


Synthesis of 5-Metalla-Spiro[4.5]Heterodecenes by [1,4]-Cycloaddition Reaction of Group 13 Diyls with 1,2-Diketones


Abstract
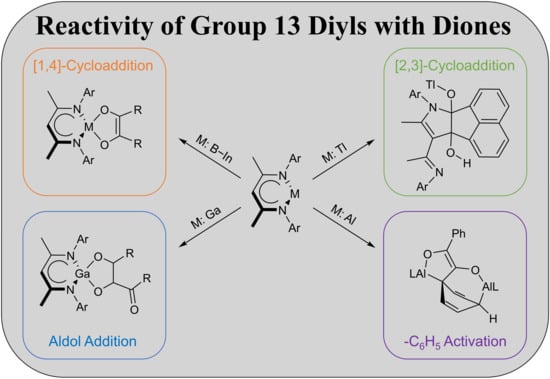
1. Introduction
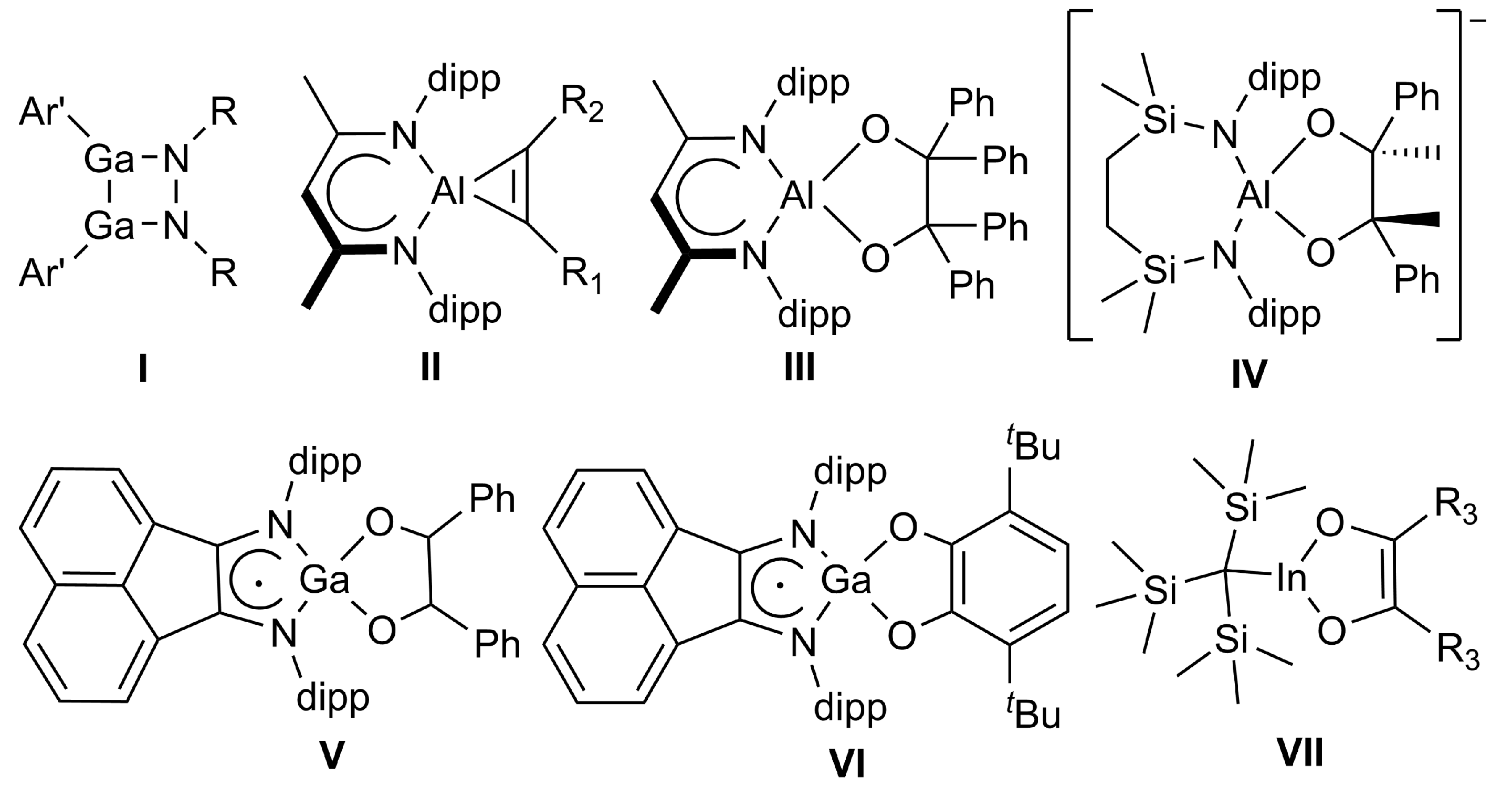
2. Results and Discussion
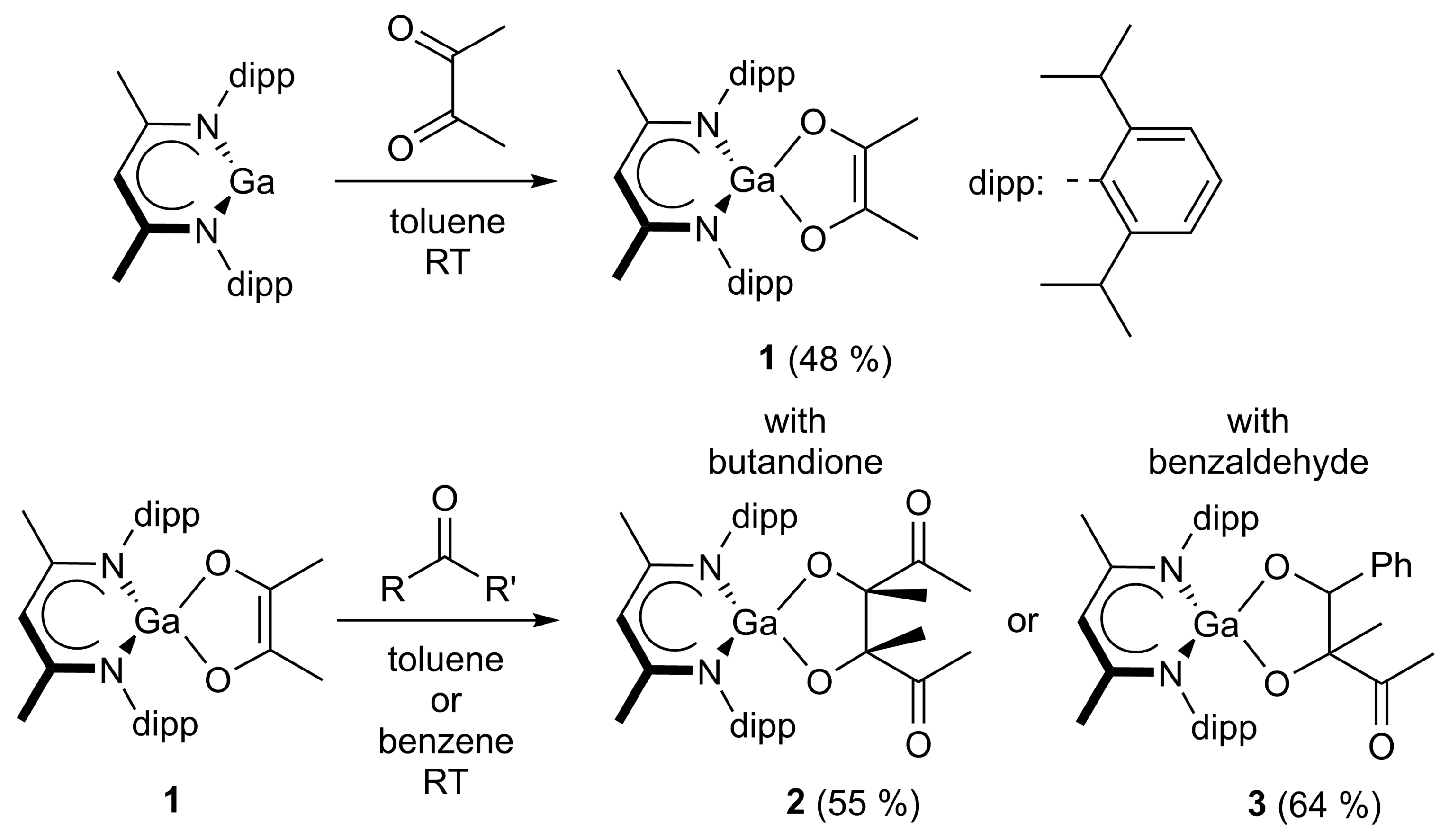
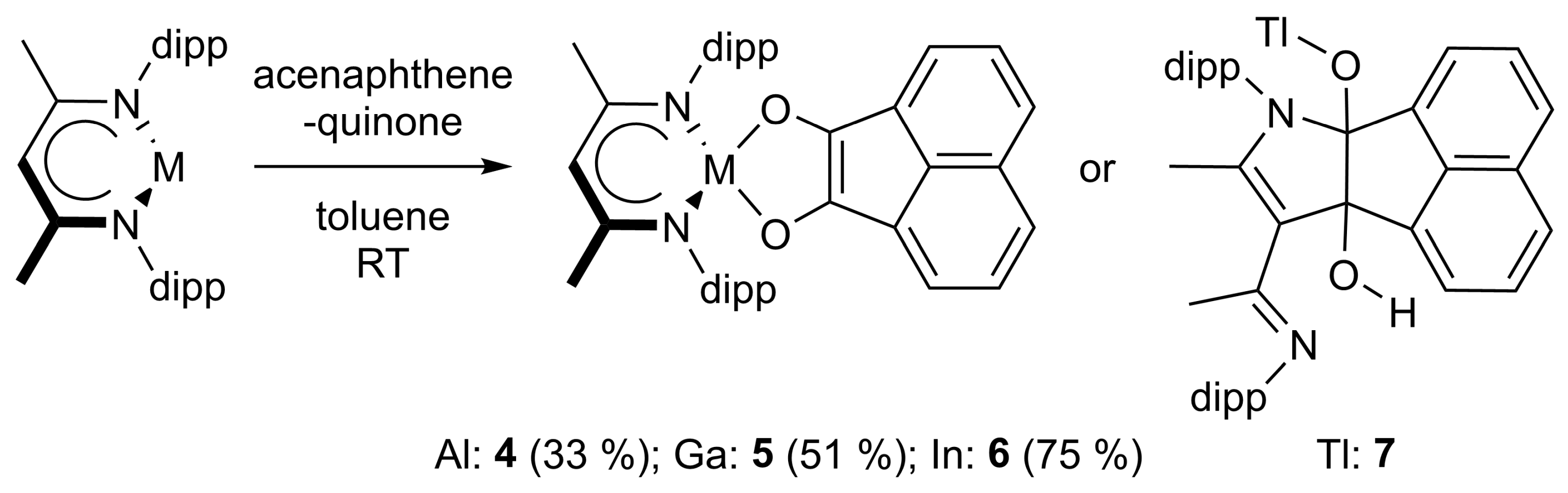
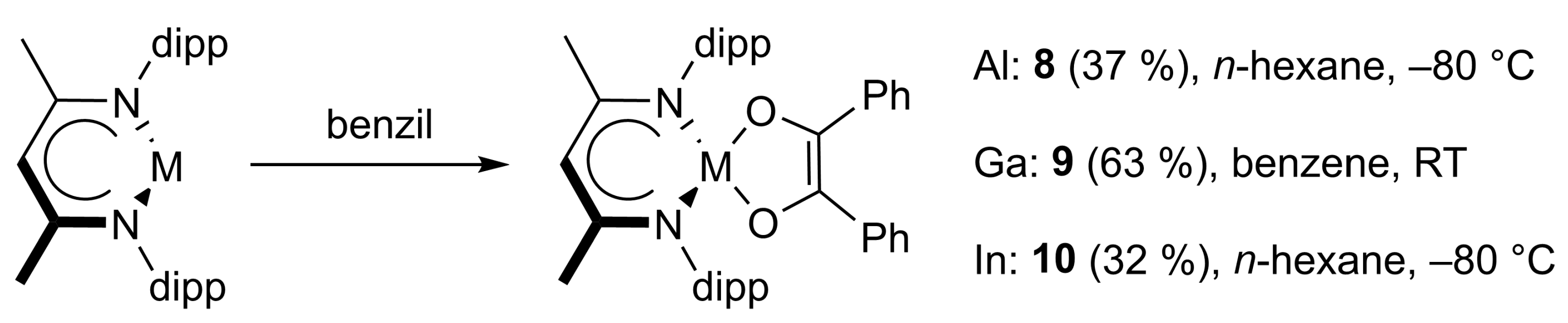
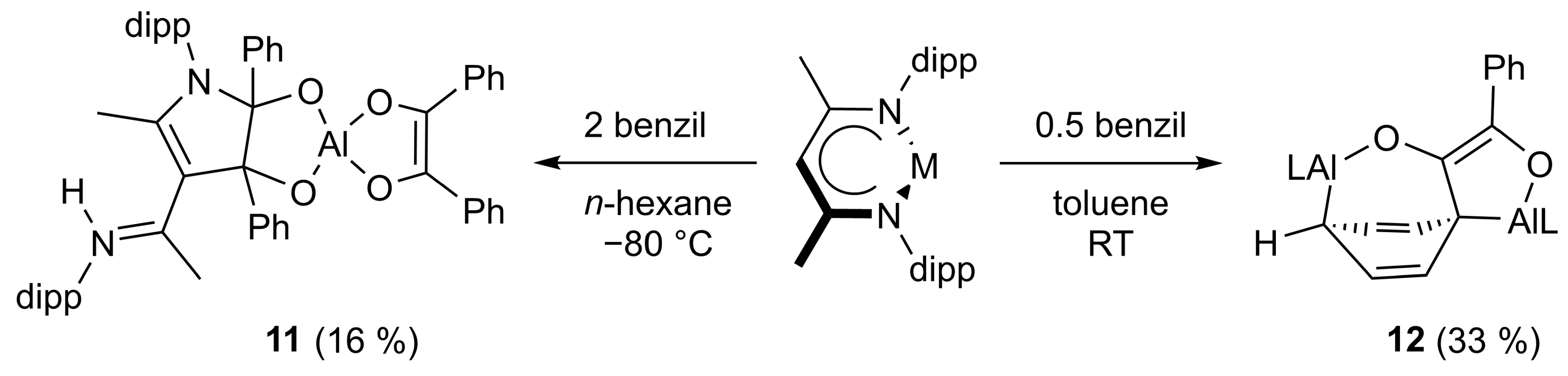
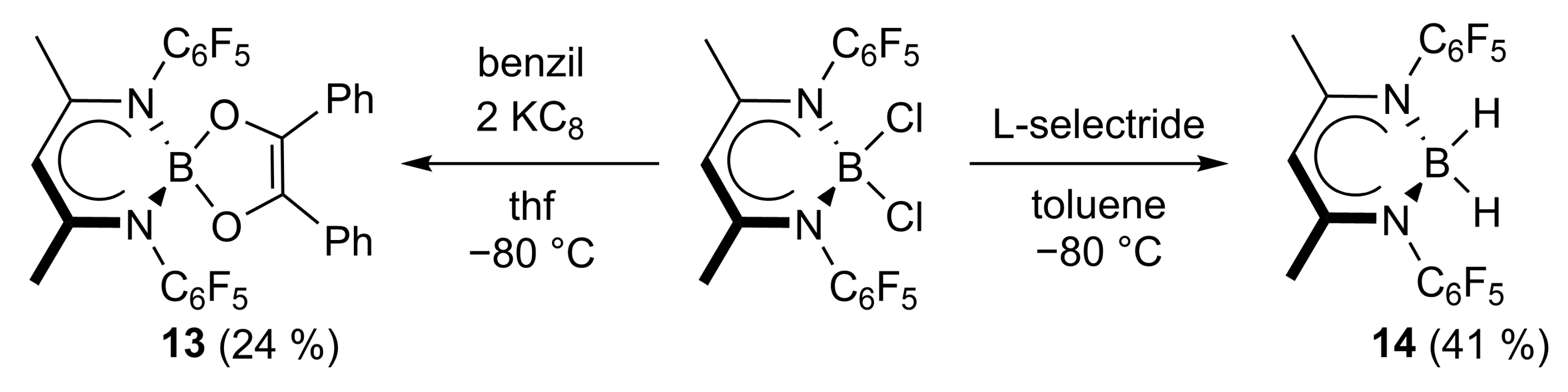
2.1. Synthesis
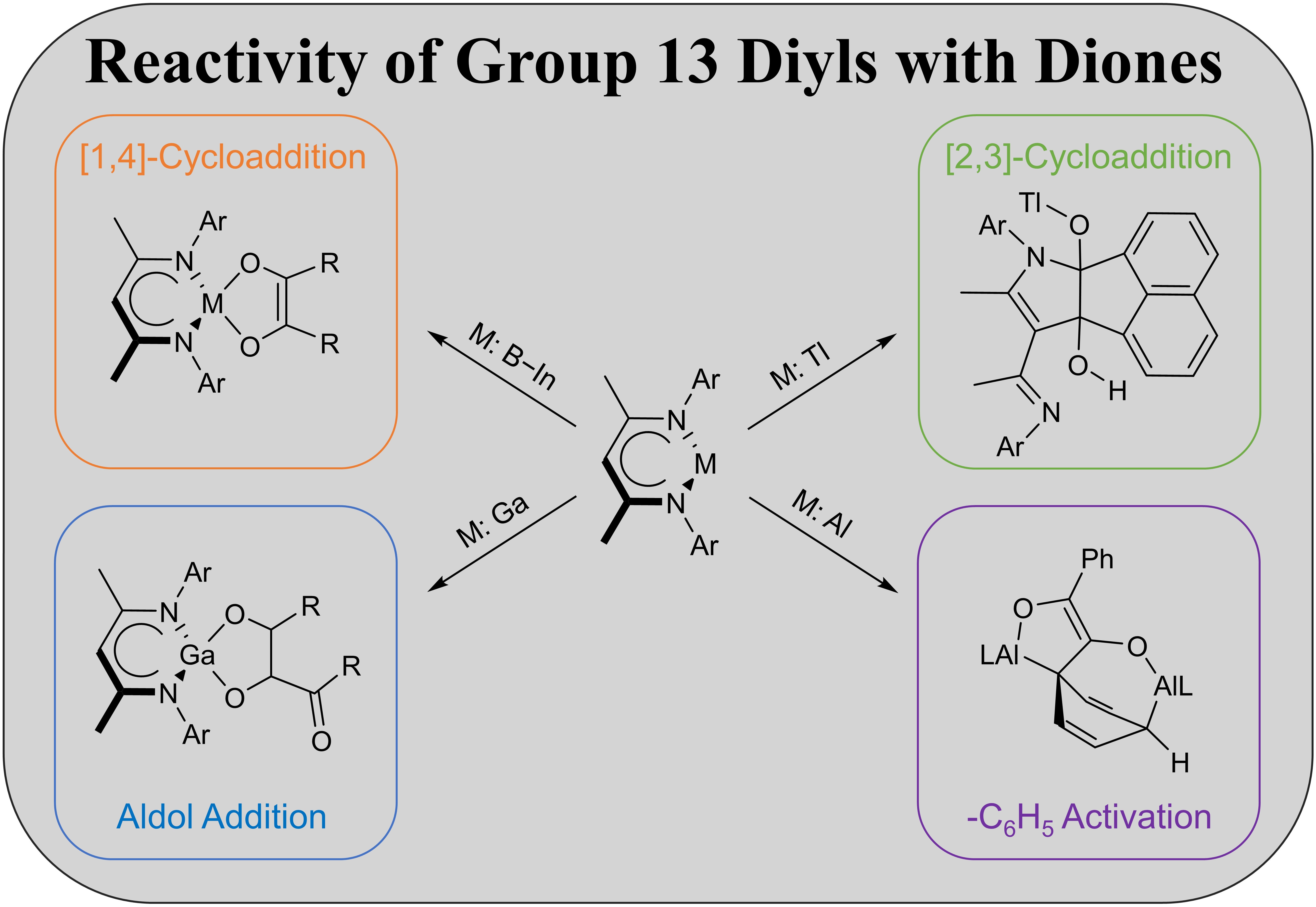
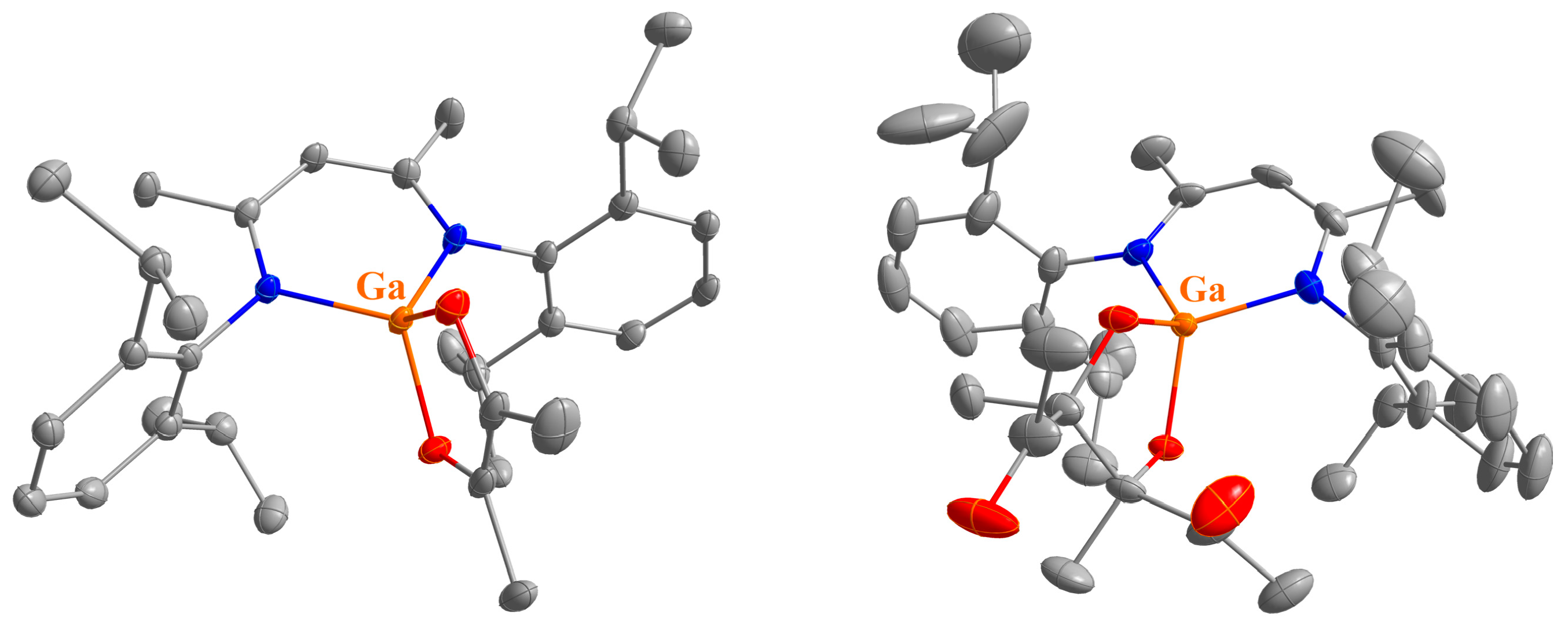
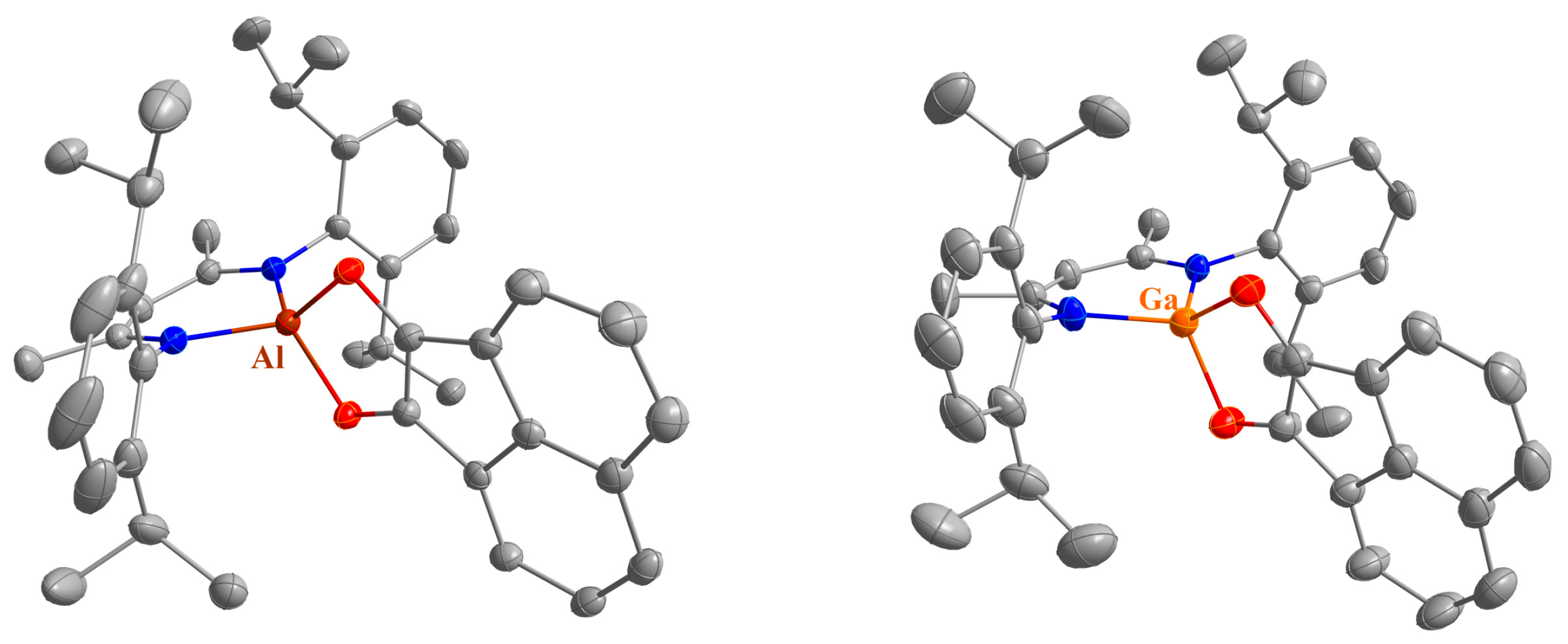
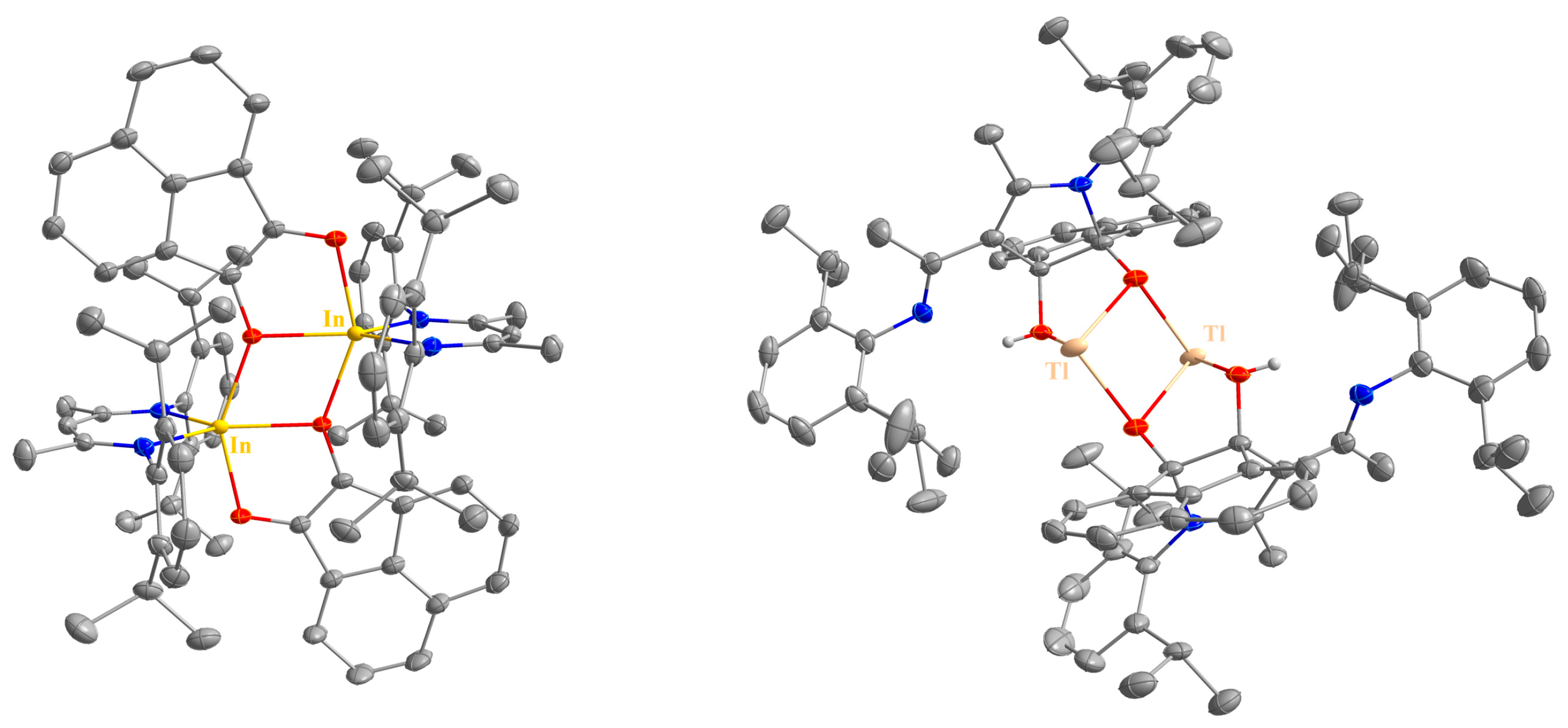
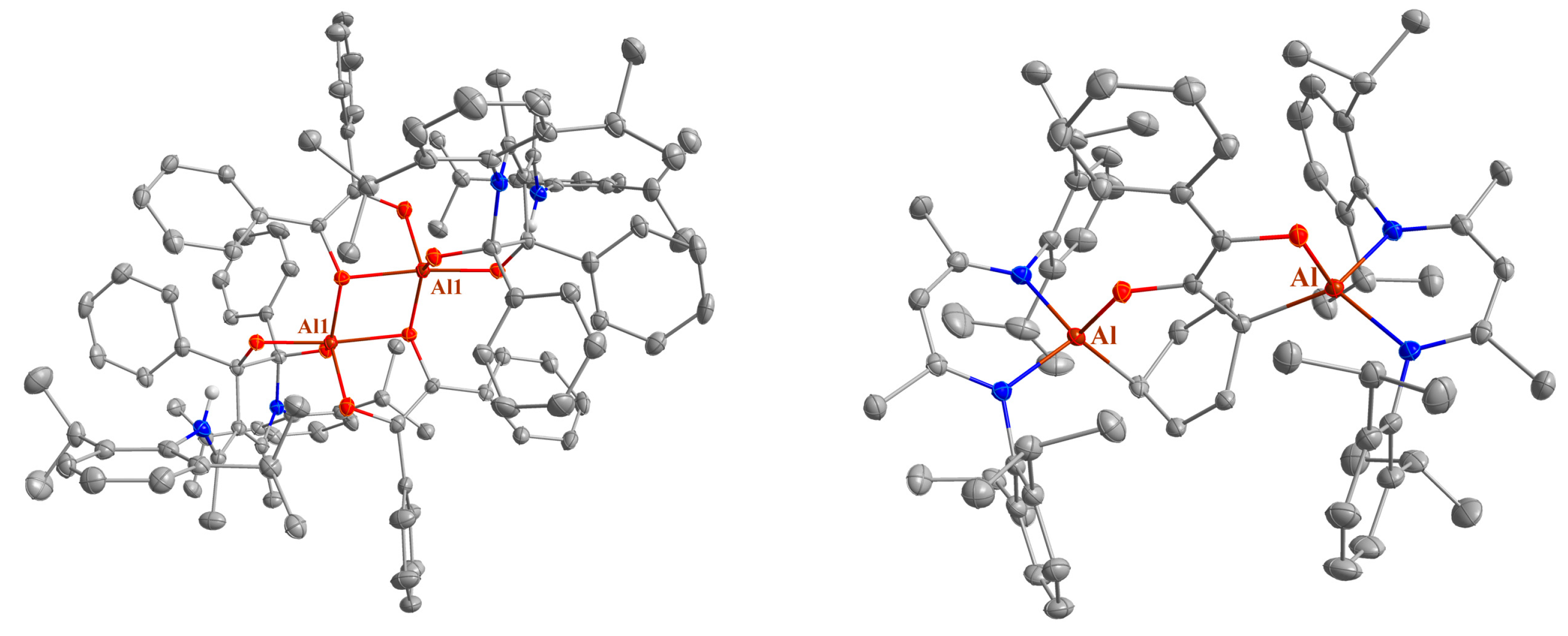
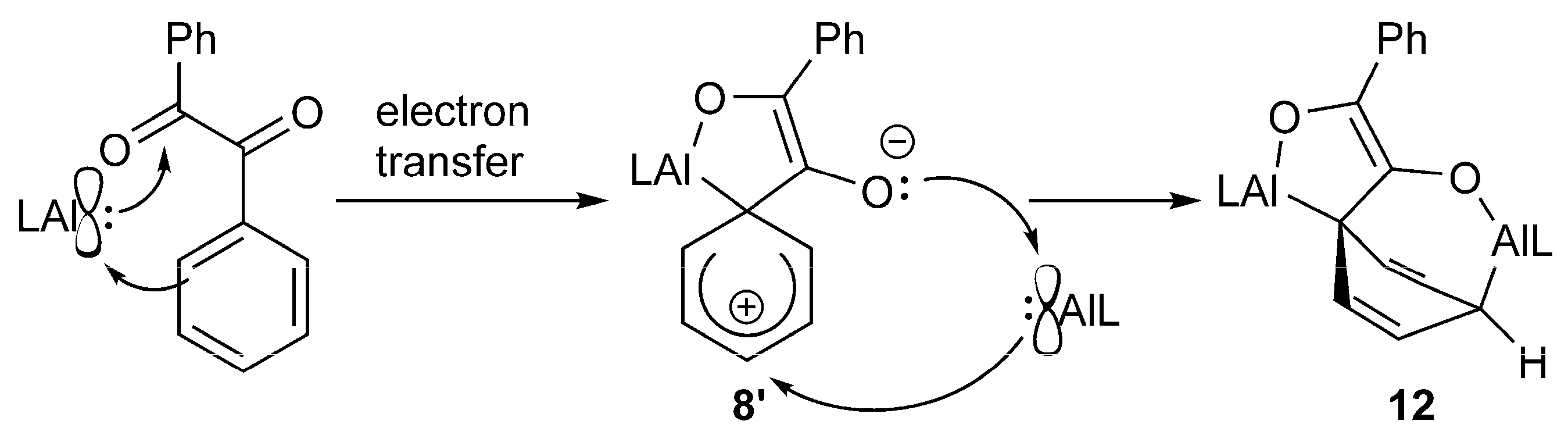
2.2. Spectroscopic Characterization and Single Crystal X-ray Structures
3. Conclusions
4. Materials and Methods
4.1. Synthesis of LGa(C4H6O2) (1)
4.2. Synthesis of LGa(C8H12O4) (2)
4.3. Synthesis of LGa(C11H12O3) (3)
4.4. Synthesis of LAl(C12H6O2) (4)
4.5. Synthesis of LGa(C12H6O2) (5)
4.6. Synthesis of LIn(C12H6O2) (6)
4.7. Synthesis of LTl(C14H10O2) (7)
4.8. Synthesis of LAl(C14H10O2) (8) and LAl(C14H10O2)2 (11)
4.9. Synthesis of LGa(C14H10O2) (9)
4.10. Synthesis of LIn(C14H10O2)·MeCN (10)
4.11. Synthesis of (LAl)2(C14H10O2) (12)
4.12. Synthesis of L’B(C14H10O2) (13)
4.13. Synthesis of L’BH2 (14)
4.14. Crystallographic Details
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cui, C.; Roesky, H.W.; Schmidt, H.-G.; Noltemeyer, M.; Hao, H.; Cimpoesu, F. Synthesis and Structure of a Monomeric Aluminum(I) Compound [{HC(CMeNAr)2}Al] (Ar=2,6–iPr2C6H3): A Stable Aluminum Analogue of a Carbene. Angew. Chem. Int. Ed. 2000, 39, 4274–4276. [Google Scholar] [CrossRef]
- Hardman, N.J.; Eichler, B.E.; Power, P.P. Synthesis and characterization of the monomer Ga{(NDippCMe)2CH} (Dipp = C6H3Pri2-2,6): A low valent gallium(I) carbene analogue. Chem. Commun. 2000, 1991–1992. [Google Scholar] [CrossRef]
- Hill, M.S.; Hitchcock, P.B. A mononuclear indium(I) carbene analogue. Chem. Commun. 2004, 1818–1819. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.S.; Hitchcock, P.B.; Pongtavornpinyo, R. Neutral carbene analogues of the heaviest Group 13 elements: Consideration of electronic and steric effects on structure and stability. Dalton Trans. 2005, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, J.; Ma, X.; Yang, Z.; Roesky, H.W. The chemistry of aluminum(I) with β-diketiminate ligands and pentamethylcyclopentadienyl-substituents: Synthesis, reactivity and applications. Coord. Chem. Rev. 2018, 374, 387–415. [Google Scholar] [CrossRef]
- Weetman, C.; Xu, H.; Inoue, S. Recent Developments in Low-Valent Aluminum Chemistry. EIBC 2020, 1–20. [Google Scholar] [CrossRef]
- Zhong, M.; Sinhababu, S.; Roesky, H.W. The unique β-diketiminate ligand in aluminum(I) and gallium(I) chemistry. Dalton Trans. 2020, 49, 1351–1364. [Google Scholar] [CrossRef]
- Hobson, K.; Carmalt, C.J.; Bakewell, C. Recent advances in low oxidation state aluminium chemistry. Chem. Sci. 2020, 11, 6942–6956. [Google Scholar] [CrossRef]
- Power, P.P. Main-group elements as transition metals. Nature 2010, 463, 171–177. [Google Scholar] [CrossRef]
- Weetman, C.; Inoue, S. The Road Travelled: After Main-Group Elements as Transition Metals. ChemCatChem 2018, 10, 4213–4228. [Google Scholar] [CrossRef]
- Chu, T.; Boyko, Y.; Korobkov, I.; Nikonov, G.I. Transition Metal-like Oxidative Addition of C–F and C–O Bonds to an Aluminum(I) Center. Organometallics 2015, 34, 5363–5365. [Google Scholar] [CrossRef]
- Bakewell, C.; White, A.J.P.; Crimmin, M.R. Reactions of Fluoroalkenes with an Aluminium(I) Complex. Angew. Chem. 2018, 130, 6748–6752. [Google Scholar] [CrossRef]
- Crimmin, M.R.; Butler, M.J.; White, A.J.P. Oxidative addition of carbon-fluorine and carbon-oxygen bonds to Al(I). Chem. Commun. 2015, 51, 15994–15996. [Google Scholar] [CrossRef] [PubMed]
- Chu, T.; Nikonov, G.I. Oxidative Addition and Reductive Elimination at Main-Group Element Centers. Chem. Rev. 2018, 118, 3608–3680. [Google Scholar] [CrossRef]
- Wright, R.J.; Brynda, M.; Fettinger, J.C.; Betzer, A.R.; Power, P.P. Quasi-isomeric gallium amides and imides GaNR2 and RGaNR (R = organic group): Reactions of the digallene, Ar’GaGaAr’ (Ar’ = C6H3-2,6-(C6H3-2,6-Pri2)2) with unsaturated nitrogen compounds. J. Am. Chem. Soc. 2006, 128, 12498–12509. [Google Scholar] [CrossRef]
- Cui, C.; Köpke, S.; Herbst-Irmer, R.; Roesky, H.W.; Noltemeyer, M.; Schmidt, H.G.; Wrackmeyer, B. Facile synthesis of cyclopropene analogues of aluminum and an aluminum pinacolate, and the reactivity of LAlη2-C2(SiMe3)2 toward unsaturated molecules (L = HC(CMe)(NAr)2, Ar = 2,6-i-Pr2C6H3). J. Am. Chem. Soc. 2001, 123, 9091–9098. [Google Scholar] [CrossRef]
- Liu, H.-Y.; Hill, M.S.; Mahon, M.F. Diverse reactivity of an Al(I)-centred anion towards ketones. Chem. Commun. 2022, 58, 6938–6941. [Google Scholar] [CrossRef]
- Sokolov, V.G.; Koptseva, T.S.; Moskalev, M.V.; Bazyakina, N.L.; Piskunov, A.V.; Cherkasov, A.V.; Fedushkin, I.L. Gallium Hydrides with a Radical-Anionic Ligand. Inorg. Chem. 2017, 56, 13401–13410. [Google Scholar] [CrossRef]
- Fedushkin, I.L.; Skatova, A.A.; Dodonov, V.A.; Chudakova, V.A.; Bazyakina, N.L.; Piskunov, A.V.; Demeshko, S.V.; Fukin, G.K. Digallane with redox-active diimine ligand: Dualism of electron-transfer reactions. Inorg. Chem. 2014, 53, 5159–5170. [Google Scholar] [CrossRef]
- Uhl, W.; Keimling, S.U.; Phol, S.; Saak, W.; Wartchow, R. Benzil Derivatives as Trapping Reagents for the Monomeric Alkylindium(i) Compound in–c(SiMe3)3. Chem. Ber. 1997, 130, 1269–1272. [Google Scholar] [CrossRef]
- Ganesamoorthy, C.; Bläser, D.; Wölper, C.; Schulz, S. Temperature-Dependent Electron Shuffle in Molecular Group 13/15 Intermetallic Complexes. Angew. Chem. 2014, 126, 11771–11775. [Google Scholar] [CrossRef]
- Ganesamoorthy, C.; Helling, C.; Wölper, C.; Frank, W.; Bill, E.; Cutsail, G.E., III; Schulz, S. From stable Sb- and Bi-centered radicals to a compound with a Ga=Sb double bond. Nat. Commun. 2018, 9, 87. [Google Scholar] [CrossRef] [PubMed]
- Helling, C.; Wölper, C.; Schulz, S. Synthesis of a gallaarsene {HC[C(Me)N-2,6-i-Pr2-C6H3]2}GaAsCp* containing a Ga=As double bond. J. Am. Chem. Soc. 2018, 140, 5053–5056. [Google Scholar] [CrossRef]
- Krüger, J.; Ganesamoorthy, C.; John, L.; Wölper, C.; Schulz, S. A General Pathway for the Synthesis of Gallastibenes containing Ga=Sb Double Bonds. Chem. Eur. J. 2018, 24, 9157–9164. [Google Scholar] [CrossRef]
- Helling, C.; Cutsail, G.E.; Weinert, H.; Wölper, C.; Schulz, S. Ligand Effects on the Electronic Structure of Heteroleptic Antimony-Centered Radicals. Angew. Chem. 2020, 132, 7631–7638. [Google Scholar] [CrossRef]
- Ganesamoorthy, C.; Schoening, J.; Wölper, C.; Song, L.; Schreiner, P.R.; Schulz, S. A silicon–carbonyl complex stable at room temperature. Nat. Chem. 2020, 12, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Weinert, H.M.; Wölper, C.; Haak, J.; Cutsail, G.E.; Schulz, S. Synthesis, structure and bonding nature of heavy dipnictene radical anions. Chem. Sci. 2021, 12, 14024–14032. [Google Scholar] [CrossRef]
- Weinert, H.M.; Wölper, C.; Schulz, S. Synthesis of distibiranes and azadistibiranes by cycloaddition reactions of distibenes with diazomethanes and azides. Chem. Sci. 2022, 13, 3775–3786. [Google Scholar] [CrossRef]
- Sharma, M.K.; Wölper, C.; Haberhauer, G.; Schulz, S. Reversible and Irreversible [2+2] Cycloaddition Reactions of Heteroallenes to a Gallaphosphene. Angew. Chem. 2021, 133, 21953–21957. [Google Scholar] [CrossRef]
- Sharma, M.K.; Wölper, C.; Haberhauer, G.; Schulz, S. Multi-talented gallaphosphene for Ga–P–Ga heteroallyl cation generation, CO2 storage and C(sp3)–H bond activation. Angew. Chem. 2021, 133, 6859–6865. [Google Scholar] [CrossRef]
- Wöhler, F.; von Liebig, J. Untersuchungen über das Radikal der Benzoesäure. Ann. Pharm. 1832, 3, 249–282. [Google Scholar] [CrossRef]
- Chen, C.-H.; Tsai, M.-L.; Su, M.-D. Theoretical Study of the Reactivities of Neutral Six-Membered Carbene Analogues of the Group 13 Elements. Organometallics 2006, 25, 2766–2773. [Google Scholar] [CrossRef]
- Hardman, N.J.; Phillips, A.D.; Power, P.P. Bonding and Reactivity of a β-Diketiminate, Gallium(I), Carbene Analogue. ACS Symp. Ser. 2002, 822, 2–15. [Google Scholar] [CrossRef]
- Vidovic, D.; Findlater, M.; Cowley, A.H. A β-diketiminate-Supported Boron Dication. J. Am. Chem. Soc. 2007, 129, 8436–8437. [Google Scholar] [CrossRef]
- Pyykkö, P.; Atsumi, M. Molecular single-bond covalent radii for elements 1-118. Chem. Eur. J. 2009, 15, 186–197. [Google Scholar] [CrossRef]
- Panda, A.; Stender, M.; Wright, R.J.; Olmstead, M.M.; Klavins, P.; Power, P.P. Synthesis and characterization of three-coordinate and related beta-diketiminate derivatives of manganese, iron, and cobalt. Inorg. Chem. 2002, 41, 3909–3916. [Google Scholar] [CrossRef]
- Qian, B.; Ward, D.L.; Smith, M.R. Synthesis, Structure, and Reactivity of β-Diketiminato Aluminum Complexes. Organometallics 1998, 17, 3070–3076. [Google Scholar] [CrossRef]
- Stender, M.; Eichler, B.E.; Hardman, N.J.; Power, P.P.; Prust, J.; Noltemeyer, M.; Roesky, H.W. Synthesis and Characterization of HC{C(Me)N(C6H3-2,6-i-Pr2)}2MX2 (M = Al, X = Cl, I; M = Ga, In, X = Me, Cl, I): Sterically Encumbered β-Diketiminate Group 13 Metal Derivatives. Inorg. Chem. 2001, 40, 2794–2799. [Google Scholar] [CrossRef]
- Stender, M.; Wright, R.J.; Eichler, B.E.; Prust, J.; Olmstead, M.M.; Roesky, H.W.; Power, P.P. The synthesis and structure of lithium derivatives of the sterically encumbered β-diketiminate ligand [{(2,6-Pri2H3C6)N(CH3)C}2CH]–, and a modified synthesis of the aminoimine precursor. Dalton Trans. 2001, 3465–3469. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Phase annealing in SHELX-90: Direct methods for larger structures. Acta Crystallogr. 1990, A46, 467–473. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXL-2014, Program for the Refinement of Crystal Structures; Univ. of Göttingen: Göttingen, Germany, 2014. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Cryst. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- van der Sluis, P.; Spek, A.L. BYPASS: An effective method for the refinement of crystal structures containing disordered solvent regions. Acta Cryst. 1990, A46, 194–201. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| O–M | O–M–O | N–M | N–M–N | |
|---|---|---|---|---|
| 1 | 1.8280(10), 1.8355(10) | 92.89(5) | 1.9122(12), 1.9176(12) | 98.53(5) |
| 2 a | 1.827(3), 1.831(3); 1.836(3), 1.831(3) | 91.88(13); 92,45(14) | 1.920(3), 1,916(3); 1.919(3), 1.903(3) | 98.84(15); 98.67(13) |
| 4 | 1.7804(9), 1.7766(8) | 97.18(4) | 1.8582(10), 1.8597(10) | 98.16(4) |
| 5 | 1.8648(14), 1.8665(13) | 94.80(5) | 1.9055(14), 19150(13) | 99.87(6) |
| 6 | 2.0903(9), 2.2310(10) | 79.12(4) | 2.1537(11), 2.1580(11) | 93.39(5) |
| 7 | 2.7463(18) b, 2.4536(15) c | 61.51(5) | / | / |
| 8 a | 1.7502(14), 1.7544(14); 1.74958(14), 1.7489(14) | 93.36(5); 92.92(5) | 1.8648(15), 1.8664(14); 1.8641(14), 1.8620(14) | 98.21(6); 98.11(7) |
| 9 a | 1.8336(10), 1.8315(10); 1.8293(10), 1.8364(10) | 91.73(4); 91.20(4) | 1.9056(12), 1.9025(11); 1.9050(11), 1.9055(11) | 99.79(5); 98.99(5) |
| 10 | 2.0684(10), 2.0474(10) | 82.29(4) | 2.1192(10), 2.1289(11) | 92.23(4) |
| 11 | 1.7830(16), 1.8087(16); 1.9530(16) d, 1.7941(17) | 88.15(7); 84.26(7) | / | / |
| 12a | 1.7393(8), 1.9885(11) e; 1.7496(8), 2.0136(11) e | 108.90(4)e; 94.06(4) e | 1.9095(9), 1.9115(9); 1.9125(9), 1.8974(9) | 95.97(4); 95.31(4) |
| 12b a | 1.7332(11), 1.9851(15) e; 1.7507(11), 2.014(14) e 1.7355(11), 1.9830(15) e; 1.7492(11), 2.0155(15) e | 108.17(6) e, 93.88(5) e 107.81(6) e, 93.83(5) e | 1.9127(13), 1.9022(13); 1.9120(13), 1.9070(13) 1.9148(13), 1.9060(13); 1.9059(13), 1.9151(13) | 95.09(6); 94.83(5) 94.76(5); 95.11(5) |
| 13 | 1.4573(13), 1.4587(13) | 105.18(8) | 1.5622(14), 1.5576(14) | 105.51(8) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weinert, H.M.; Wölper, C.; Schulz, S. Synthesis of 5-Metalla-Spiro[4.5]Heterodecenes by [1,4]-Cycloaddition Reaction of Group 13 Diyls with 1,2-Diketones. Chemistry 2023, 5, 948-964. https://doi.org/10.3390/chemistry5020064
Weinert HM, Wölper C, Schulz S. Synthesis of 5-Metalla-Spiro[4.5]Heterodecenes by [1,4]-Cycloaddition Reaction of Group 13 Diyls with 1,2-Diketones. Chemistry. 2023; 5(2):948-964. https://doi.org/10.3390/chemistry5020064
Chicago/Turabian StyleWeinert, Hanns M., Christoph Wölper, and Stephan Schulz. 2023. "Synthesis of 5-Metalla-Spiro[4.5]Heterodecenes by [1,4]-Cycloaddition Reaction of Group 13 Diyls with 1,2-Diketones" Chemistry 5, no. 2: 948-964. https://doi.org/10.3390/chemistry5020064
APA StyleWeinert, H. M., Wölper, C., & Schulz, S. (2023). Synthesis of 5-Metalla-Spiro[4.5]Heterodecenes by [1,4]-Cycloaddition Reaction of Group 13 Diyls with 1,2-Diketones. Chemistry, 5(2), 948-964. https://doi.org/10.3390/chemistry5020064