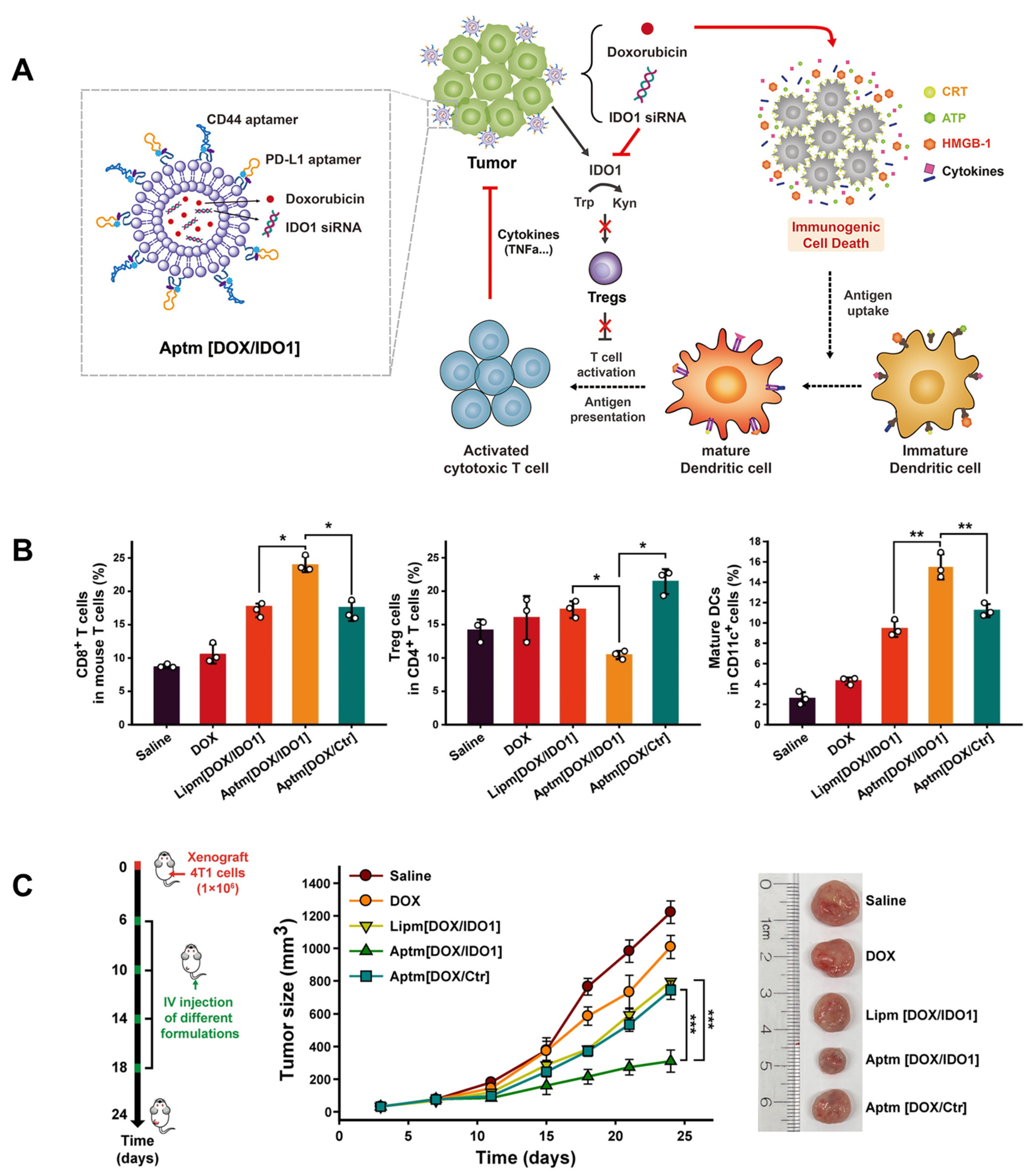
3.3.1. Aptamer–Liposome Conjugates
Direct Conjugation Strategy
Pre-Conjugation Strategy

Post-Insertion Strategy
3.3.2. Aptamer–Hydrogel Conjugates
DNA/RNA Hydrogels
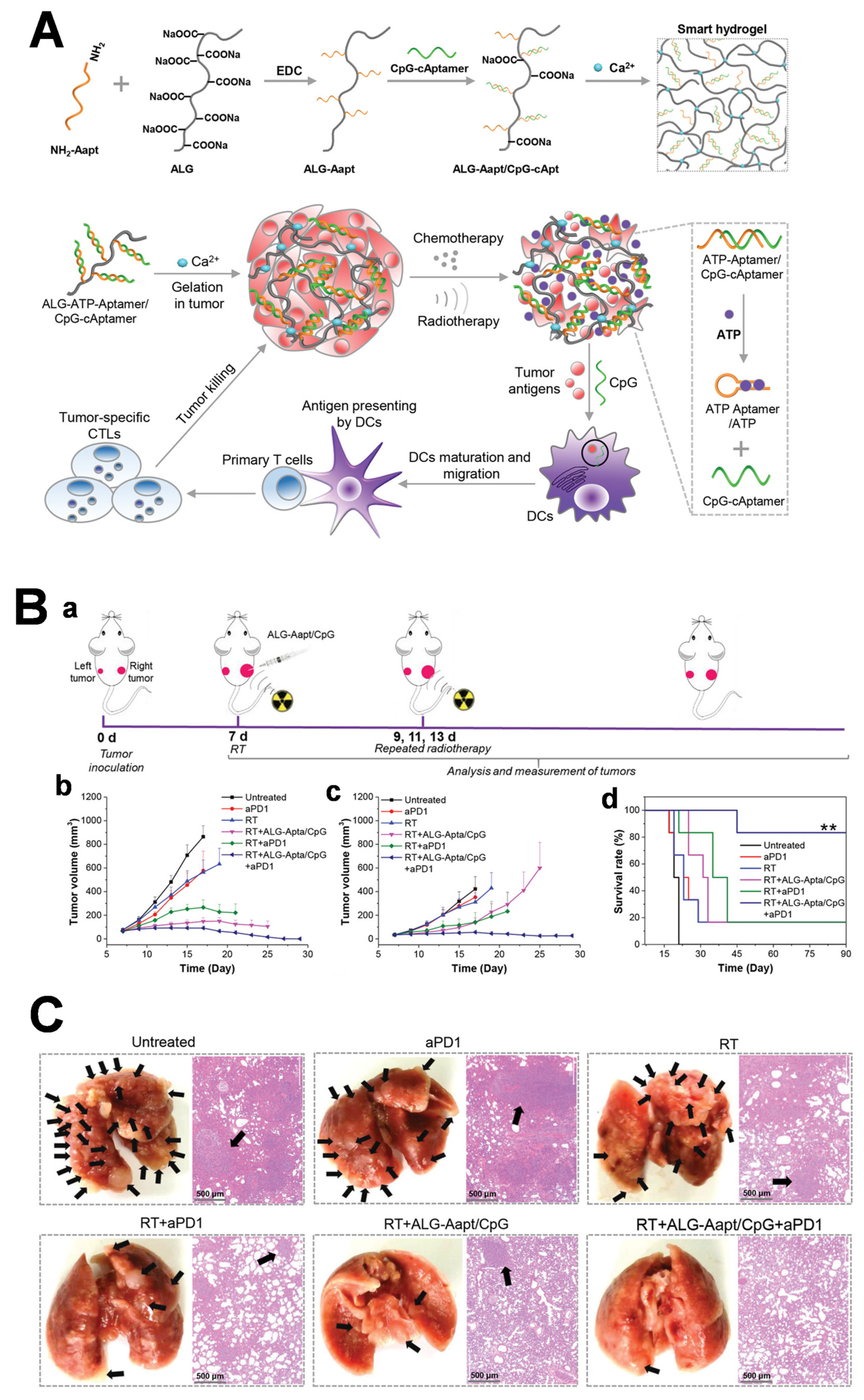
Alginate Hydrogels
3.4. Aptamer-Engineered Immune Cells
3.4.1. Aptamer–NK Cell Assemblies
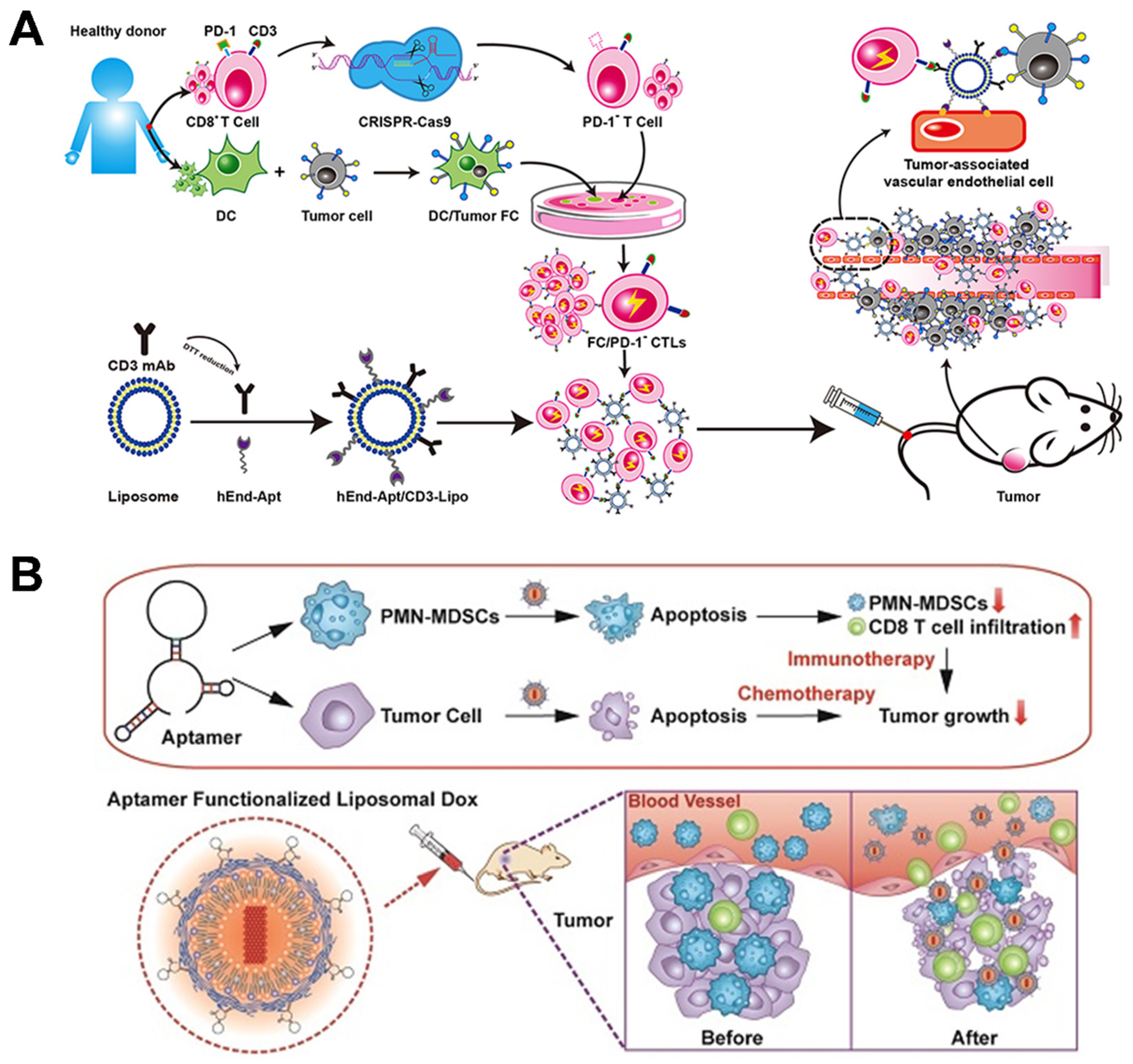
3.4.2. Tunable Aptamer–Antibody Nano-Assembly for Precise T Cell Immunotherapy
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Varadé, J.; Magadán, S.; González-Fernández, Á. Human immunology and immunotherapy: Main achievements and challenges. Cell. Mol. Immunol. 2021, 18, 805–828. [Google Scholar] [CrossRef]
- Kimiz-Gebologlu, I.; Gulce-Iz, S.; Biray-Avci, C. Monoclonal antibodies in cancer immunotherapy. Mol. Biol. Rep. 2018, 45, 2935–2940. [Google Scholar] [CrossRef]
- Lote, H.; Chau, I. Emerging HER2-directed therapeutic agents for gastric cancer in early phase clinical trials. Expert Opin. Investig. Drugs 2022, 31, 59–78. [Google Scholar] [CrossRef]
- Zhu, G.; Niu, G.; Chen, X. Aptamer-drug conjugates. Bioconjugate Chem. 2015, 26, 2186–2197. [Google Scholar] [CrossRef]
- Pastor, F. Aptamers: A new technological platform in cancer immunotherapy. Pharmaceuticals 2016, 9, 64. [Google Scholar] [CrossRef]
- Kalaora, S.; Nagler, A.; Wargo, J.A.; Samuels, Y. Mechanisms of immune activation and regulation: Lessons from melanoma. Nat. Rev. Cancer 2022, 22, 195–207. [Google Scholar] [CrossRef]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef]
- Xuan, W.; Peng, Y.; Deng, Z.; Peng, T.; Kuai, H.; Li, Y.; He, J.; Jin, C.; Liu, Y.; Wang, R.; et al. A basic insight into aptamer-drug conjugates (ApDCs). Biomaterials 2018, 182, 216–226. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, X. Investigations upon the bioconjugation-based construction technologies and applications of aptamer-drug conjugates. Chem. J. Chin. Univ. Chin. 2021, 42, 3367–3378. [Google Scholar]
- Xiang, D.; Zheng, C.; Zhou, S.-F.; Qiao, S.; Tran, P.H.L.; Pu, C.; Li, Y.; Kong, L.; Kouzani, A.Z.; Lin, J.; et al. Superior performance of aptamer in tumor penetration over antibody: Implication of aptamer-based theranostics in solid tumors. Theranostics 2015, 5, 1083–1097. [Google Scholar] [CrossRef]
- Prodeus, A.; Abdul-Wahid, A.; Fischer, N.W.; Huang, E.H.B.; Cydzik, M.; Gariépy, J. Targeting the PD-1/PD-L1 immune evasion axis with DNA aptamers as a novel therapeutic strategy for the treatment of disseminated cancers. Mol. Ther. Nucleic Acids 2015, 4, e237. [Google Scholar] [CrossRef]
- Pastor, F.; Soldevilla, M.M.; Villanueva, H.; Kolonias, D.; Inoges, S.; de Cerio, A.L.; Kandzia, R.; Klimyuk, V.; Gleba, Y.; Gilboa, E.; et al. CD28 aptamers as powerful immune response modulators. Mol. Ther. Nucleic Acids 2013, 2, e98. [Google Scholar] [CrossRef]
- Sung, H.J.; Choi, S.; Lee, J.W.; Ok, C.Y.; Bae, Y.S.; Kim, Y.H.; Lee, W.; Heo, K.; Kim, I.H. Inhibition of human neutrophil activity by an RNA aptamer bound to interleukin-8. Biomaterials 2014, 35, 578–589. [Google Scholar] [CrossRef]
- Xiong, H.; Liu, L.; Wang, Y.; Jiang, H.; Wang, X. Engineered aptamer-organic amphiphile self-assemblies for biomedical applications: Progress and challenges. Small 2022, 18, 2104341. [Google Scholar] [CrossRef]
- Tian, L.; Shao, M.; Gong, Y.; Chao, Y.; Wei, T.; Yang, K.; Chen, Q.; Liu, Z. Albumin-binding lipid-aptamer conjugates for cancer immunoimaging and immunotherapy. Sci. China Chem. 2022, 65, 574–583. [Google Scholar] [CrossRef]
- Gong, H.; Dai, Q.; Peng, P. Cell-Membrane-Anchored DNA Logic-Gated Nanoassemblies for In Situ Extracellular Bioimaging. ACS Appl. Mater. Interfaces 2022, 14, 43026–43034. [Google Scholar] [CrossRef]
- Song, W.; Hu, J.J.; Song, S.J.; Xu, Y.; Yang, H.; Yang, F.; Zhou, Y.; Yu, T.; Qiu, W.X. Aptamer-gold nanocage composite for photoactivated immunotherapy. Acs Appl. Mater. Interfaces 2022, 14, 42931–42939. [Google Scholar] [CrossRef]
- Geng, Z.; Wang, L.; Liu, K.; Liu, J.; Tan, W. Enhancing anti-PD-1 immunotherapy by nanomicelles self-assembled from multivalent aptamer drug conjugates. Angew. Chem. Int. Ed. 2021, 60, 15459–15465. [Google Scholar] [CrossRef]
- Sun, H.; Zhu, X.; Lu, P.Y.; Rosato, R.R.; Tan, W.; Zu, Y. Oligonucleotide aptamers: New tools for targeted cancer therapy. Mol. Ther. Nucleic Acids 2014, 3, e182. [Google Scholar] [CrossRef]
- Santulli-Marotto, S.; Nair, S.K.; Rusconi, C.; Sullenger, B.; Gilboa, E. Multivalent RNA aptamers that inhibit CTLA-4 and enhance tumor immunity. Cancer Res. 2003, 63, 7483–7489. [Google Scholar]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef]
- Li, L.; Xu, S.; Yan, H.; Li, X.; Yazd, H.S.; Li, X.; Huang, T.; Cui, C.; Jiang, J.; Tan, W. Nucleic acid aptamers for molecular diagnostics and therapeutics: Advances and perspectives. Angew. Chem. (Int. Ed. Engl.) 2021, 60, 2221–2231. [Google Scholar] [CrossRef]
- Xiong, H.; Yan, J.; Cai, S.; He, Q.; Peng, D.; Liu, Z.; Liu, Y. Cancer protein biomarker discovery based on nucleic acid aptamers. Int. J. Biol. Macromol. 2019, 132, 190–202. [Google Scholar] [CrossRef]
- Huang, B.T.; Lai, W.Y.; Chang, Y.C.; Wang, J.W.; Yeh, S.D.; Lin, E.P.; Yang, P.C. A CTLA-4 antagonizing DNA aptamer with antitumor effect. Mol. Ther. Nucleic Acids 2017, 8, 520–528. [Google Scholar] [CrossRef]
- Yan, J.; Xiong, H.; Cai, S.; Wen, N.; He, Q.; Liu, Y.; Peng, D.; Liu, Z. Advances in aptamer screening technologies. Talanta 2019, 200, 124–144. [Google Scholar] [CrossRef]
- Lai, H.C.; Wang, C.H.; Liou, T.M.; Lee, G.B. Influenza A virus-specific aptamers screened by using an integrated microfluidic system. Lab A Chip 2014, 14, 2002–2013. [Google Scholar] [CrossRef]
- Mosing, R.K.; Bowser, M.T. Isolating aptamers using capillary electrophoresis-SELEX (CE-SELEX). Methods Mol. Biol. 2009, 535, 33–43. [Google Scholar]
- Duan, N.; Gong, W.; Wu, S.; Wang, Z. An ssDNA library immobilized SELEX technique for selection of an aptamer against ractopamine. Anal. Chim. Acta 2017, 961, 100–105. [Google Scholar] [CrossRef]
- Yang, J.; Bowser, M.T. Capillary Electrophoresis–SELEX selection of catalytic DNA aptamers for a small-molecule porphyrin target. Anal. Chem. 2013, 85, 1525–1530. [Google Scholar] [CrossRef]
- Chang, D.; Wang, Z.; Flynn, C.D.; Mahmud, A.; Labib, M.; Wang, H.; Geraili, A.; Li, X.; Zhang, J.; Sargent, E.H.; et al. A high-dimensional microfluidic approach for selection of aptamers with programmable binding affinities. Nat. Chem. 2023, 15, 773–780. [Google Scholar] [CrossRef]
- Hirose, K.; Tsuchida, M.; Asakura, H.; Wakui, K.; Yoshimoto, K.; Iida, K.; Sato, M.; Shibukawa, M.; Suganuma, M.; Saito, S. A single-round selection of selective DNA aptamers for mammalian cells by polymer-enhanced capillary transient isotachophoresis. Analyst 2017, 142, 4030–4038. [Google Scholar] [CrossRef] [PubMed]
- Mehan, M.R.; Ostroff, R.; Wilcox, S.K.; Steele, F.; Schneider, D.; Jarvis, T.C.; Baird, G.S.; Gold, L.; Janjic, N. Highly multiplexed proteomic platform for biomarker discovery, diagnostics, and therapeutics. Adv. Exp. Med. Biol. 2013, 735, 283–300. [Google Scholar]
- Wang, D.R.; Wu, X.L.; Sun, Y.L. Therapeutic targets and biomarkers of tumor immunotherapy: Response versus non-response. Signal Transduct. Target. Ther. 2022, 7, 331. [Google Scholar]
- Lv, B.; Wang, Y.; Ma, D.; Cheng, W.; Liu, J.; Yong, T.; Chen, H.; Wang, C. Immunotherapy: Reshape the tumor immune microenvironment. Front. Immunol. 2022, 13, 844142. [Google Scholar] [CrossRef]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef]
- Kubli, S.P.; Berger, T.; Araujo, D.V.; Siu, L.L.; Mak, T.W. Beyond immune checkpoint blockade: Emerging immunological strategies. Nat. Rev. Drug Discov. 2021, 20, 899–919. [Google Scholar] [CrossRef]
- Gao, T.; Pei, R. Isolation of DNA aptamer targeting PD-1 with an antitumor immunotherapy effect. Acs Appl. Bio Mater. 2020, 3, 7080–7086. [Google Scholar] [CrossRef]
- Lai, W.Y.; Huang, B.T.; Wang, J.W.; Lin, P.Y.; Yang, P.C. A novel PD-L1-targeting antagonistic DNA aptamer with antitumor effects. Mol. Ther. Nucleic Acids 2016, 5, e397. [Google Scholar] [CrossRef]
- Wang, H.; Lam, C.H.; Li, X.; West, D.L.; Yang, X. Selection of PD1/PD-L1 X-aptamers. Biochimie 2018, 145, 125–130. [Google Scholar] [CrossRef]
- Li, X.; Li, Z.; Yu, H. Selection of threose nucleic acid aptamers to block PD-1/PD-L1 interaction for cancer immunotherapy. Chem. Commun. 2020, 56, 14653–14656. [Google Scholar] [CrossRef] [PubMed]
- Hervas-Stubbs, S.; Soldevilla, M.M.; Villanueva, H.; Mancheño, U.; Bendandi, M.; Pastor, F. Identification of TIM3 2’-fluoro oligonucleotide aptamer by HT-SELEX for cancer immunotherapy. Oncotarget 2016, 7, 4522–4530. [Google Scholar] [CrossRef] [PubMed]
- Soldevilla, M.M.; Hervas, S.; Villanueva, H.; Lozano, T.; Rabal, O.; Oyarzabal, J.; Lasarte, J.J.; Bendandi, M.; Inoges, S.; López-Díaz de Cerio, A.; et al. Identification of LAG3 high affinity aptamers by HT-SELEX and Conserved Motif Accumulation (CMA). PLoS ONE 2017, 12, e0185169. [Google Scholar] [CrossRef] [PubMed]
- Capece, D.; Verzella, D.; Fischietti, M.; Zazzeroni, F.; Alesse, E. Targeting costimulatory molecules to improve antitumor immunity. J. Biomed. Biotechnol. 2012, 926321. [Google Scholar] [CrossRef]
- McNamara, J.O., II; Kolonias, D.; Pastor, F.; Mittler, R.S.; Chen, L.; Giangrande, P.H.; Sullenger, B.; Gilboa, E. Multivalent 4-1BB binding aptamers costimulate CD8+ T cells and inhibit tumor growth in mice. J. Clin. Investig. 2008, 118, 376–386. [Google Scholar] [CrossRef]
- Pastor, F.; Kolonias, D.; Giangrande, P.H.; Gilboa, E. Induction of tumour immunity by targeted inhibition of nonsense-mediated mRNA decay. Nature 2010, 465, 227–230. [Google Scholar] [CrossRef]
- Benaduce, A.P.; Brenneman, R.; Schrand, B.; Pollack, A.; Gilboa, E.; Ishkanian, A. 4-1BB aptamer-based immunomodulation enhances the therapeutic index of radiation therapy in murine tumor models. Int. J. Radiat. Oncol. Biol. Phys. 2016, 96, 458–461. [Google Scholar] [CrossRef]
- Dollins, C.M.; Nair, S.; Boczkowski, D.; Lee, J.; Layzer, J.M.; Gilboa, E.; Sullenger, B.A. Assembling OX40 aptamers on a molecular scaffold to create a receptor-activating aptamer. Chem. Biol. 2008, 15, 675–682. [Google Scholar] [CrossRef]
- Pratico, E.D.; Sullenger, B.A.; Nair, S.K. Identification and characterization of an agonistic aptamer against the t cell costimulatory receptor, OX40. Nucleic Acid Ther. 2013, 23, 35–43. [Google Scholar] [CrossRef]
- Soldevilla, M.M.; Villanueva, H.; Bendandi, M.; Inoges, S.; López-Díaz de Cerio, A.; Pastor, F. 2-fluoro-RNA oligonucleotide CD40 targeted aptamers for the control of B lymphoma and bone-marrow aplasia. Biomaterials 2015, 67, 274–285. [Google Scholar] [CrossRef]
- Atallah-Yunes, S.A.; Robertson, M.J. Cytokine based immunotherapy for cancer and lymphoma: Biology, challenges and future perspectives. Front. Immunol. 2022, 13, 872010. [Google Scholar] [CrossRef]
- Rallis, K.S.; Corrigan, A.E.; Dadah, H.; George, A.M.; Keshwara, S.M.; Sideris, M.; Szabados, B. Cytokine-based cancer immunotherapy: Challenges and opportunities for IL-10. Anticancer Res. 2021, 41, 3247–3252. [Google Scholar] [CrossRef] [PubMed]
- Rallis, K.S.; Corrigan, A.E.; Dadah, H.; Stanislovas, J.; Zamani, P.; Makker, S.; Szabados, B.; Sideris, M. IL-10 in cancer: An essential thermostatic regulator between homeostatic immunity and inflammation—A comprehensive review. Future Oncol. 2022, 18, 3349–3365. [Google Scholar] [CrossRef]
- Ni, G.; Zhang, L.; Yang, X.; Li, H.; Ma, B.; Walton, S.; Wu, X.; Yuan, J.; Wang, T.; Liu, X. Targeting interleukin-10 signalling for cancer immunotherapy, a promising and complicated task. Hum. Vaccines Immunother. 2020, 16, 2328–2332. [Google Scholar] [CrossRef]
- Berezhnoy, A.; Stewart, C.A.; McNamara, J.O., 2nd; Thiel, W.; Giangrande, P.; Trinchieri, G.; Gilboa, E. Isolation and optimization of murine IL-10 receptor blocking oligonucleotide aptamers using high-throughput sequencing. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 1242–1250. [Google Scholar] [CrossRef]
- Burns, J.M.; Summers, B.C.; Wang, Y.; Melikian, A.; Berahovich, R.; Miao, Z.; Penfold, M.E.; Sunshine, M.J.; Littman, D.R.; Kuo, C.J.; et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J. Exp. Med. 2006, 203, 2201–2213. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Luker, K.E.; Summers, B.C.; Berahovich, R.; Bhojani, M.S.; Rehemtulla, A.; Kleer, C.G.; Essner, J.J.; Nasevicius, A.; Luker, G.D.; et al. CXCR7 (RDC1) promotes breast and lung tumor growth in vivo and is expressed on tumor-associated vasculature. Proc. Natl. Acad. Sci. USA 2007, 104, 15735–15740. [Google Scholar] [CrossRef] [PubMed]
- Azab, A.K.; Runnels, J.M.; Pitsillides, C.; Moreau, A.S.; Azab, F.; Leleu, X.; Jia, X.; Wright, R.; Ospina, B.; Carlson, A.L.; et al. CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood 2009, 113, 4341–4351. [Google Scholar] [CrossRef]
- Eulberg, D.; Klussmann, S. Spiegelmers: Biostable aptamers. ChemBioChem 2003, 4, 979–983. [Google Scholar] [CrossRef]
- Hoellenriegel, J.; Zboralski, D.; Maasch, C.; Rosin, N.Y.; Wierda, W.G.; Keating, M.J.; Kruschinski, A.; Burger, J.A. The Spiegelmer NOX-A12, a novel CXCL12 inhibitor, interferes with chronic lymphocytic leukemia cell motility and causes chemosensitization. Blood 2014, 123, 1032–1039. [Google Scholar] [CrossRef]
- Vater, A.; Sahlmann, J.; Kröger, N.; Zöllner, S.; Lioznov, M.; Maasch, C.; Buchner, K.; Vossmeyer, D.; Schwoebel, F.; Purschke, W.G.; et al. Hematopoietic stem and progenitor cell mobilization in mice and humans by a first-in-class mirror-image oligonucleotide inhibitor of CXCL12. Clin. Pharmacol. Ther. 2013, 94, 150–157. [Google Scholar] [CrossRef]
- Shigdar, S.; Schrand, B.; Giangrande, P.H.; de Franciscis, V. Aptamers: Cutting edge of cancer therapies. Mol. Ther. 2021, 29, 2396–2411. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Lee, M.S.; Copland, J.A.; Luxon, B.A.; Gorenstein, D.G. Combinatorial selection of a single stranded DNA thioaptamer targeting TGF-β1 protein. Bioorganic Med. Chem. Lett. 2008, 18, 1835–1839. [Google Scholar] [CrossRef] [PubMed]
- Marro, M.L.; Daniels, D.A.; McNamee, A.; Andrew, D.P.; Chapman, T.D.; Jiang, M.S.; Wu, Z.; Smith, J.L.; Patel, K.K.; Gearing, K.L. Identification of potent and selective RNA antagonists of the IFN-γ-inducible CXCL10 chemokine. Biochemistry 2005, 44, 8449–8460. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.J.; Porciani, D.; Burke, D.H. Cancer immunomodulation using bispecific aptamers. Mol. Ther. Nucleic Acids 2022, 27, 894–915. [Google Scholar] [CrossRef]
- Vandghanooni, S.; Eskandani, M.; Barar, J.; Omidi, Y. Bispecific therapeutic aptamers for targeted therapy of cancer: A review on cellular perspective. J. Mol. Med. 2018, 96, 885–902. [Google Scholar] [CrossRef] [PubMed]
- Boltz, A.; Piater, B.; Toleikis, L.; Guenther, R.; Kolmar, H.; Hock, B. Bi-specific aptamers mediating tumor cell lysis. J. Biol. Chem. 2011, 286, 21896–21905. [Google Scholar] [CrossRef]
- Yang, Y.; Sun, X.; Xu, J.; Cui, C.; Safari Yazd, H.; Pan, X.; Zhu, Y.; Chen, X.; Li, X.; Li, J.; et al. Circular bispecific aptamer-mediated artificial intercellular recognition for targeted t cell immunotherapy. ACS Nano 2020, 14, 9562–9571. [Google Scholar] [CrossRef]
- Kuai, H.; Zhao, Z.; Mo, L.; Liu, H.; Hu, X.; Fu, T.; Zhang, X.; Tan, W. Circular bivalent aptamers enable in vivo stability and recognition. J. Am. Chem. Soc. 2017, 139, 9128–9131. [Google Scholar] [CrossRef]
- Du, Y.; Zhang, D.; Wang, Y.; Wu, M.; Zhang, C.; Zheng, Y.; Zheng, A.; Liu, X. A highly stable multifunctional aptamer for enhancing antitumor immunity against hepatocellular carcinoma by blocking dual immune checkpoints. Biomater. Sci. 2021, 9, 4159–4168. [Google Scholar] [CrossRef]
- Zheng, A.; Du, Y.; Wang, Y.; Zheng, Y.; Ning, Z.; Wu, M.; Zhang, C.; Zhang, D.; Liu, J.; Liu, X. CD16/PD-L1 bi-specific aptamer for cancer immunotherapy through recruiting NK cells and acting as immunocheckpoint blockade. Mol. Ther. Nucleic Acids 2022, 27, 998–1009. [Google Scholar] [CrossRef]
- Sun, Y.; Mo, L.; Hu, X.; Yu, D.; Xie, S.; Li, J.; Zhao, Z.; Fang, X.; Ye, M.; Qiu, L.; et al. Bispecific aptamer-based recognition-then-conjugation strategy for PD1/PDL1 axis blockade and enhanced immunotherapy. ACS Nano 2022, 16, 21129–21138. [Google Scholar] [CrossRef] [PubMed]
- Pastor, F.; Kolonias, D.; McNamara Ii, J.O.; Gilboa, E. Targeting 4-1BB costimulation to disseminated tumor lesions with bi-specific oligonucleotide aptamers. Mol. Ther. 2011, 19, 1878–1886. [Google Scholar] [CrossRef] [PubMed]
- Schrand, B.; Berezhnoy, A.; Brenneman, R.; Williams, A.; Levay, A.; Kong, L.Y.; Rao, G.; Zhou, S.; Heimberger, A.B.; Gilboa, E. Targeting 4-1BB costimulation to the tumor stroma with bispecific aptamer conjugates enhances the therapeutic index of tumor immunotherapy. Cancer Immunol. Res. 2014, 2, 867–877. [Google Scholar] [CrossRef]
- Wei, C.Y.; Wang, L.; Zhu, M.X.; Deng, X.Y.; Wang, D.H.; Zhang, S.M.; Ying, J.H.; Yuan, X.; Wang, Q.; Xuan, T.F.; et al. TRIM44 activates the AKT/mTOR signal pathway to induce melanoma progression by stabilizing TLR4. J. Exp. Clin. Cancer Res. 2019, 38, 137. [Google Scholar] [CrossRef] [PubMed]
- Soldevilla, M.M.; Villanueva, H.; Casares, N.; Lasarte, J.J.; Bendandi, M.; Inoges, S.; López-Díaz de Cerio, A.; Pastor, F. MRP1-CD28 bi-specific oligonucleotide aptamers: Target costimulation to drug-resistant melanoma cancer stem cells. Oncotarget 2016, 7, 23182–23196. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Hu, Y.; An, Y.; Duan, J.; Li, X.; Yang, X.D. Novel bispecific aptamer enhances immune cytotoxicity against MUC1-positive tumor cells by MUC1-CD16 dual targeting. Molecules 2019, 24, 478. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yan, H.; Liu, Y.; Chang, Y. Targeted cell–cell interactions by DNA nanoscaffold-templated multivalent bispecific aptamers. Small 2011, 7, 1673–1682. [Google Scholar] [CrossRef]
- Etter, E.L.; Mei, K.C.; Nguyen, J. Delivering more for less: Nanosized, minimal-carrier and pharmacoactive drug delivery systems. Adv. Drug Deliv. Rev. 2021, 179, 113944. [Google Scholar] [CrossRef]
- Lee, B.K.; Yun, Y.H.; Park, K. Smart nanoparticles for drug delivery: Boundaries and opportunities. Chem. Eng. Sci. 2015, 125, 158–164. [Google Scholar] [CrossRef]
- Filipczak, N.; Pan, J.; Yalamarty, S.S.K.; Torchilin, V.P. Recent advancements in liposome technology. Adv. Drug Deliv. Rev. 2020, 156, 4–22. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, C.; Wang, C.; Jankovic, K.E.; Dong, Y. Lipids and lipid derivatives for RNA delivery. Chem Rev 2021, 121, 12181–12277. [Google Scholar] [CrossRef]
- Wang, C.; Liu, B.; Lu, J.; Zhang, G.; Lu, A. Strategies for combination of aptamer and targeted drug delivery. J. Nanosci. Nanotechnol. 2014, 14, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Castro Bravo, K.M.; Liu, J. Targeted liposomal drug delivery: A nanoscience and biophysical perspective. Nanoscale Horiz. 2021, 6, 78–94. [Google Scholar] [CrossRef]
- Moosavian, S.A.; Sahebkar, A. Aptamer-functionalized liposomes for targeted cancer therapy. Cancer Lett. 2019, 448, 144–154. [Google Scholar] [CrossRef]
- Odeh, F.; Nsairat, H.; Alshaer, W.; Ismail, M.A.; Esawi, E.; Qaqish, B.; Bawab, A.A.; Ismail, S.I. Aptamers chemistry: Chemical modifications and conjugation strategies. Molecules 2019, 25, 3. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Hou, X.; Yang, W.; Shi, W.; Yang, X.; Duan, S.; Mo, F.; Liu, A.; Wang, W.; Lu, X. Endoglin-aptamer-functionalized liposome-equipped PD-1-silenced T cells enhance antitumoral immunotherapeutic effects. Int. J. Nanomed. 2021, 16, 6017–6034. [Google Scholar] [CrossRef]
- Hong, S.; Ding, P.; Luo, Y.; Gao, T.; Zhang, Y.; Pei, R. Aptamer-integrated α-Gal liposomes as bispecific agents to trigger immune response for killing tumor cells. J. Biomed. Mater. Res. Part A 2019, 107, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 2005, 5, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Mai, J.; Shen, J.; Wolfram, J.; Li, Z.; Zhang, G.; Xu, R.; Li, Y.; Mu, C.; Zu, Y.; et al. A novel DNA aptamer for dual targeting of polymorphonuclear myeloid-derived suppressor cells and tumor cells. Theranostics 2018, 8, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Iden, D.L.; Allen, T.M. A combinatorial approach to producing sterically stabilized (Stealth) immunoliposomal drugs. FEBS Lett. 1999, 460, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, N.; Wu, J.; Xu, X.; Kamaly, N.; Farokhzad, O.C. Cancer nanotechnology: The impact of passive and active targeting in the era of modern cancer biology. Adv. Drug Deliv. Rev. 2014, 66, 2–25. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Lee, J.S.; Kim, W.; Lee, J.H.; Jun, B.H.; Kim, K.S.; Kim, D.E. Aptamer-conjugated nano-liposome for immunogenic chemotherapy with reversal of immunosuppression. J. Control. Release Off. J. Control. Release Soc. 2022, 348, 893–910. [Google Scholar] [CrossRef]
- Muir, V.G.; Burdick, J.A. Chemically modified biopolymers for the formation of biomedical hydrogels. Chem. Rev. 2021, 121, 10908–10949. [Google Scholar] [CrossRef]
- Bao, Z.; Xian, C.; Yuan, Q.; Liu, G.; Wu, J. Natural polymer-based hydrogels with enhanced mechanical performances: Preparation, structure, and property. Adv. Healthc. Mater. 2019, 8, 1900670. [Google Scholar] [CrossRef]
- Li, J.; Mo, L.; Lu, C.H.; Fu, T.; Yang, H.H.; Tan, W. Functional nucleic acid-based hydrogels for bioanalytical and biomedical applications. Chem. Soc. Rev. 2016, 45, 1410–1431. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, R.; Yang, S.; Li, S.; Gao, Z. Design and application of stimuli-responsive DNA hydrogels: A review. Mater. Today Bio 2022, 16, 100430. [Google Scholar] [CrossRef]
- Gacanin, J.; Synatschke, C.V.; Weil, T. Biomedical applications of DNA-based hydrogels. Adv. Funct. Mater. 2020, 30, 1906253. [Google Scholar] [CrossRef]
- Lee, J.; Le, Q.V.; Yang, G.; Oh, Y.K. Cas9-edited immune checkpoint blockade PD-1 DNA polyaptamer hydrogel for cancer immunotherapy. Biomaterials 2019, 218, 119359. [Google Scholar] [CrossRef]
- Wei, H.; Zhao, Z.; Wang, Y.; Zou, J.; Lin, Q.; Duan, Y. One-step self-assembly of multifunctional DNA nanohydrogels: An enhanced and harmless strategy for guiding combined antitumor therapy. Acs Appl. Mater. Interfaces 2019, 11, 46479–46489. [Google Scholar] [CrossRef]
- Wang, W.; Liu, X.; Ding, L.; Jin, H.J.; Li, X. RNA hydrogel combined with MnO2 nanoparticles as a nano-vaccine to treat triple negative breast cancer. Front. Chem. 2021, 9, 797094. [Google Scholar] [CrossRef]
- Tan, J.; Luo, Y.; Guo, Y.; Zhou, Y.; Liao, X.; Li, D.; Lai, X.; Liu, Y. Development of alginate-based hydrogels: Crosslinking strategies and biomedical applications. Int. J. Biol. Macromol. 2023, 239, 124275. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Shen, F.; Tian, L.; Tao, H.; Xiong, Z.; Xu, J.; Liu, Z. ATP-responsive smart hydrogel releasing immune adjuvant synchronized with repeated chemotherapy or radiotherapy to boost antitumor immunity. Adv. Mater. 2021, 33, 2007910. [Google Scholar] [CrossRef]
- Xiong, X.; Liu, H.; Zhao, Z.; Altman, M.B.; Lopez-Colon, D.; Yang, C.J.; Chang, L.-J.; Liu, C.; Tan, W. DNA aptamer-mediated cell targeting. Angew. Chem. Int. Ed. 2013, 52, 1472–1476. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wen, J.; Li, H.; Xu, L.; Liu, Y.; Zhao, N.; Zeng, Z.; Qi, J.; Jiang, W.; Han, W.; et al. Aptamer-engineered natural killer cells for cell-specific adaptive immunotherapy. Small 2019, 15, e1900903. [Google Scholar] [CrossRef] [PubMed]
- Gong, N.; Sheppard, N.C.; Billingsley, M.M.; June, C.H.; Mitchell, M.J. Nanomaterials for T-cell cancer immunotherapy. Nat. Nanotechnol. 2021, 16, 25–36. [Google Scholar] [CrossRef]
- Tang, R.; Fu, Y.H.; Gong, B.; Fan, Y.Y.; Wang, H.H.; Huang, Y.; Nie, Z.; Wei, P. A chimeric conjugate of antibody and programmable DNA nanoassembly smartly activates T Cells for precise cancer cell targeting. Angew. Chem. (Int. Ed. Engl.) 2022, 61, e202205902. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Types | Targets | Immune Function | Reference |
|---|---|---|---|
| Aptamers against immune checkpoint (IC) | PD1 | antagonist | [12,38,40] |
| PD-L1 | antagonist | [39,40,41] | |
| CTLA-4 | antagonist | [21,25] | |
| TIM-3 | antagonist | [42] | |
| LAG3 | antagonist | [43] | |
| Aptamers against immune costimulatory molecules (CM) | CD28 | agonist | [13] |
| CD40 | agonist | [50] | |
| 4-1BB | agonist | [45] | |
| Aptamers against cytokines | IL-10 | antagonist | [55] |
| IL-8 | antagonist | [14] | |
| TGF-β1 | antagonist | [63] | |
| CXCL-10 | antagonist | [64] | |
| CXCL-12 | antagonist | [60] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiong, H.; Liu, L.; Liu, X.; Jiang, H.; Wang, X. Aptamer-Based Immune Drug Systems (AptIDCs) Potentiating Cancer Immunotherapy. Chemistry 2023, 5, 1656-1680. https://doi.org/10.3390/chemistry5030114
Xiong H, Liu L, Liu X, Jiang H, Wang X. Aptamer-Based Immune Drug Systems (AptIDCs) Potentiating Cancer Immunotherapy. Chemistry. 2023; 5(3):1656-1680. https://doi.org/10.3390/chemistry5030114
Chicago/Turabian StyleXiong, Hongjie, Liu Liu, Xiaohui Liu, Hui Jiang, and Xuemei Wang. 2023. "Aptamer-Based Immune Drug Systems (AptIDCs) Potentiating Cancer Immunotherapy" Chemistry 5, no. 3: 1656-1680. https://doi.org/10.3390/chemistry5030114
APA StyleXiong, H., Liu, L., Liu, X., Jiang, H., & Wang, X. (2023). Aptamer-Based Immune Drug Systems (AptIDCs) Potentiating Cancer Immunotherapy. Chemistry, 5(3), 1656-1680. https://doi.org/10.3390/chemistry5030114