
Short I⋯O Interactions in the Crystal Structures of Two 2-Iodo-Phenyl Methyl-Amides as Substrates for Radical Translocation Reactions



Abstract
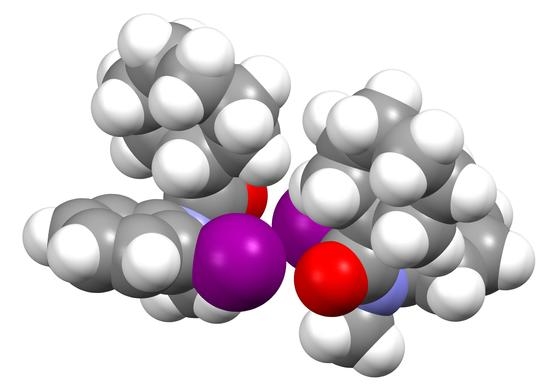
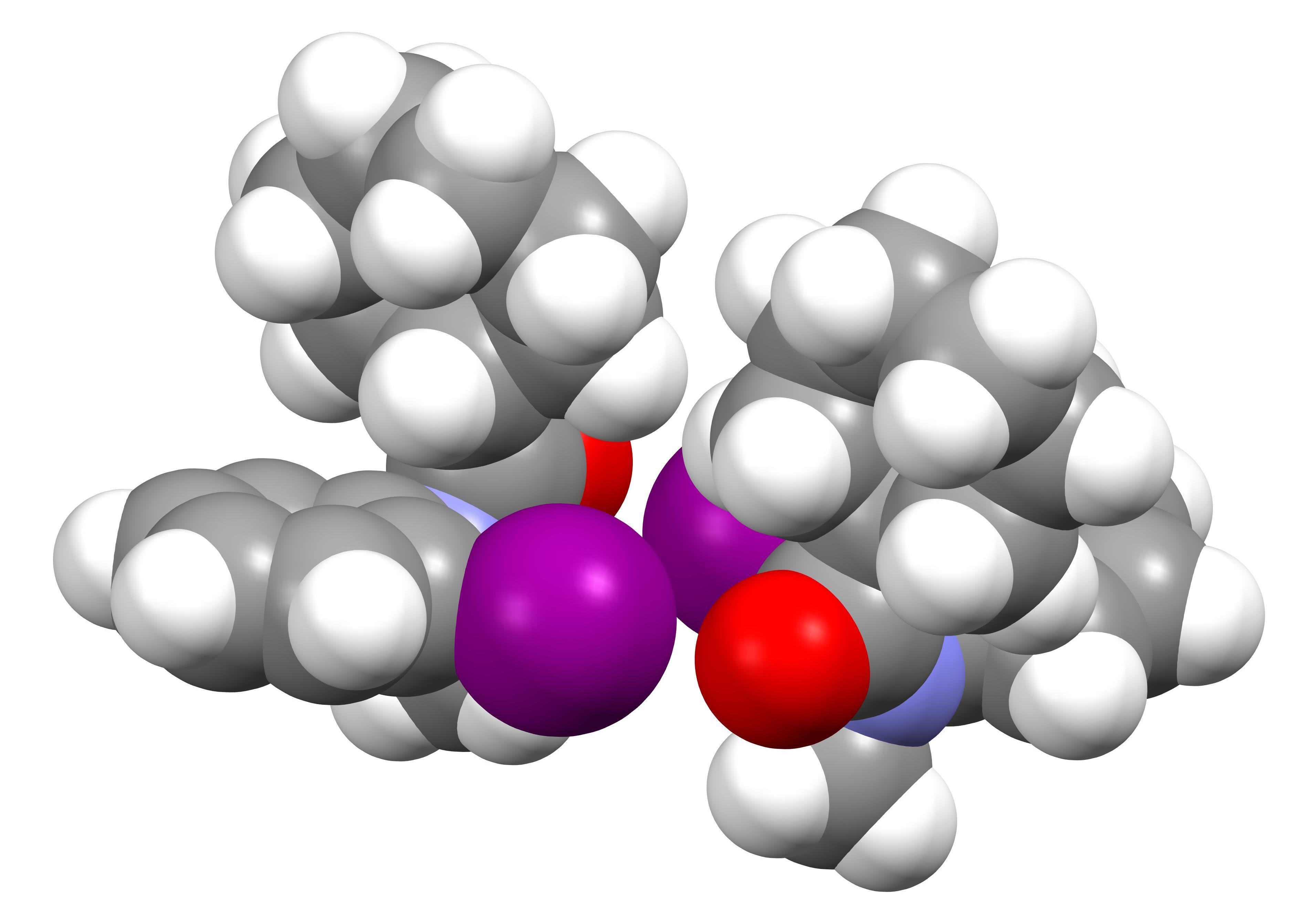
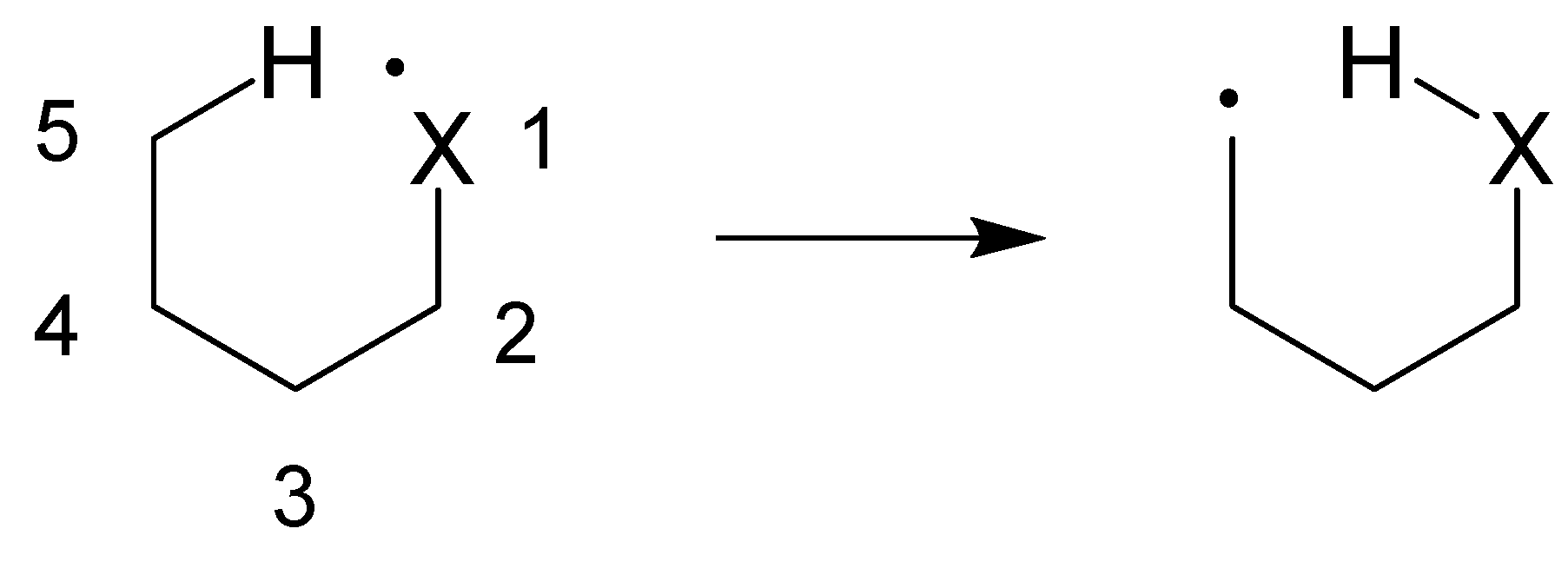
1. Introduction
2. Materials and Methods
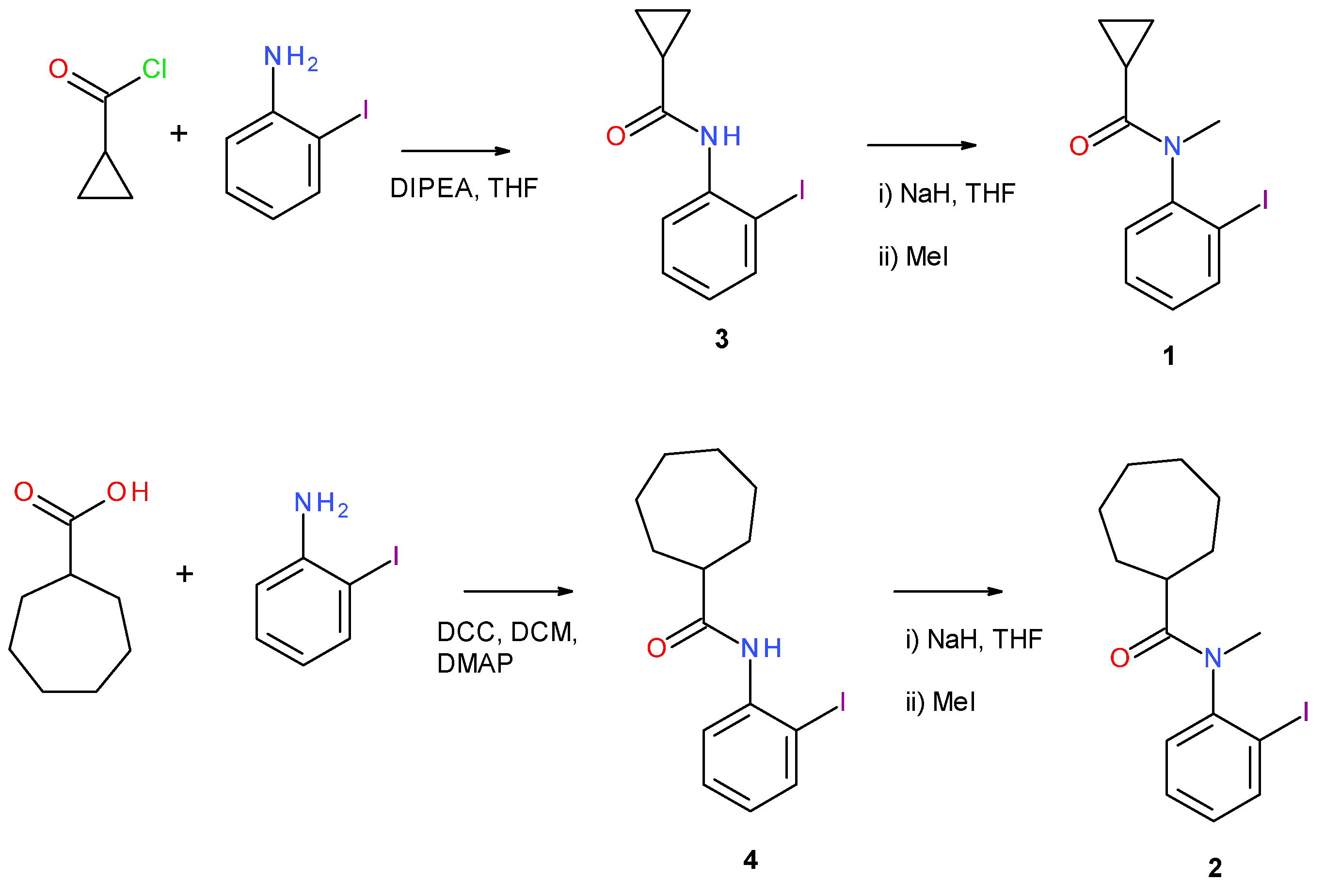
2.1. Synthesis of 1
2.2. Synthesis of 2
2.3. X-ray Data Collection and Refinement
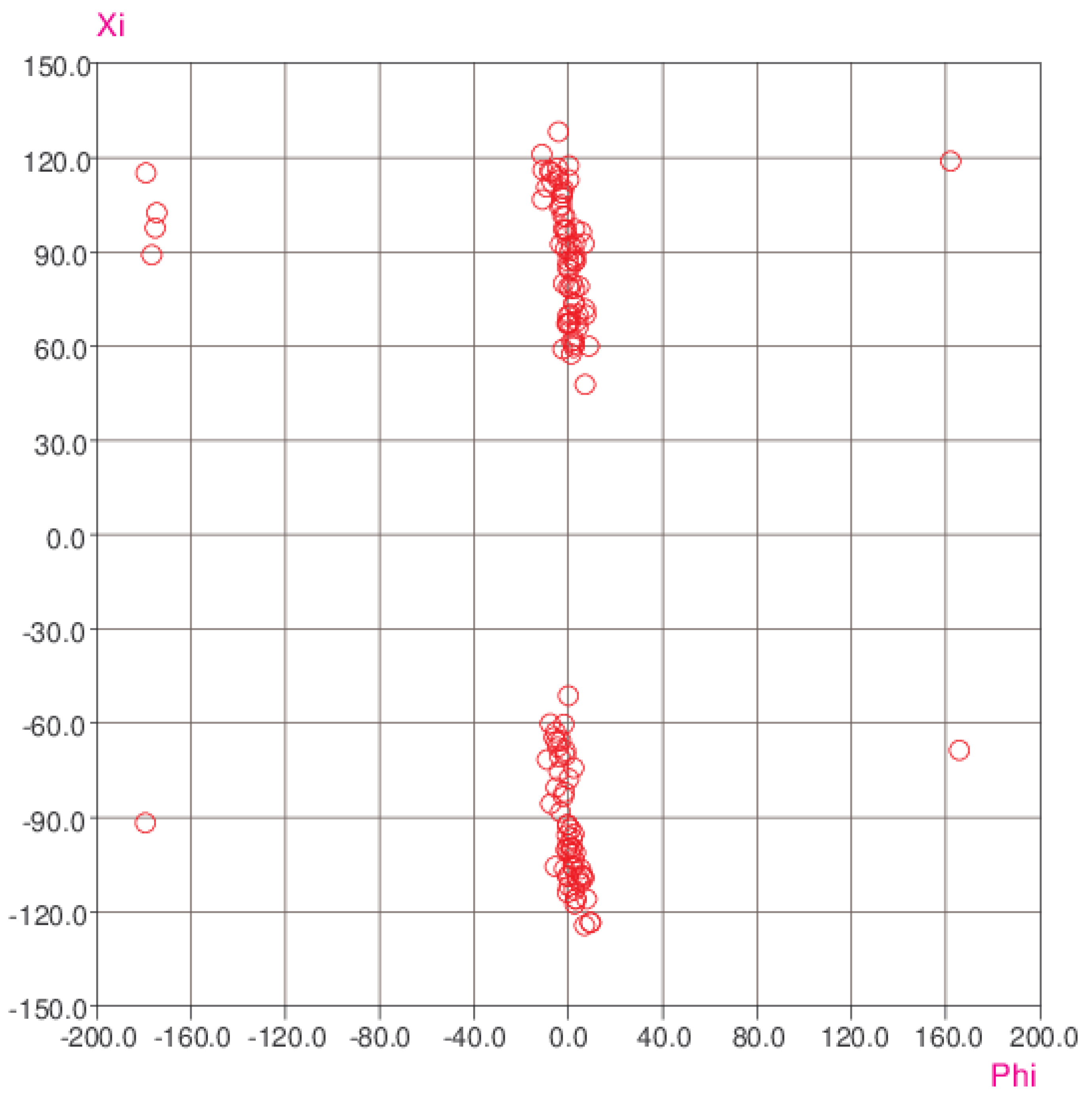
3. Results and Discussion
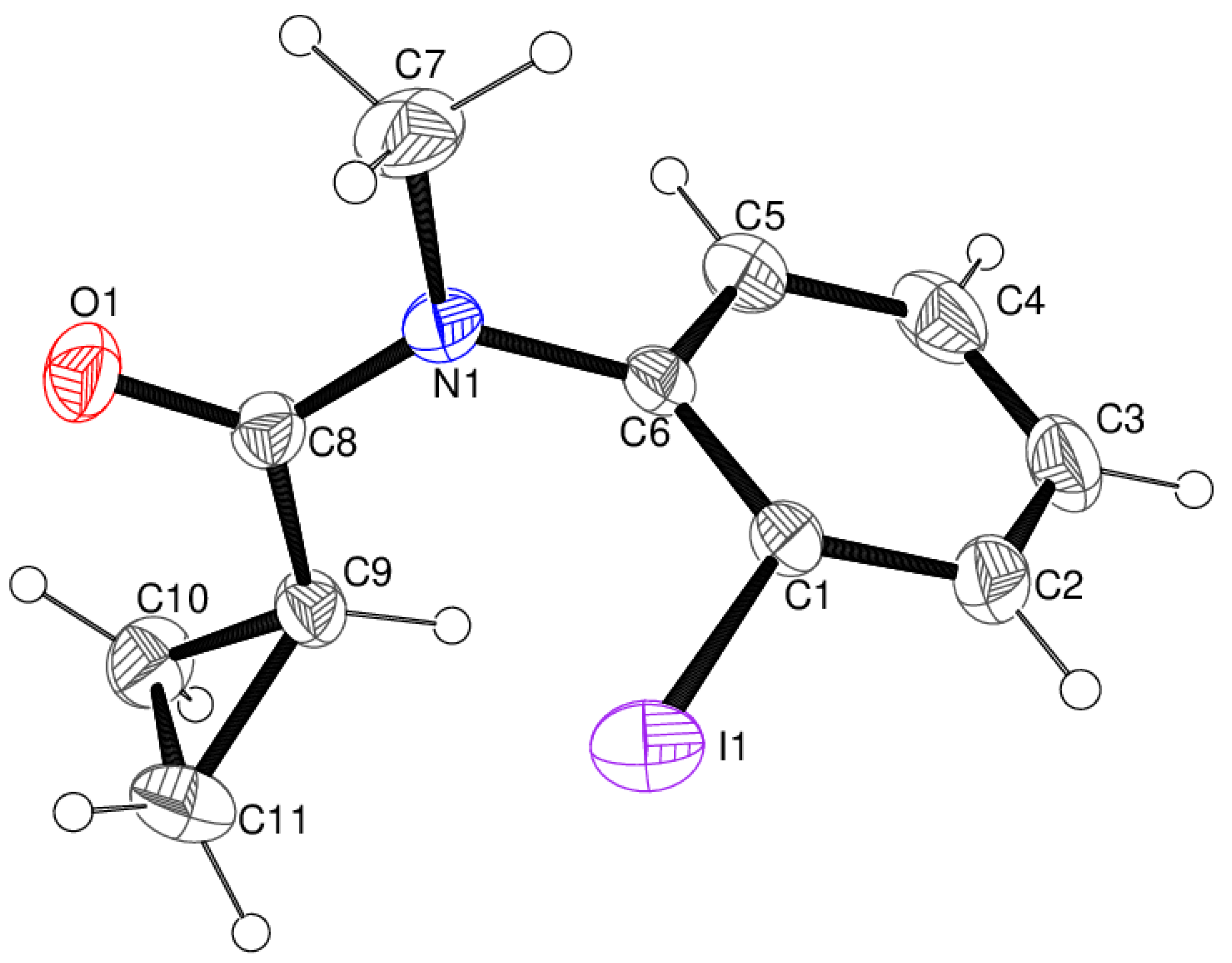
3.1. Structure of C11H12INO (1)
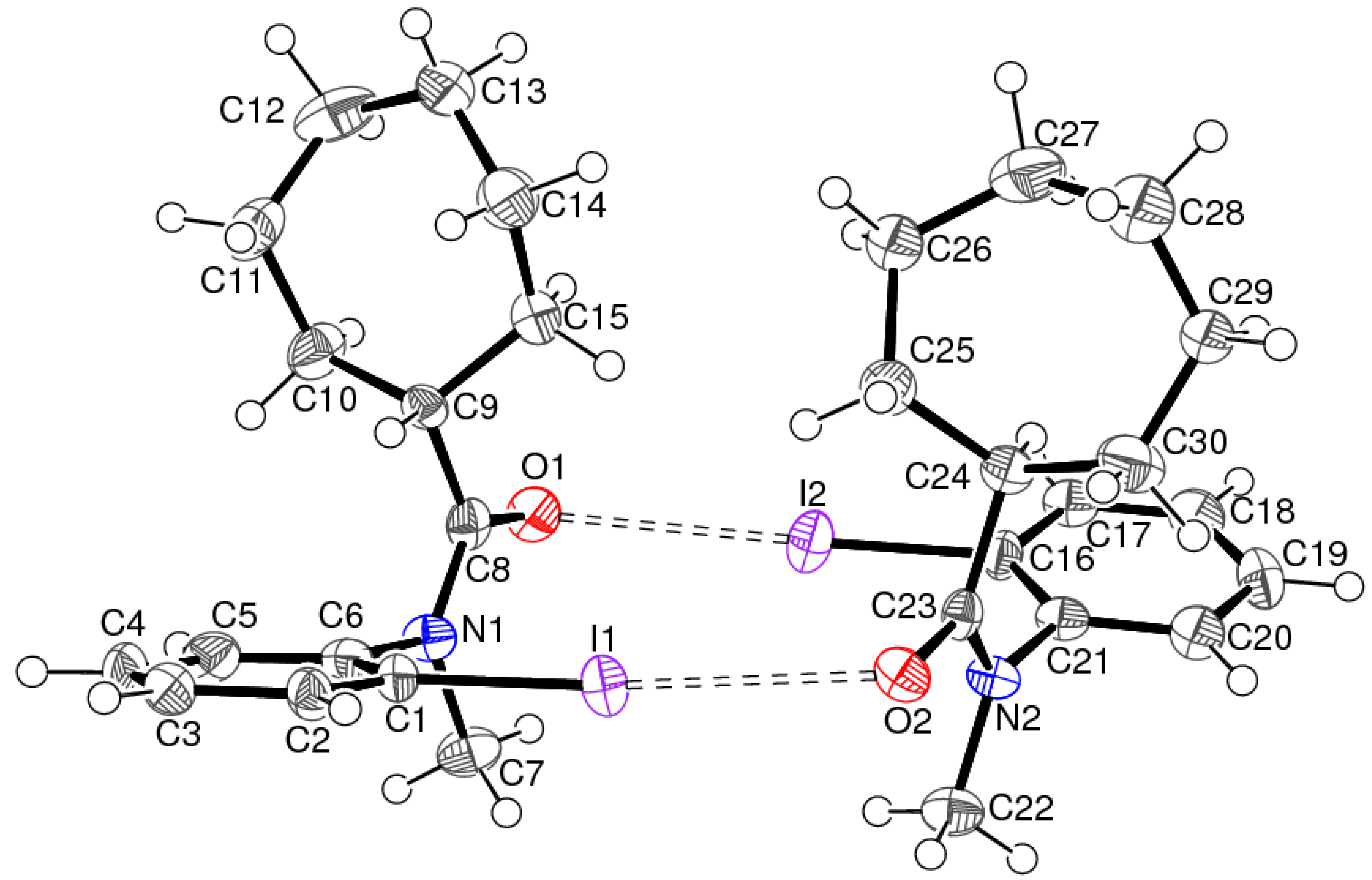
3.2. Structure of C15H14INO (2)
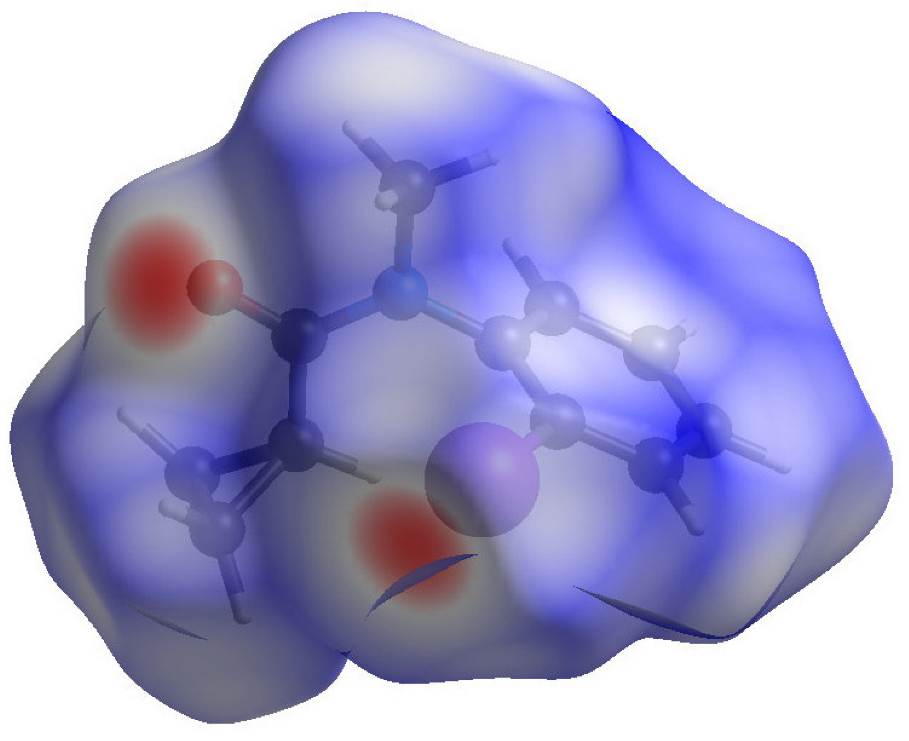
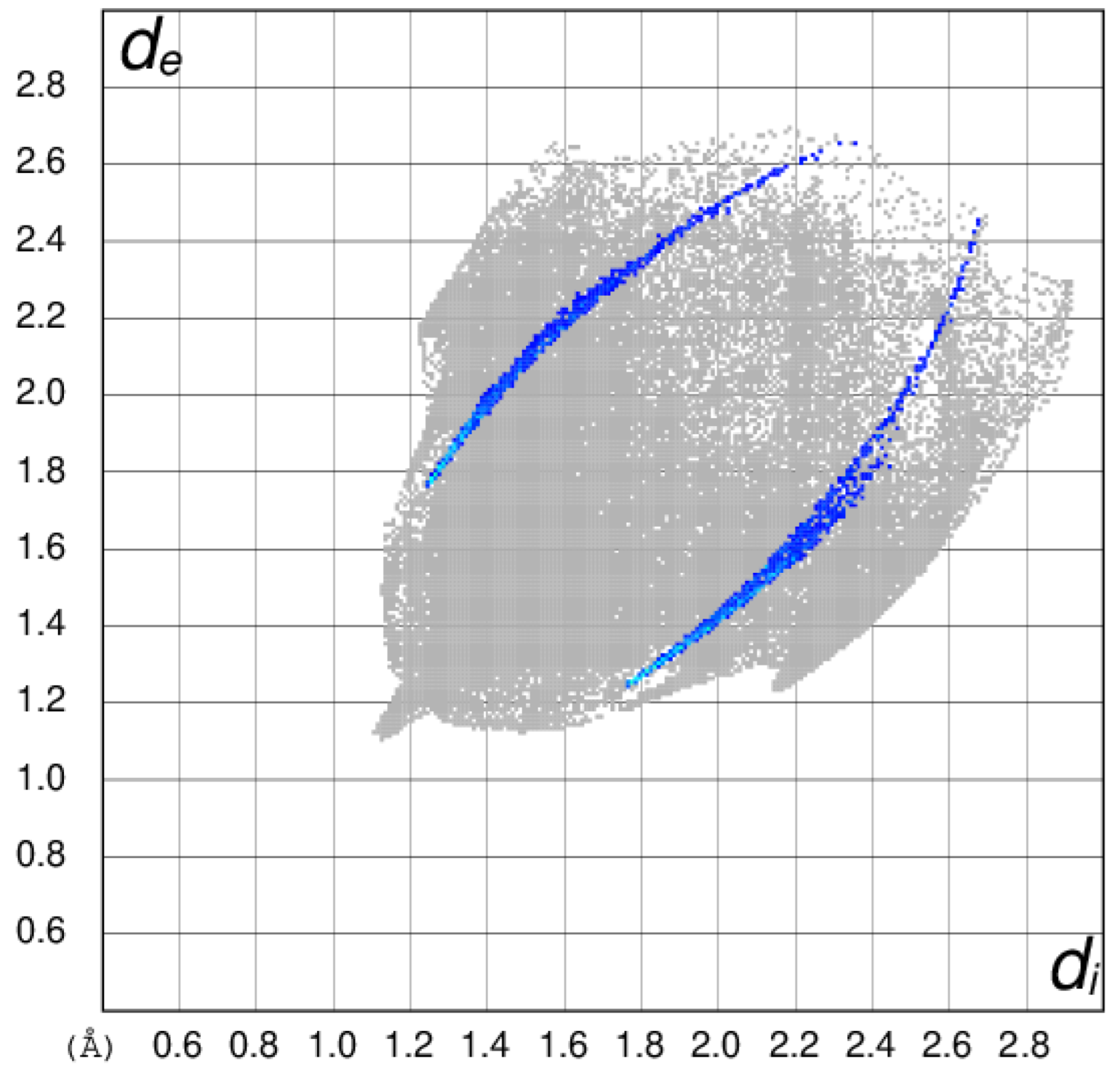
3.3. Hirshfeld Surface Analyses
3.4. Comparison with Related Structures
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Renauld, P.; Sibi, M.P. Radicals in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2002; Volume 1–2. [Google Scholar]
- Takeda, Y.; Nakabayashi, T.; Shirai, A.; Fukumoto, D.; Kiguchi, T.; Naito, T. A formal synthesis of martinelline via a combination of two types of radical reactions. Tetrahedron Lett. 2004, 45, 3481–3484. [Google Scholar] [CrossRef]
- Chakraborty, T.K.; Chattopadhyay, A.K.; Ghosh, S. Total synthesis of (+)-blastmycinone and formal synthesis of (+)-antimycin A(3b). Tetrahedron Lett. 2007, 48, 1139–1142. [Google Scholar] [CrossRef]
- Montevecchi, P.C.; Navacchia, M.L. Rearrangements and cyclizations in vinyl radicals. Unusual example of 1,4-radical translocation. Tetrahedron Lett. 1996, 36, 6583–6586. [Google Scholar] [CrossRef]
- Van Dort, P.C.; Fuchs, P.L. Free radical self-immolative 1,2-elimination and reductive desulfonylation of aryl sulfones promoted by intramolecular reactions with ortho-attached carbon-centered radicals. J. Org. Chem. 1997, 62, 7142–7147. [Google Scholar] [CrossRef] [PubMed]
- Curran, D.P.; Shen, W. Radical translocation reactions of vinyl radicals–substituent effects on 1,5-hydrogen transfer reactions. J. Am. Chem. Soc. 1993, 115, 6051–6059. [Google Scholar] [CrossRef]
- Jones, K.; Storey, J.M.D. Aryl radical cyclizations involving an amide group in the linking chain. J. Chem. Soc. Chem. Commun. 1992, 1766–1767. [Google Scholar] [CrossRef]
- Dorigo, A.E.; Houk, K.N. Transition structures for intramolecular hydrogen-atom transfers—The energetic advantage of 7-membered over 6-membered transition structures. J. Am. Chem. Soc. 1987, 109, 2195–2197. [Google Scholar] [CrossRef]
- Penn, R.E.; Boggs, J.E. Substituent-induced asymmetry of the cyclopropane ring. J. Chem. Soc. Chem. Commun. 1972, 666–667. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Shing Ho, P.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond. Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals’ volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Mallada, B.; Gallardo, A.; Lamanec, M.; De La Torre, B.; Spirko, V.; Hobza, P.; Jelinek, P. Real-space imaging of anisotropic charge of σ-hole by means of Kelvin probe force microscopy. Science 2021, 374, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Metrangolo, P.; Neukirch, H.; Pilati, T.; Resnati, G. Halogen bonding based recognition processes: a world parallel to hydrogen bonding. Acc. Chem. Res. 2005, 38, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Etter, M.C. Encoding and decoding hydrogen-bond patters of organic-compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Turner, M.J.; Mckinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Crystal Explorer 17; The University of Western Australia: Crawley, WA, Australia, 2017. [Google Scholar]
- Tan, S.L.; Jotani, M.M.; Tiekink, E.R.T. Utilizing Hirshfeld surface calculations, non-covalent interaction (NCI) plots and the calculation of interaction energies in the analysis of molecular packing. Acta Cryst. 2019, E75, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Allen, F.H.; Motherwell, W.D.S. Applications of the Cambridge Structural Database in organic chemistry and crystal chemistry. Acta Cryst. 2002, B58, 407–422. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2A | 2B | |
|---|---|---|---|
| C1—I1 | 2.112 (4) | 2.101 (7) | 2.096 (7) |
| C6—N1 | 1.435 (4) | 1.428 (9) | 1.431 (10) |
| C8—N1 | 1.357 (4) | 1.359 (10) | 1.370 (9) |
| C8—O1 | 1.226 (4) | 1.213 (9) | 1.236 (8) |
| C8—C9 | 1.479 (5) | 1.527 (10) | 1.533 (10) |
| O1—C8—N1 | 121.1 (3) | 121.8 (7) | 120.6 (7) |
| C6—N1—C8 | 124.0 (3) | 124.3 (6) | 123.5 (6) |
| C6—N1—C7 | 117.3 (3) | 115.4 (6) | 117.0 (6) |
| C7—N1—C8 | 118.7 (3) | 120.3 (6) | 119.5 (6) |
| C1—C6—N1—C8 (φ) | −87.2 (4) | −89.5 (7) | −90.3 (7) |
| C7—N1—C8—O1 (ξ) | 2.5 (5) | 0.8 (8) | 2.9 (7) |
| C6—N1—C8—C9 (ψ) | 2.6 (5) | −0.2 (7) | 0.4 (7) |
| I⋯O | 3.012 (2) | 3.024 (4) | 3.057 (4) |
| C—I⋯O | 171.78 (9) | 171.71 (17) | 175.98 (16) |
| I⋯O=C | 135.3 (2) | 114.3 (3) | 113.1 (3) |
| C—I⋯O=C | −7.8 (4) | −118.4 (7) | −165.5 (7) |
| I⋯O=C—N | −87.6 (4) | −82.3 (7) | −85.2 (7) |
| Contact Type | 1 | 2A | 2B * |
|---|---|---|---|
| H⋅⋅⋅H | 58.6 | 67.5 | 67.2 |
| H⋅⋅⋅O/O⋅⋅⋅H | 15.3 | 6.5 | 5.5 |
| H⋅⋅⋅I/I⋅⋅⋅H | 14.7 | 12.0 | 12.3 |
| H⋅⋅⋅C/C⋅⋅⋅H | 6.8 | 10.3 | 11.5 |
| O⋅⋅⋅I/I⋅⋅⋅O | 4.1 | 2.6 | 2.6 |
| Others | 0.5 | 1.1 | 0.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ishaq, A.; Storey, J.M.D.; Harrison, W.T.A. Short I⋯O Interactions in the Crystal Structures of Two 2-Iodo-Phenyl Methyl-Amides as Substrates for Radical Translocation Reactions. Chemistry 2023, 5, 1233-1242. https://doi.org/10.3390/chemistry5020083
Ishaq A, Storey JMD, Harrison WTA. Short I⋯O Interactions in the Crystal Structures of Two 2-Iodo-Phenyl Methyl-Amides as Substrates for Radical Translocation Reactions. Chemistry. 2023; 5(2):1233-1242. https://doi.org/10.3390/chemistry5020083
Chicago/Turabian StyleIshaq, Ahtsham, John M. D. Storey, and William T. A. Harrison. 2023. "Short I⋯O Interactions in the Crystal Structures of Two 2-Iodo-Phenyl Methyl-Amides as Substrates for Radical Translocation Reactions" Chemistry 5, no. 2: 1233-1242. https://doi.org/10.3390/chemistry5020083
APA StyleIshaq, A., Storey, J. M. D., & Harrison, W. T. A. (2023). Short I⋯O Interactions in the Crystal Structures of Two 2-Iodo-Phenyl Methyl-Amides as Substrates for Radical Translocation Reactions. Chemistry, 5(2), 1233-1242. https://doi.org/10.3390/chemistry5020083