


Improved Synthesis and Coordination Behavior of 1H-1,2,3-Triazole-4,5-dithiolates (tazdt2−) with NiII, PdII, PtII and CoIII


Abstract
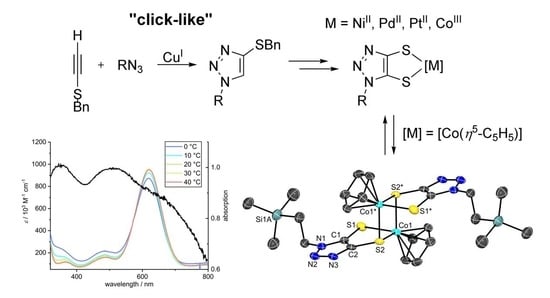
1. Introduction
2. Materials and Methods
2.1. Chemical Reagents and Instruments
2.2. Synthetic Protocols
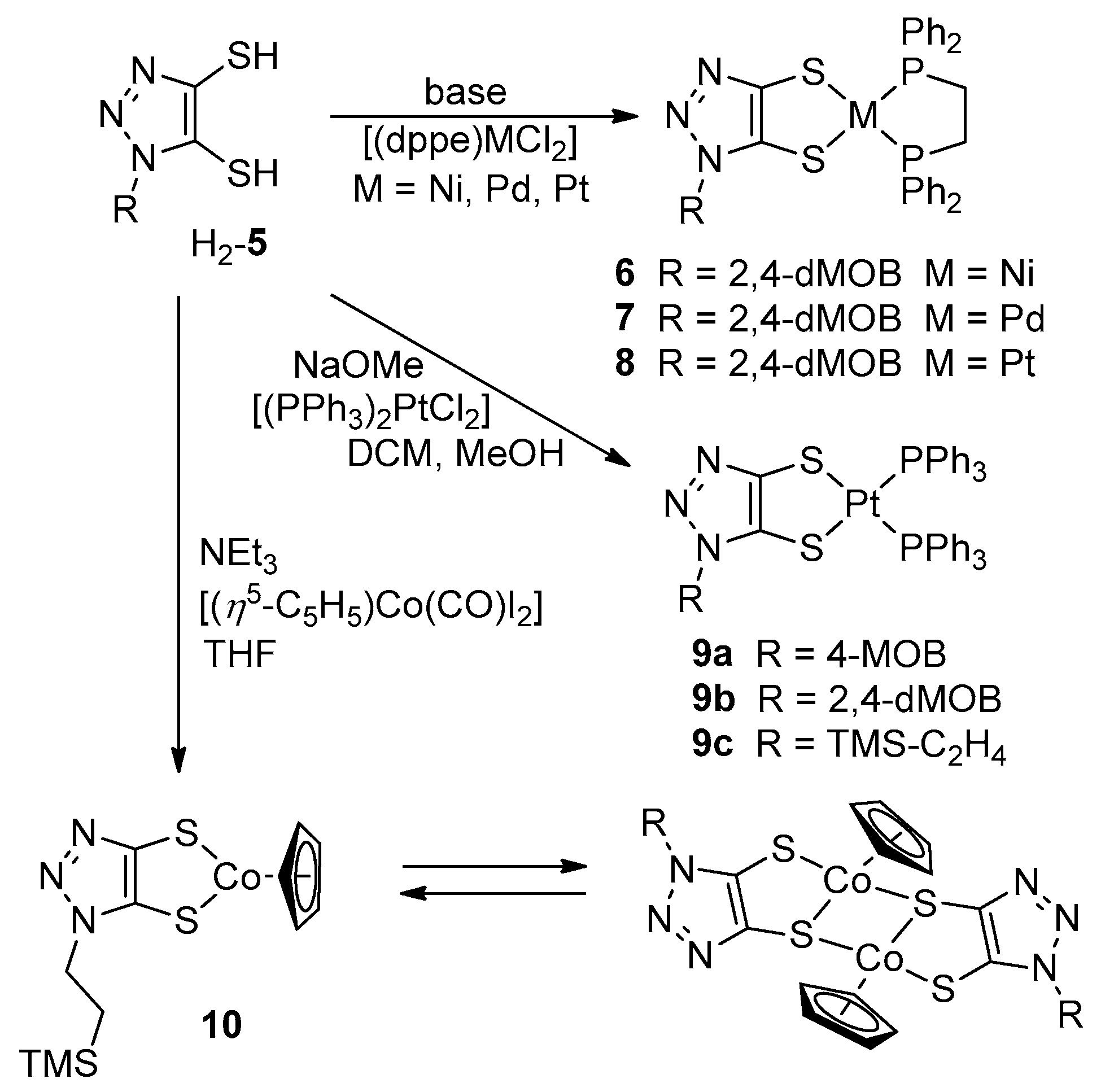
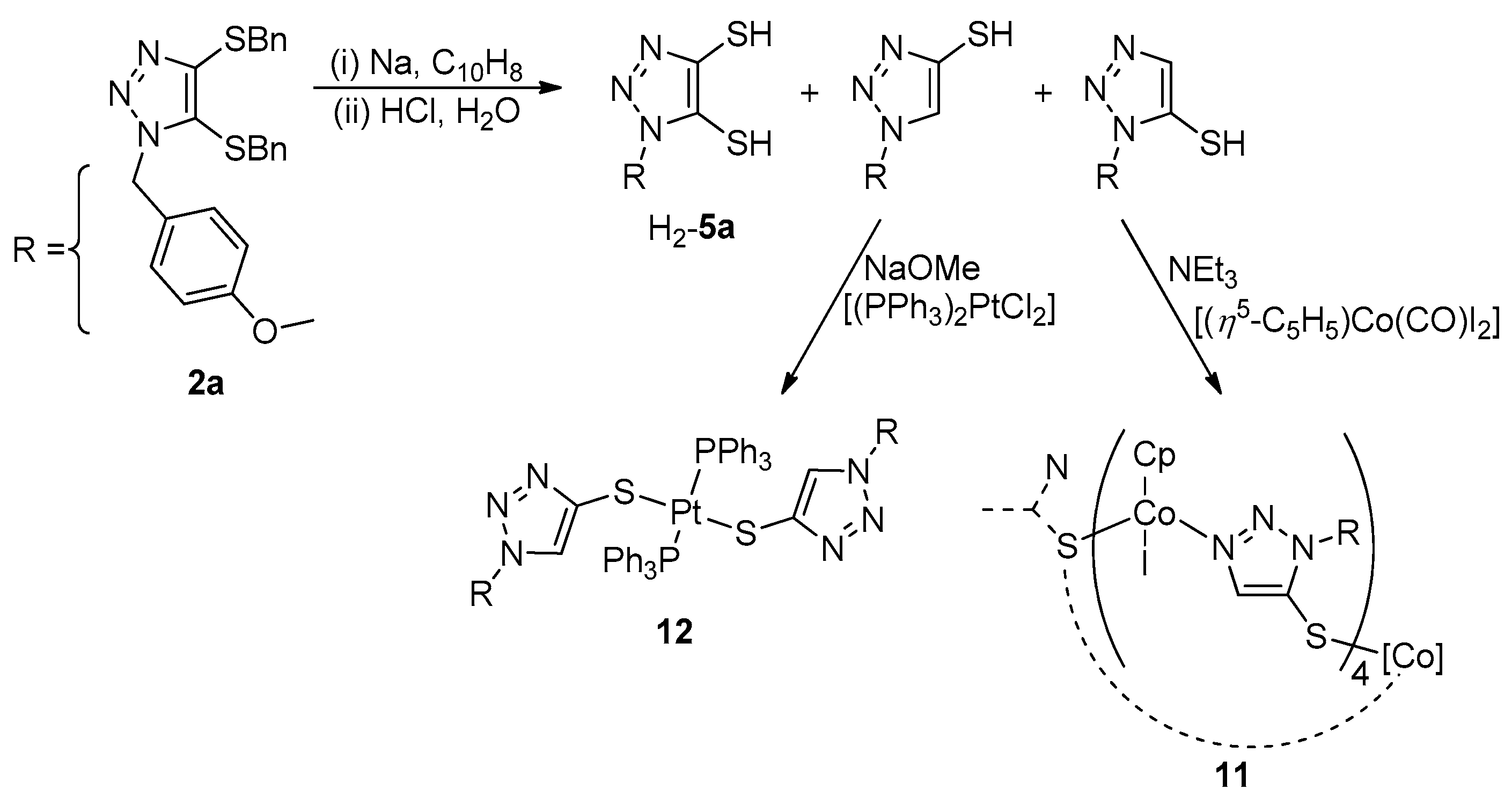
2.2.1. General Synthesis of 5
2.2.2. General Synthesis of the Metal Complexes 6 and 7
2.2.3. Synthesis of [(dppe)Pt(5b)] (8)
2.2.4. General Synthesis of 9
2.2.5. Synthesis of 10
2.2.6. Synthesis of 11
3. Results and Discussion
3.1. Ligand Synthesis
3.2. Synthesis of Metal Complexes
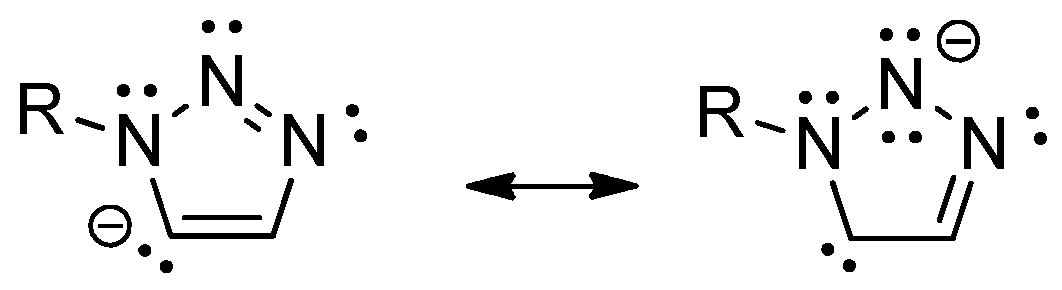
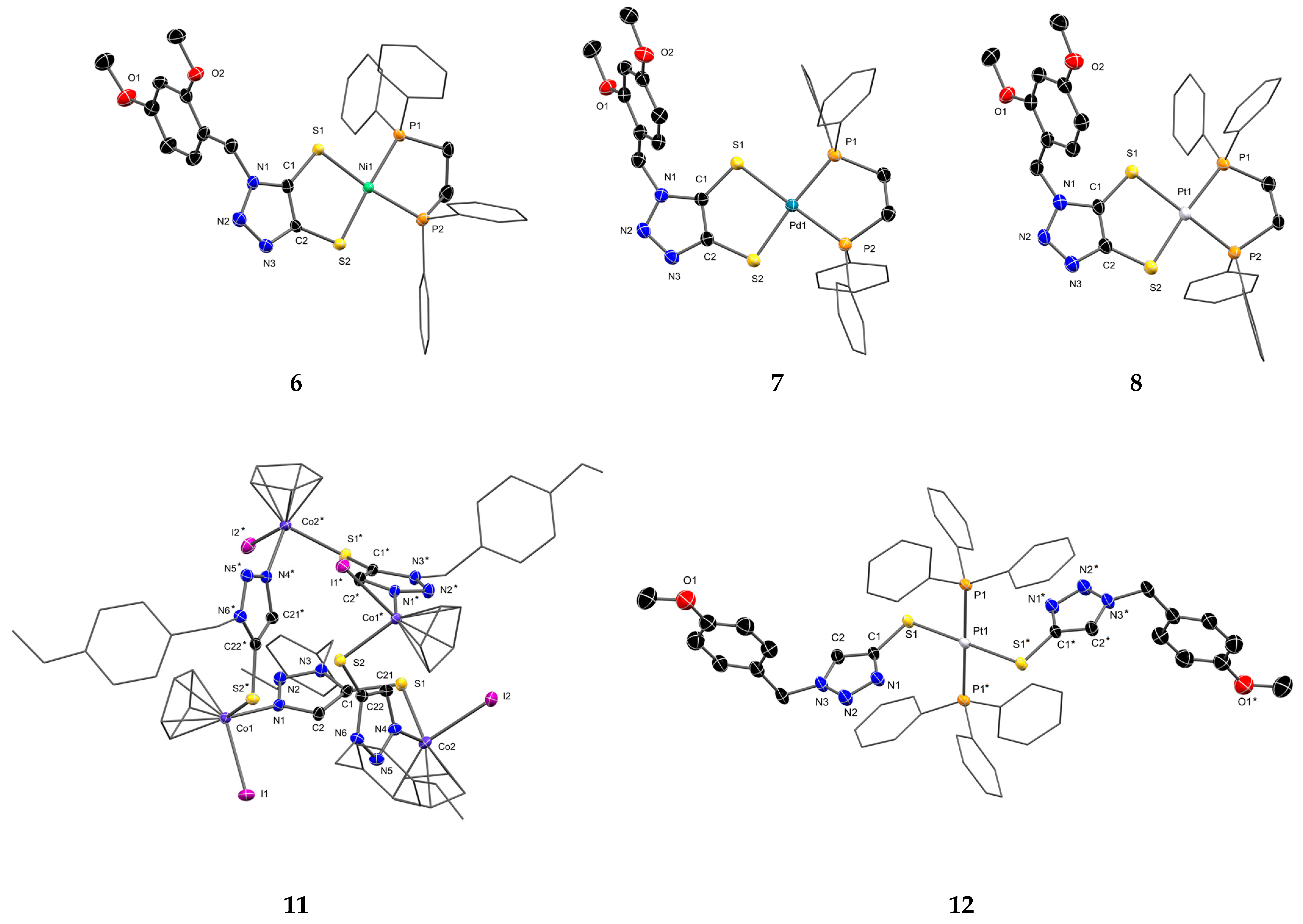
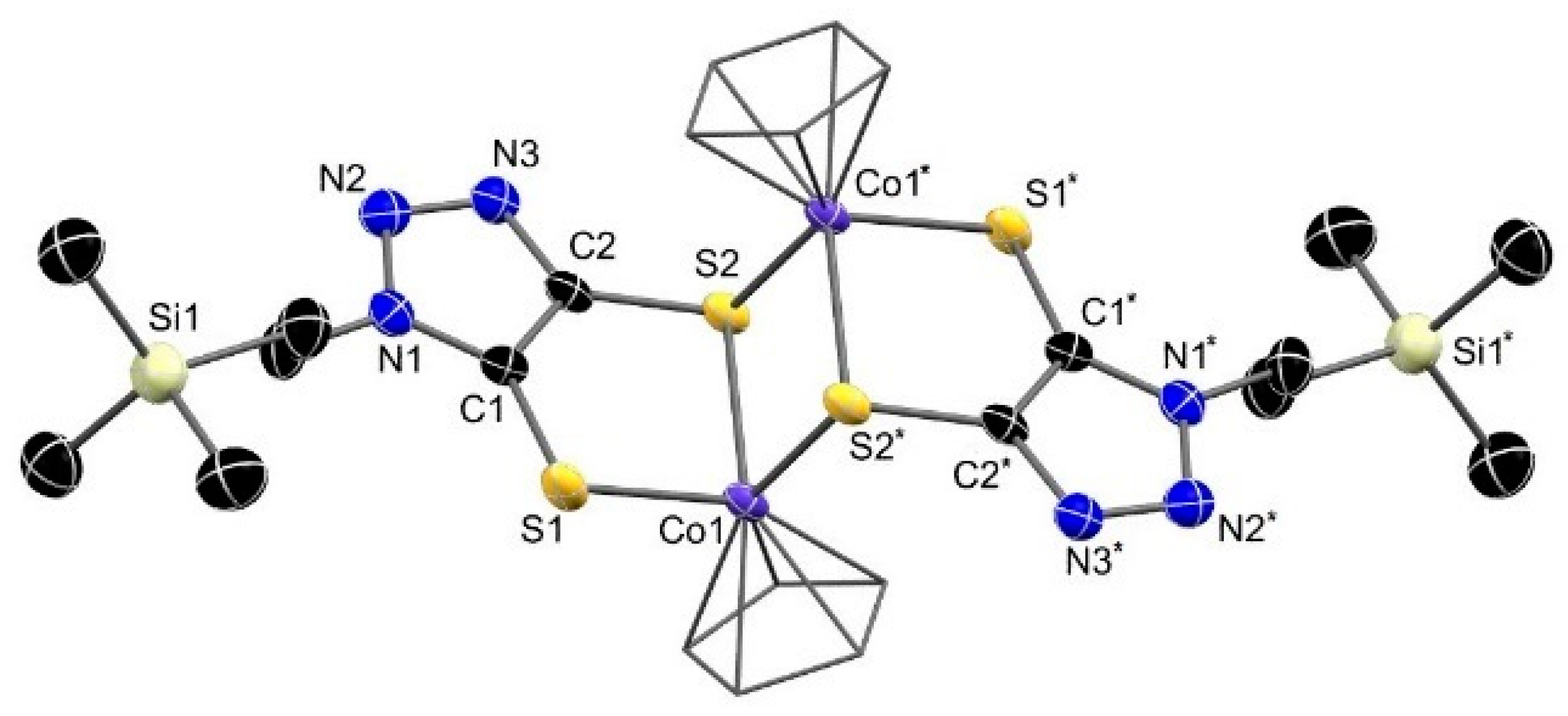
3.3. Molecular Structure of the Complexes
3.4. NMR Spectroscopy of Metal Complexes
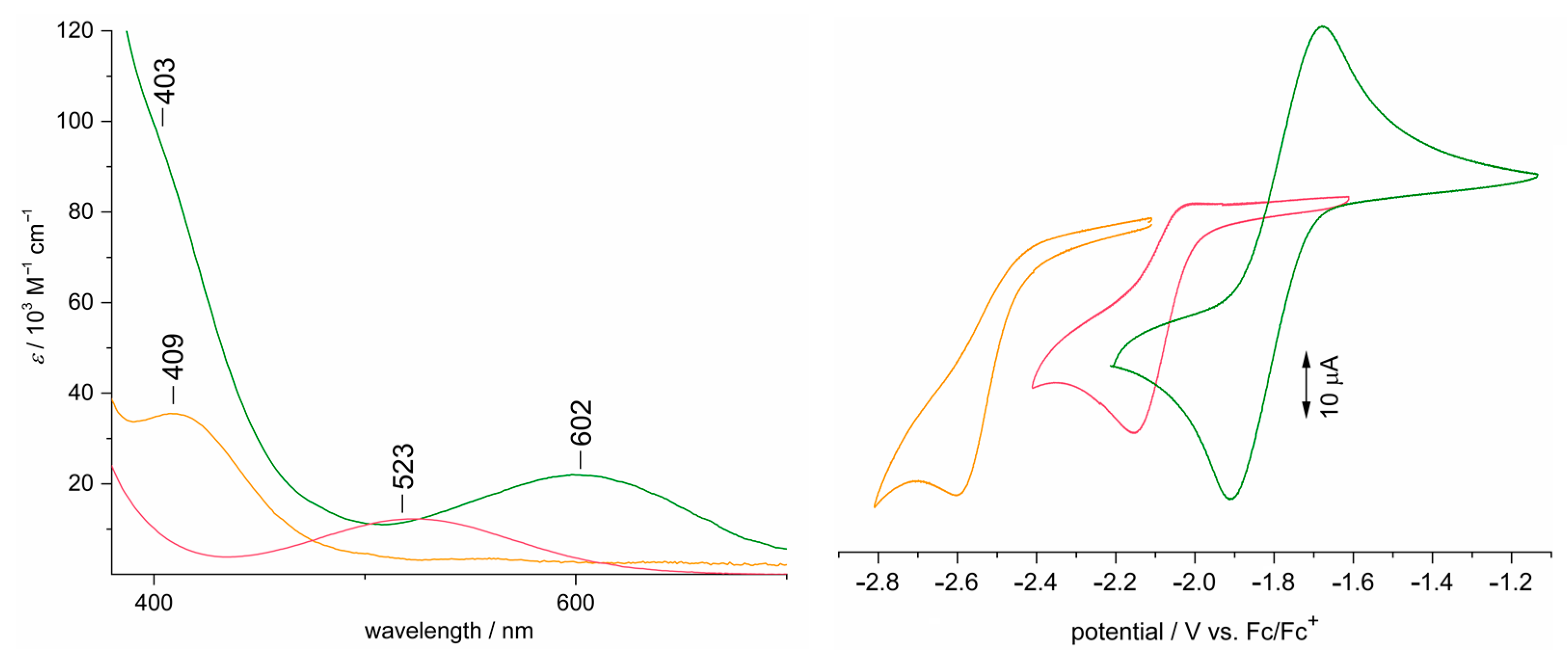
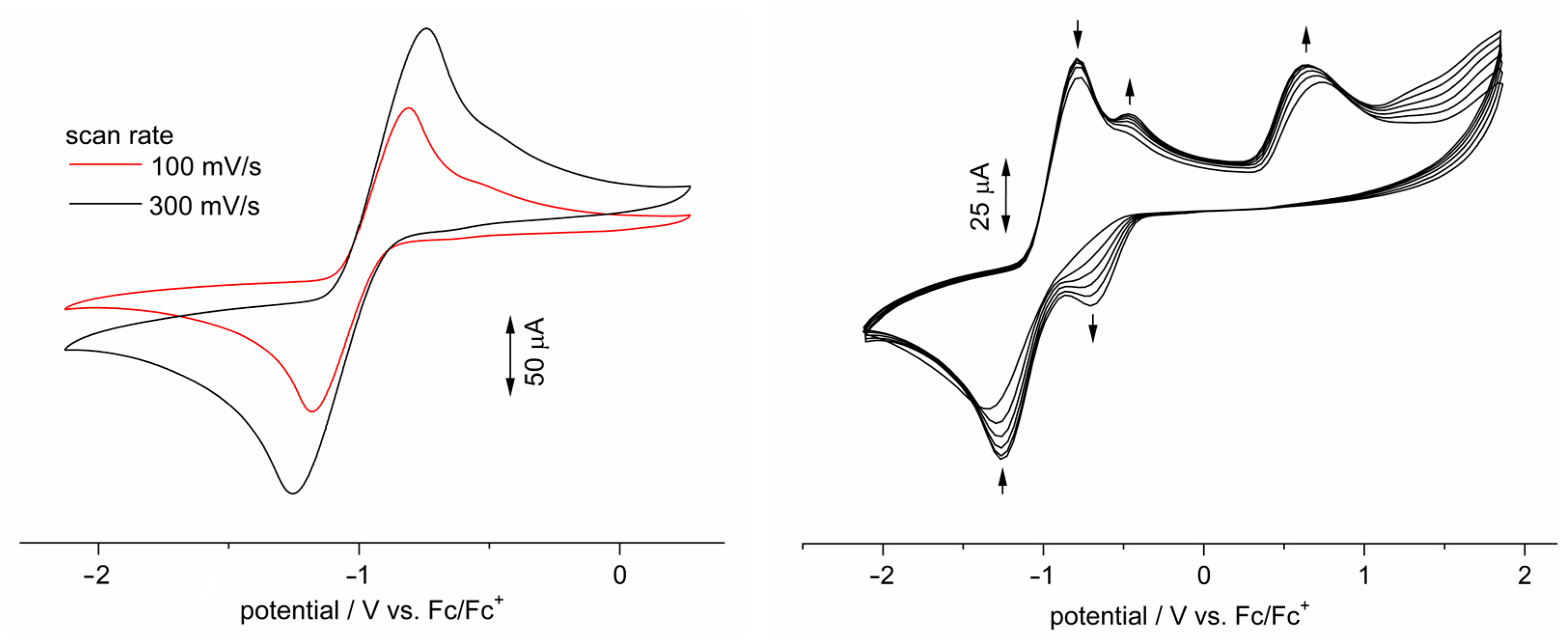
3.5. Electronic Structure Elucidation
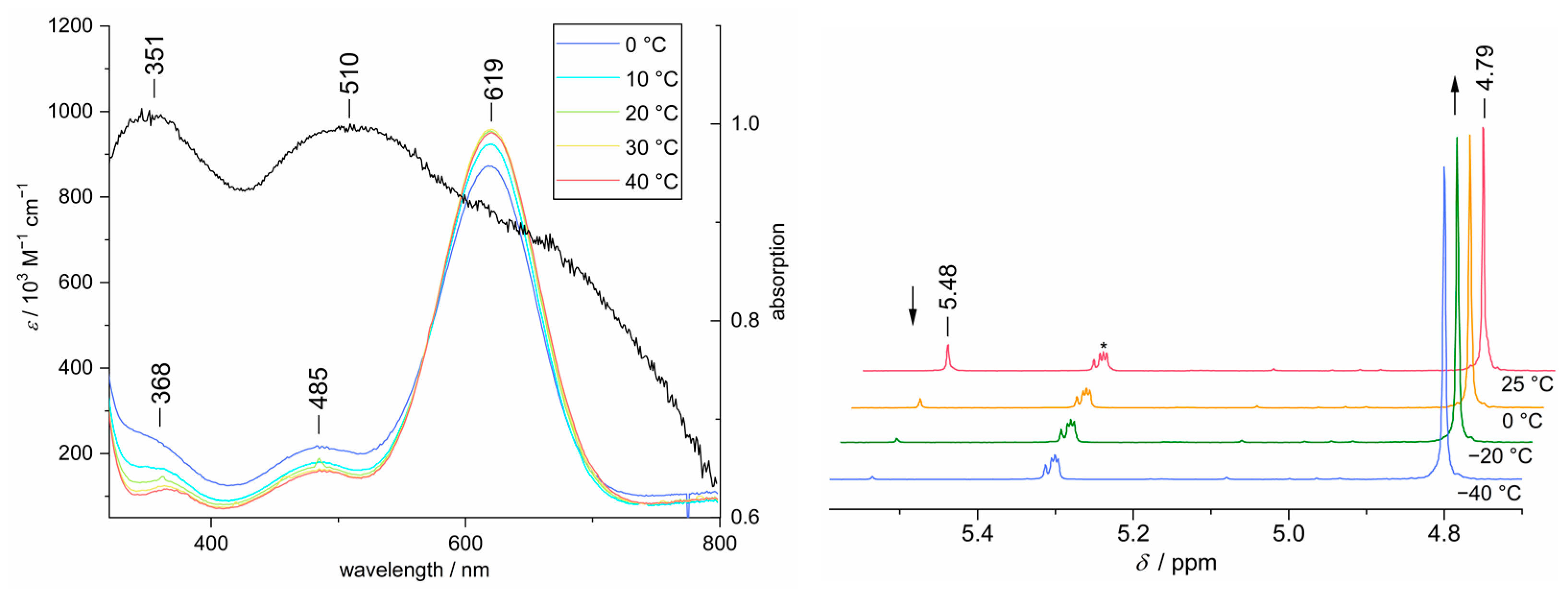
3.6. Investigation of Dimerization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Peplow, M. Click and bioorthogonal chemistry win 2022 Nobel Prize in Chemistry. Chem. Eng. News 2022, 36, 3. [Google Scholar]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Lewis, W.G.; Green, L.G.; Grynszpan, F.; Radić, Z.; Carlier, P.R.; Taylor, P.; Finn, M.G.; Sharpless, K.B. Click Chemistry In Situ: Acetylcholinesterase as a Reaction Vessel for the Selective Assembly of a Femtomolar Inhibitor from an Array of Building Blocks. Angew. Chem. Int. Ed. 2002, 41, 1053–1057. [Google Scholar] [CrossRef]
- Ramachary, D.B.; Barbas, C.F. Towards Organo-Click Chemistry: Development of Organocatalytic Multicomponent Reactions Through Combinations of Aldol, Wittig, Knoevenagel, Michael, Diels-Alder and Huisgen Cycloaddition Reactions. Chem. Eur. J. 2004, 10, 5323–5331. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Clarke, D.; Mares, R.W.; McNab, H. Preparation and pyrolisis of 1-(pyrazol-5-yl)-1,2,3-triazoles and related compounds. J. Chem. Soc. Parkin Trans. 1 1997, 1799–1804. [Google Scholar] [CrossRef]
- Huisgen, R. 1,3-Dipolar Cycloadditions. Past and Future. Angew. Chem. Int. Ed. Engl. 1963, 2, 565–598. [Google Scholar] [CrossRef]
- Qin, A.; Jim, C.K.W.; Lu, W.; Lam, J.W.Y.; Häussler, M.; Dong, Y.; Sung, H.H.Y.; Williams, I.D.; Wong, G.K.L.; Tang, B.Z. Click Polymerization: Facile Synthesis of Functional Poly(aroyltriazole)s by Metal-Free, Regioselective 1,3-Dipolar Polycycloaddition. Macromolecules 2007, 40, 2308–2317. [Google Scholar] [CrossRef]
- Jasiński, R. Nitroacetylene as dipolarophile in 2 + 3 cycloaddition reactions with allenyl-type three-atom components: DFT computational study. Monatsh. Chem. 2015, 146, 591–599. [Google Scholar] [CrossRef]
- Hein, J.E.; Fokin, V.V. Copper-catalyzed azide-alkyne cycloaddition (CuAAC) and beyond: New reactivity of copper(I) acetylides. Chem. Soc. Rev. 2010, 39, 1302–1315. [Google Scholar] [CrossRef]
- Beerhues, J.; Aberhan, H.; Streit, T.-N.; Sarkar, B. Probing Electronic Properties of Triazolylidenes through Mesoionic Selones, Triazolium Salts, and Ir-Carbonyl-Triazolylidene Complexes. Organometallics 2020, 39, 4557–4564. [Google Scholar] [CrossRef]
- Tran, H.-V.; Haghdoost, M.M.; Poulet, S.; Tcherkawsky, P.; Castonguay, A. Exploiting exo and endo furan-maleimide Diels-Alder linkages for the functionalization of organoruthenium complexes. Dalton Trans. 2022, 51, 2214–2218. [Google Scholar] [CrossRef]
- Crowley, J.D.; Bandeen, P.H.; Hanton, L.R. A one pot multi-component CuAAC “click” approach to bidentate and tridentate pyridyl-1,2,3-triazole ligands: Synthesis, X-ray structures and copper(II) and silver(I) complexes. Polyhedron 2010, 29, 70–83. [Google Scholar] [CrossRef]
- Struthers, H.; Mindt, T.L.; Schibli, R. Metal chelating systems synthesized using the copper(I) catalyzed azide-alkyne cycloaddition. Dalton Trans. 2010, 39, 675–696. [Google Scholar] [CrossRef]
- Haas, A.; Krächter, H.-U. Darstellung und Reaktionen Trifluormethylchalkogenyl-substituierter Alkine. Chem. Ber. 1988, 121, 1833–1840. [Google Scholar] [CrossRef]
- Schallenberg, D.; Pardemann, N.; Villinger, A.; Seidel, W.W. Synthesis and coordination behaviour of 1H-1,2,3-triazole-4,5-dithiolates. Dalton Trans. 2022, 51, 13681–13691. [Google Scholar] [CrossRef]
- Szilagyi, R.K.; Lim, B.S.; Glaser, T.; Holm, R.H.; Hedman, B.; Hodgson, K.O.; Solomon, E.I. Description of the Ground State Wave Functions of Ni Dithiolenes Using Sulfur K-edge X-ray Absorption Spectroscopy. J. Am. Chem. Soc. 2003, 125, 9158–9169. [Google Scholar] [CrossRef]
- Lim, B.S.; Fomitchev, D.V.; Holm, R.H. Nickel Dithiolenes Revisited: Structures and Electron Distribution from Density Functional Theory for the Three-Member Electron-Transfer Series [Ni(S2C2Me2)2]0,1−,2−. Inorg. Chem. 2001, 40, 4257–4262. [Google Scholar] [CrossRef]
- Aragoni, M.C.; Caltagirone, C.; Lippolis, V.; Podda, E.; Slawin, A.M.Z.; Woollins, J.D.; Pintus, A.; Arca, M. Diradical Character of Neutral Heteroleptic Bis(1,2-dithiolene) Metal Complexes: Case Study of [Pd(Me2timdt)(mnt)] (Me2timdt = 1,3-Dimethyl-2,4,5-trithioxoimidazolidine; mnt2- = 1,2-Dicyano-1,2-ethylenedithiolate). Inorg. Chem. 2020, 59, 17385–17401. [Google Scholar] [CrossRef]
- Schallenberg, D. Der Aufbau Polynuklearer Komplexe auf der Basis N-Heterozyklischer Dithiolene; Universität Rostock: Rostock, Germany, 2013; pp. 1–49. [Google Scholar]
- Ding, S.; Jia, G.; Sun, J. Iridium-Catalyzed Intermolecular Azide-Alkyne Cycloaddition of Internal Thioalkynes under Mild Conditions. Angew. Chem. Int. Ed. 2014, 53, 1877–1880. [Google Scholar] [CrossRef]
- Haldón, E.; Nicasio, M.C.; Pérez, P.J. Copper-catalysed azide-alkyne cycloadditions (CuAAC): An update. Org. Biomol. Chem. 2015, 13, 9528–9550. [Google Scholar] [CrossRef] [PubMed]
- Kappe, C.O.; Van der Eycken, E. Click chemistry under non-classical reaction conditions. Chem. Soc. Rev. 2010, 39, 1280–1290. [Google Scholar] [CrossRef] [PubMed]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Seidel, W.W.; Meel, M.J.; Schaffrath, M.; Pape, T. In Pursuit of an Acetylenedithiolate Synthesis. Eur. J. Org. Chem. 2007, 2007, 3526–3532. [Google Scholar] [CrossRef]
- Rasmussen, L.K.; Boren, B.C.; Fokin, V.V. Ruthenium-Catalyzed Cycloaddition of Aryl Azides and Alkynes. Org. Lett. 2007, 9, 5337–5339. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, X.; Xue, P.; Sun, H.H.Y.; Williams, I.D.; Sharpless, K.B.; Fokin, V.V.; Jia, G. Ruthenium-Catalyzed Cycloaddition of Alkynes and Organic Azides. J. Am. Chem. Soc. 2005, 127, 15998–15999. [Google Scholar] [CrossRef]
- Boren, B.C.; Narayan, S.; Rasmussen, L.K.; Zhang, L.; Zhao, H.; Lin, Z.; Jia, G.; Fokin, V.V. Ruthenium-Catalyzed Azide-Alkyne Cycloaddition: Scope and Mechanism. J. Am. Chem. Soc. 2008, 130, 8923–8930. [Google Scholar] [CrossRef]
- Landis, K.G.; Hunter, A.D.; Wagner, T.R.; Curtin, L.S.; Filler, F.L.; Jansen-Varnum, S.A. The synthesis and characterisation of Ni, Pd and Pt maleonitriledithiolate complexes: X-ray crystal structure of the isomorphous Ni, Pd and Pt (Ph2PCH2CH2PPh2)M(maleonitriledithiolate) congeners. Inorg. Chim. Acta 1998, 282, 155–162. [Google Scholar] [CrossRef]
- Wrixon, J.D.; Hayward, J.J.; Raza, O.; Rawson, J.M. Oxidative addition chemistry of tetrathiocines: Synthesis, structures and properties of group 10 dithiolate complexes. Dalton Trans. 2014, 43, 2134–2139. [Google Scholar] [CrossRef]
- Wrixon, J.D.; Ahmed, Z.S.; Anwar, M.U.; Beldjoudi, Y.; Hamidouche, N.; Hayward, J.J.; Rawson, J.M. Oxidative addition of bis-(dimethoxybenzo)-1,2,5,6-tetrathiocins to Pt(PPh3)4: Synthesis and structures of mono- and di-metallic platinum dithiolate complexes, (dmobdt)Pt(PPh3)2 and [(dmobdt)Pt(PPh3)]2. Polyhedron 2016, 108, 115–121. [Google Scholar] [CrossRef]
- Miller, E.J.; Brill, T.B.; Rheingold, A.L.; Fultz, W.C. A Reversible Chemical Reaction in a Single Crystal. The Dimerization of (η5-C5H5)Co(S2C6H4). J. Am. Chem. Soc. 1983, 105, 7580–7584. [Google Scholar] [CrossRef]
- Nomura, M.; Sasao, T.; Hashimoto, T.; Sugiyama, T.; Kajitani, M. Structures and electrochemistry of monomeric and dimeric CpCo(dithiolene) complexes with substituted benzene-1,2-dithiolate ligand. Inorg. Chim. Acta 2010, 363, 3647–3653. [Google Scholar] [CrossRef]
- Vicente, R.; Ribas, J.; Solans, X.; Font-Alaba, M.; Mari, A.; de Loth, P.; Cassoux, P. Electrochemical, EPR, and crystal structure studies on mixed-ligand 4,5-Dimercapto-1,2-dithia-2-thione phosphine complexes of nickel, palladium and platinum, M(dmit)(dppe) and Pt(dmit)(PPh3)2. Inorg. Chim. Acta 1987, 132, 229–236. [Google Scholar] [CrossRef]
- Lee, S.-K.; Shin, K.-S.; Noh, D.-Y.; Jeannin, O.; Barrière, F.; Bergamini, J.-F.; Fourmigué, M. Redox Multifunctionality in a Series of PtII Dithiolene Complexes of a Tetrathiafulvalene-Based Diphosphine Ligand. Chem. Asian J. 2010, 5, 169–176. [Google Scholar] [CrossRef]
- Keefer, C.E.; Purrington, S.T.; Bereman, R.D.; Knight, B.W.; Bedgood, D.R., Jr.; Boyle, P.D. The synthesis and characterization of platinum monodithiolene complexes containing 1,3-dithiole-2-oxo-4,5-dithiolate (dmid2−) and 1,3-dithiole-2-thione-4,5-dithiolate (dmit2−). Inorg. Chim. Acta 1998, 282, 200–208. [Google Scholar] [CrossRef]
- Habe, S.; Yamada, T.; Nankawa, T.; Mizutani, J.; Murata, M.; Nishihara, H. Synthesis, Structure, and Dissociation Equilibrium of Co(η5-C5H5)(Se2C6H4)2, a Novel Metalladiselenolene Complex. Inorg. Chem. 2003, 42, 1952–1955. [Google Scholar] [CrossRef]
- Nomura, M.; Fourmigué, M. Isostructural diamagnetic cobalt(III) and paramegnetic nickel(III) dithiolene complexes with an extended benzdithiolate core [CpMIII(bdtodt)] (M = Co and Ni). J. Organomet. Chem. 2007, 692, 2491–2499. [Google Scholar] [CrossRef]
- Nomura, M.; Kondo, S.; Yamashita, S.; Suzuki, E.; Toyota, Y.; Alea, G.V.; Janairo, G.C.; Fujita-Takayama, C.; Sugiyama, T.; Kajitani, M. Sulfur-rich CpCo(dithiolene) complexes: Isostructural or non-isostructural couples of CpCo(III) with CpNi(III) dithiolene complexes. J. Organomet. Chem. 2010, 695, 2366–2375. [Google Scholar] [CrossRef]
- Watanabe, L.K.; Ahmed, Z.S.; Hayward, J.J.; Heyer, E.; Macdonald, C.; Rawson, J.M. Oxidative addition of 1,2,5,6-Tetrathiocins to Co(I): A Re-Examination of Crown Ether Functionalized Benzene Dithiolate Cobalt(III) Complexes. Organometallics 2022, 41, 226–234. [Google Scholar] [CrossRef]
- Yang, L.; Powell, D.R.; Houser, R.P. Structural variation in copper(I) complexes with pyridylmethylamide ligands: Structural analysis with a new four-coordinate geometry index, τ4. Dalton Trans. 2007, 955–964. [Google Scholar] [CrossRef]
- Keefer, C.E.; Bereman, R.D.; Purrington, S.T.; Knight, B.W.; Boyle, P.D. The 195Pt NMR of L2Pt(1,2-dithiolene) Complexes. Inorg. Chem. 1999, 38, 2294–2302. [Google Scholar] [CrossRef]
- Kimura, T.; Nakahodo, T.; Fujihara, H. Preparation and reactivity of benzo-1,2-dichalcogenete derivatives and their bis(triphenylphosphine)platinum complexes. Heteroat. Chem. 2018, 29, e21472. [Google Scholar] [CrossRef]
- Takeo, A.; Yoshihiro, W.; Akiko, M.; Toru, K.; Hirobumi, U.; Masatsugu, K.; Kunio, S.; Akira, S. Photochemical Reduction of (η5-Cyclopentadienyl)(1,2-disubstituted 1,2-ethylenedichalcogenolato)cobalt(III) and (η5-Cyclopentadienyl)(1,2-benzenedithiolato)cobalt(III) Complexes in the Presence of Triethanolamine. Bull. Chem. Soc. Jpn. 1992, 65, 1047–1051. [Google Scholar] [CrossRef]
- Matsuo, Y.; Ogumi, K.; Maruyama, M.; Nakagawa, T. Divergent Synthesis and Tuning of the Electronic Structures of Cobalt–Dithiolene–Fullerene Complexes for Organic Solar Cells. Organometallics 2014, 33, 659–664. [Google Scholar] [CrossRef]
- Murata, M.; Habe, S.; Araki, S.; Namiki, K.; Yamada, T.; Nakagawa, N.; Nankawa, T.; Nihei, M.; Mizutani, J.; Kurihara, M. Synthesis of Heterometal Cluster Complexes by the Reaction of Cobaltadichalcogenolato Complexes with Groups 6 and 8 Metal Carbonyls. Inorg. Chem. 2006, 45, 1108–1116. [Google Scholar] [CrossRef]
- Tsukada, S.; Kondo, M.; Sato, H.; Gunji, T. Fine electronic state tuning of cobaltadithiolene complexes by substituent groups on the benzene ring. Polyhedron 2016, 117, 265–272. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXS-2013, Program for Solution of Crystal Structure; Universität Göttingen: Göttingen, Germany, 2013. [Google Scholar]
- Sheldrick, G.M. SHELXL-2013. Program for Refinement of Crystal Structure; Universität Göttingen: Göttingen, Germany, 2013. [Google Scholar]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Kitano, H.; Choi, J.-H.; Ueda, A.; Ito, H.; Hagihara, S.; Kan, T.; Kawagishi, H.; Itami, K. Discovery of Plant Growth Stimulants by C-H Arylation of 2-Azahypoxanthine. Org. Lett. 2018, 20, 5684–5687. [Google Scholar] [CrossRef]
- Tran, T.K.; Bricaud, Q.; Ocafrain, M.; Blanchard, P.; Roncali, J.; Lenfant, S.; Godey, S.; Vuillaume, D.; Rondeau, D. Thiolate chemistry: A powerful and versatile synthetic tool for immobilization/functionalization of oligothiophenes on a gold surface. Chem. Eur. J. 2011, 17, 5628–5640. [Google Scholar] [CrossRef]
- Matake, R.; Niwa, Y.; Matsubara, H. Phase-vanishing method with acetylene evolution and its utilization in several organic syntheses. Org. Lett. 2015, 17, 2354–2357. [Google Scholar] [CrossRef]
- Spencer, L.P.; Altwer, R.; Wei, P.; Gelmini, L.; Gauld, J.; Stephan, D.W. Pyridine− and Imidazole−Phosphinimine Bidentate Ligand Complexes: Considerations for Ethylene Oligomerization Catalysts. Organometallics 2003, 22, 3841–3854. [Google Scholar] [CrossRef]
- Zanato, C.; Cascio, M.G.; Lazzari, P.; Pertwee, R.; Testa, A.; Zanda, M. Tricyclic Fused Pyrazoles with a ‘Click’ 1,2,3-Triazole Substituent in Position 3 Are Nanomolar CB1 Receptor Ligands. Synthesis 2015, 47, 817–826. [Google Scholar] [CrossRef]
- Howell, S.J.; Spencer, N.; Philp, D. Recognition-mediated regiocontrol of a dipolar cycloaddition reaction. Tetrahedron 2001, 57, 4945–4954. [Google Scholar] [CrossRef]
- Wang, T.; Wang, C.; Zhou, S.; Xu, J.; Jiang, W.; Tan, L.; Fu, J. Nanovalves-Based Bacteria-Triggered, Self-Defensive Antibacterial Coating: Using Combination Therapy, Dual Stimuli-Responsiveness, and Multiple Release Modes for Treatment of Implant-Associated Infections. Chem. Mater. 2017, 29, 8325–8337. [Google Scholar] [CrossRef]
- Kumar, A.S.; Ghule, V.D.; Subrahmanyam, S.; Sahoo, A.K. Synthesis of thermally stable energetic 1,2,3-triazole derivatives. Chem. Eur. J. 2013, 19, 509–518. [Google Scholar] [CrossRef]
- Standley, E.A.; Smith, S.J.; Müller, P.; Jamison, T.F. A Broadly Applicable Strategy for Entry into Homogeneous Nickel(0) Catalysts from Air-Stable Nickel(II) Complexes. Organometallics 2014, 33, 2012–2018. [Google Scholar] [CrossRef]
- Lobana, T.S.; Bawa, G.; Hundal, G.; Butcher, R.J.; Castineiras, A. The Influence of Substituents at C 2 Carbon of Thiosemicarbazones on the Bonding Pattern of Bis(diphenylphopshano)alkanes in Palladium(II) Complexes. Z. Anorg. Allg. Chem. 2009, 635, 1447–1453. [Google Scholar] [CrossRef]
- Li, X.; Zha, M.-Q.; Gao, S.-Y.; Low, P.-J.; Wu, Y.-Z.; Gan, N.; Cao, R. Synthesis, photoluminescence, catalysis and multilayer film assembly of an ethynylpyridine platinum compound. CrystEngComm 2011, 13, 920–926. [Google Scholar] [CrossRef]
- Ramos-Lima, F.J.; Quiroga, A.G.; Pérez, J.M.; Font-Bardia, M. Synthesis and Characterization of New Transplatinum Complexes Containing Phosphane Groups—Cytotoxic Studies in Cisplatin-Resistant Cells. Eur. J. Inorg. Chem. 2003, 2003, 1591–1598. [Google Scholar] [CrossRef]
- Shapley, J.R. Inorganic Syntheses; Wiley-Interscience: Hoboken, NJ, USA, 2004; Volume 34. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R | 1 | 2 | ||
|---|---|---|---|---|
| a |  | 4-MOB | 97% | 76% |
| b |  | 2,4-dMOB | 83% | 89% |
| c |  | TMS-C2H4 | 93% | 48% |
| d |  | Xy | 49% | 38% |
| e |  | Bn | 80% | 65% |
| f |  | 3,4-dMOB | 36% | 76% |
| g |  | 2-Pic | 91% | - |
| C–S | C–S | C–C | M–S1 | M–S2/M–S2* | S1–M–S2 | |
|---|---|---|---|---|---|---|
| 6 | 1.726(4) | 1.747(4) | 1.368(6) | 2.199(1) | 2.187(1) | 95.80(4) |
| [(dppe)Ni(tazdt-Bn)] [16] | 1.719(3) | 1.750(3) | 1.370(4) | 2.1982(8) | 2.1925(8) | 96.09(3) |
| 7 | 1.725(5) | 1.748(6) | 1.381(5) | 2.354(2) | 2.334(1) | 92.99(5) |
| [(dppe)Pd(tazdt-Bn)] [16] | 1.7333(15) | 1.7400(17) | 1.377(2) | 2.3475(4) | 2.3397(4) | 92.90(1) |
| 8 | 1.741(6) | 1.736(4) | 1.369(5) | 2.349(1) | 2.335(1) | 92.15(4) |
| [(dppe)Pt(dmit)] [34] | 1.710(11) | 1.716(11) | 1.366(16) | 2.315(3) | 2.308(3) | 90.0(1) |
| [(dppe)Pt(dddt)] [35] | - | - | - | 2.3157(13) | 2.3235(15) | 88.25(5) |
| 9a | 1.724(3) | 1.739(3) | 1.373(3) | 2.3344(7) | 2.3487(7) | 90.82(2) |
| 9b | 1.725(5) | 1.752(3) | 1.369(6) | 2.3536(9) | 2.336(1) | 91.08(4) |
| [(PPh3)2Pt(dmit)] [36] | 1.722(4) | 1.750(4) | 1.349(6) | 2.3319(11) | 2.3192(11) | 89.22(4) |
| 10 | 1.718(5) | 1.751(7) | 1.380(8) | 2.278(2) | 2.271(1)/2.269(1) | 93.81(5) |
| [(η5-C5H5)Co(Cl3bdt)] [33] | 1.734(11) | 1.765(11) | 1.384(18) | 2.211(2) | 2.214(3)/2.270(3) | 89.61(12) |
| [(η5-C5H5)Co(bdt)] [32] | 1.757(4) | 1.783(3) | 1.382 | 2.246(1) | 2.230(1)/2.272(1) | 89.73(4) |
| 11 | 1.739(2) | 1.739(2) | 1.739(2)/1.379(3) | 2.2601(7) | 2.2721(6) | - |
| 12 | 1.747(3) | 1.379(4) | 2.3274(7) | - | ||
| M | δ [ppm] | JP,P [Hz] | JP,Pt [Hz] | |
|---|---|---|---|---|
| 6 | Ni | 60.5/58.7 | 47.9 | - |
| 7 | Pd | 58.4/56.1 | 18.0 | - |
| 8 | Pt | 45.7/45.4 | 10.5 | 2778/2760 |
| 9a | Pt | 17.2/17.1 | 21.0 | 2915/2943 |
| 9b | Pt | 17.7/16.7 | 21.0 | 2862/2998 |
| 9c | Pt | 17.4/16.9 | 21.0 | 2861/2988 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pardemann, N.; Villinger, A.; Seidel, W.W. Improved Synthesis and Coordination Behavior of 1H-1,2,3-Triazole-4,5-dithiolates (tazdt2−) with NiII, PdII, PtII and CoIII. Chemistry 2023, 5, 1271-1287. https://doi.org/10.3390/chemistry5020086
Pardemann N, Villinger A, Seidel WW. Improved Synthesis and Coordination Behavior of 1H-1,2,3-Triazole-4,5-dithiolates (tazdt2−) with NiII, PdII, PtII and CoIII. Chemistry. 2023; 5(2):1271-1287. https://doi.org/10.3390/chemistry5020086
Chicago/Turabian StylePardemann, Nils, Alexander Villinger, and Wolfram W. Seidel. 2023. "Improved Synthesis and Coordination Behavior of 1H-1,2,3-Triazole-4,5-dithiolates (tazdt2−) with NiII, PdII, PtII and CoIII" Chemistry 5, no. 2: 1271-1287. https://doi.org/10.3390/chemistry5020086
APA StylePardemann, N., Villinger, A., & Seidel, W. W. (2023). Improved Synthesis and Coordination Behavior of 1H-1,2,3-Triazole-4,5-dithiolates (tazdt2−) with NiII, PdII, PtII and CoIII. Chemistry, 5(2), 1271-1287. https://doi.org/10.3390/chemistry5020086