


Natural Products as Mcl-1 Inhibitors: A Comparative Study of Experimental and Computational Modelling Data


Abstract
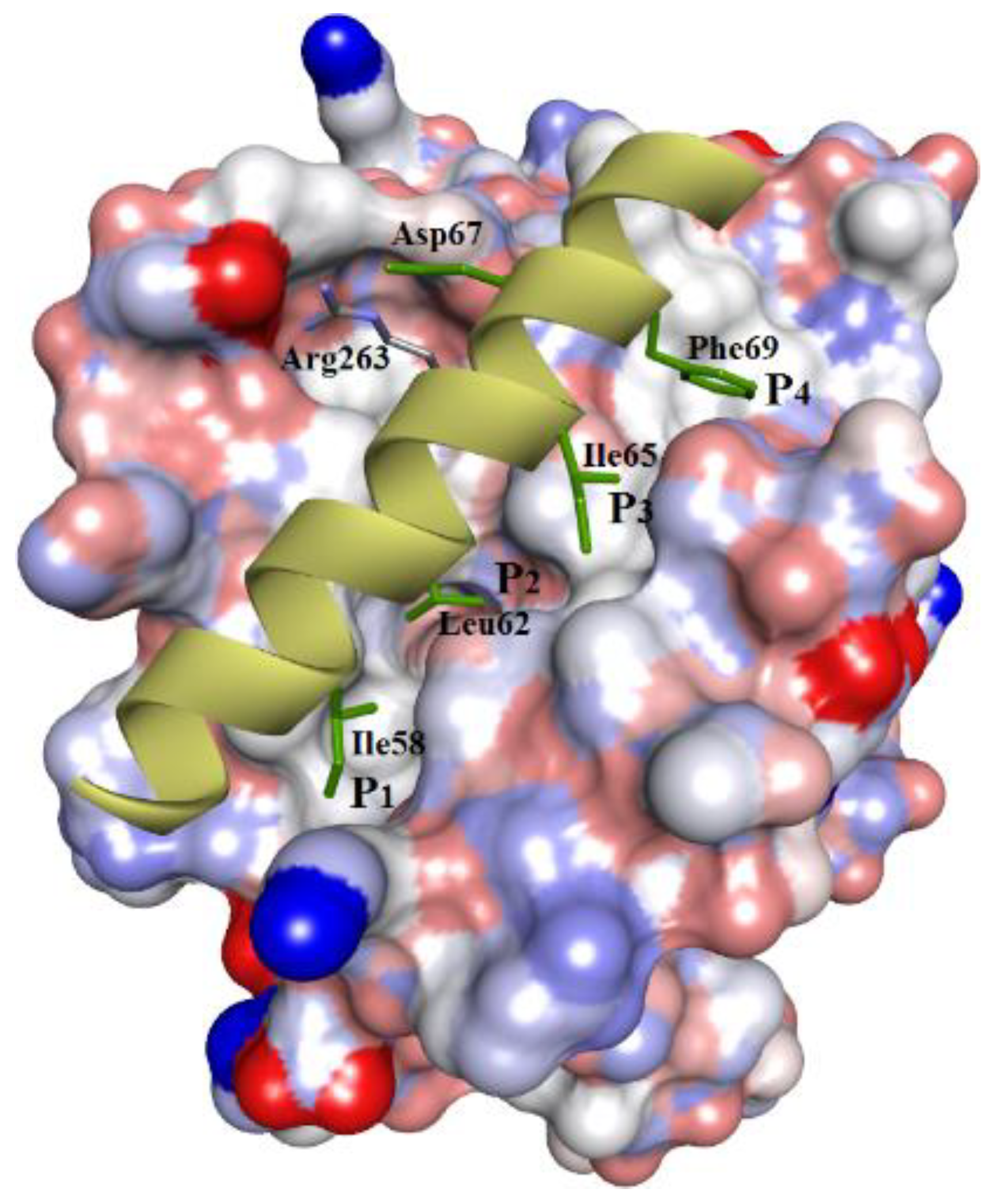
:1. Introduction
2. Materials and Methods
2.1. Protein Structure Preparation for Modeling
2.2. Ligand Structure Preparation
2.3. Superposition and Docking Protocols and Analysis
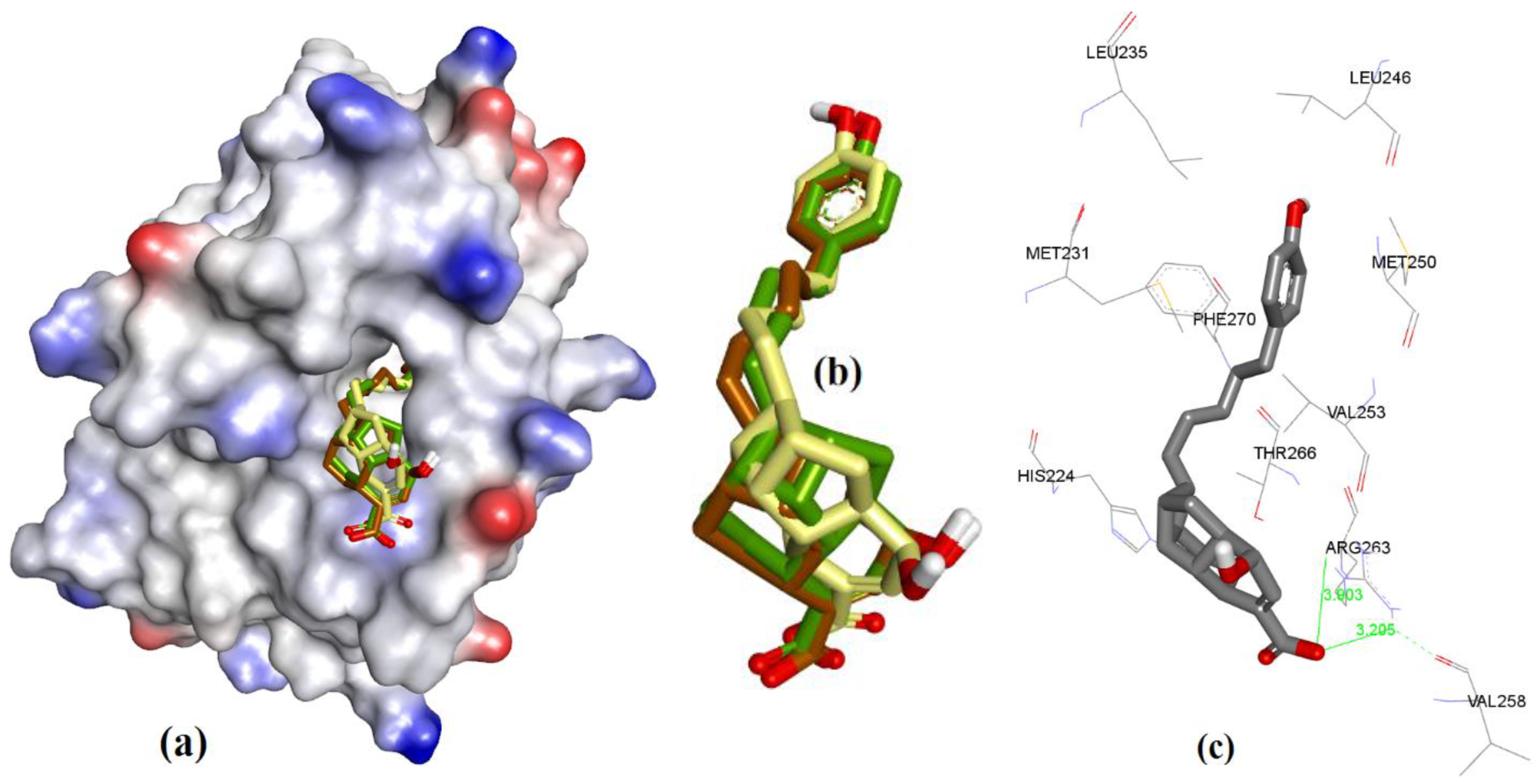
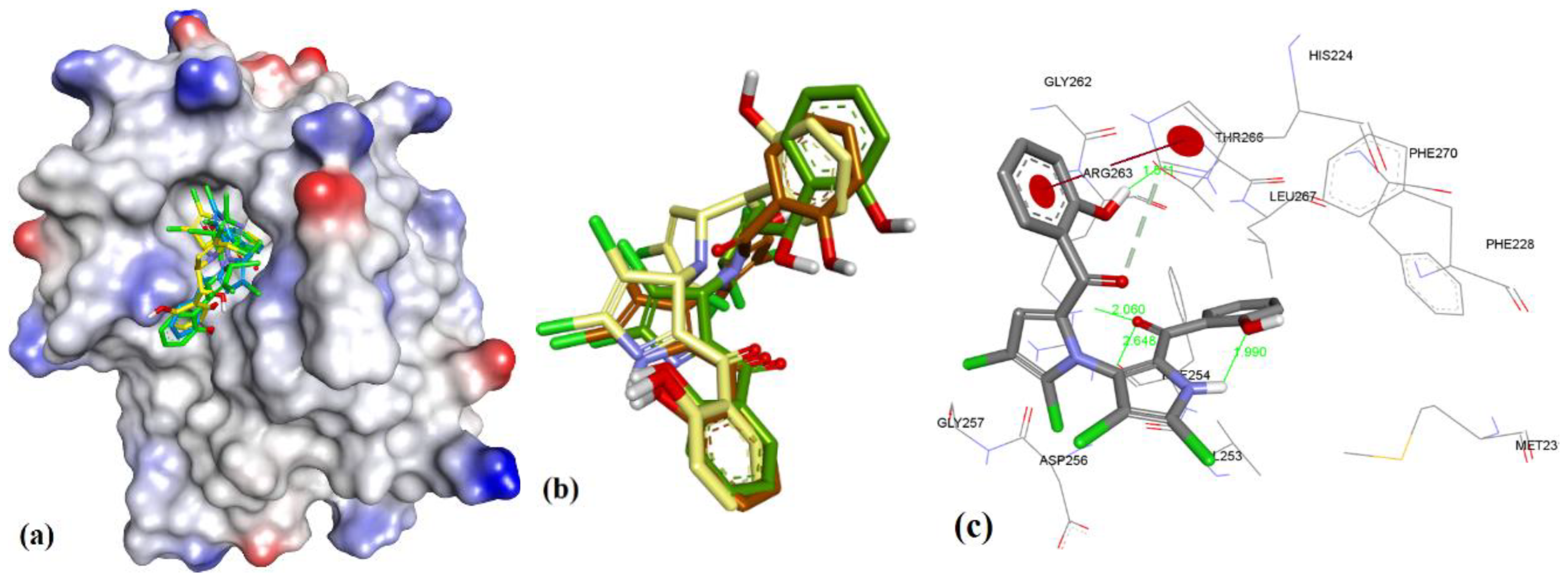
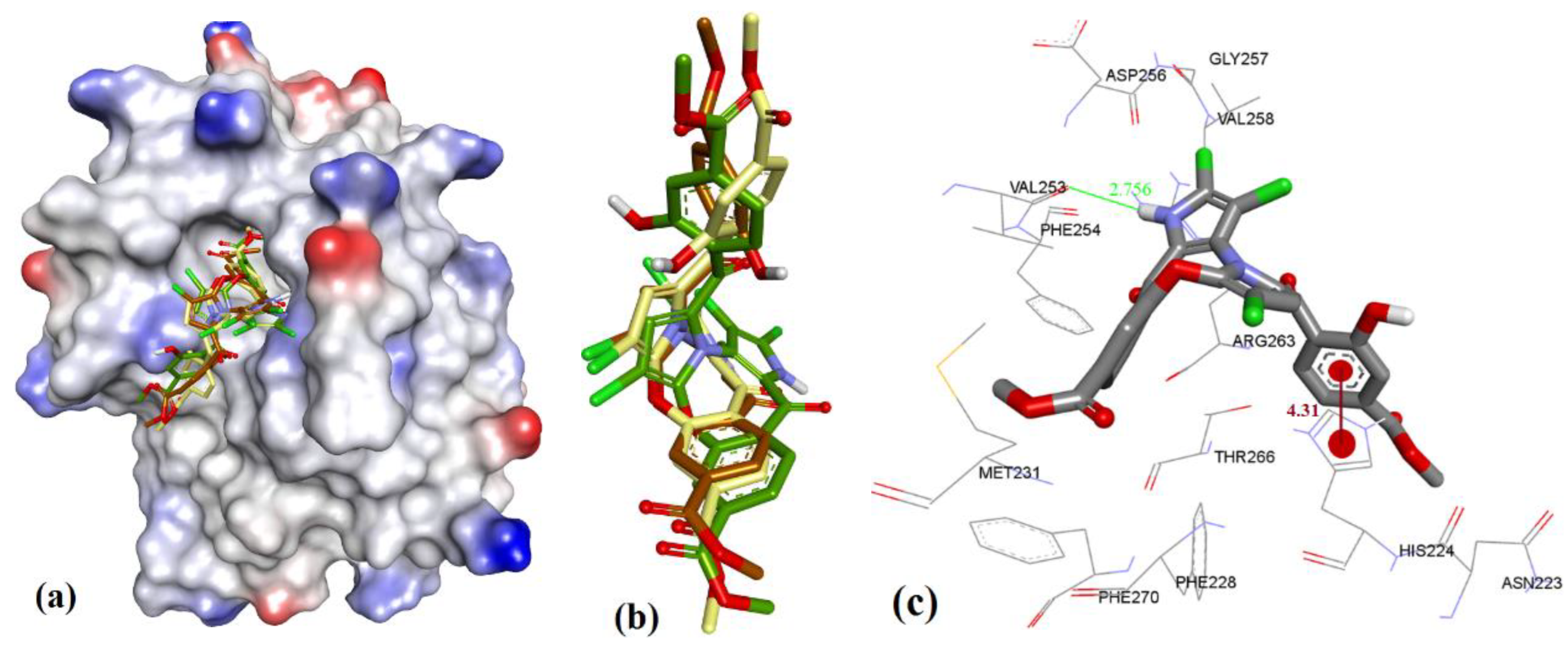
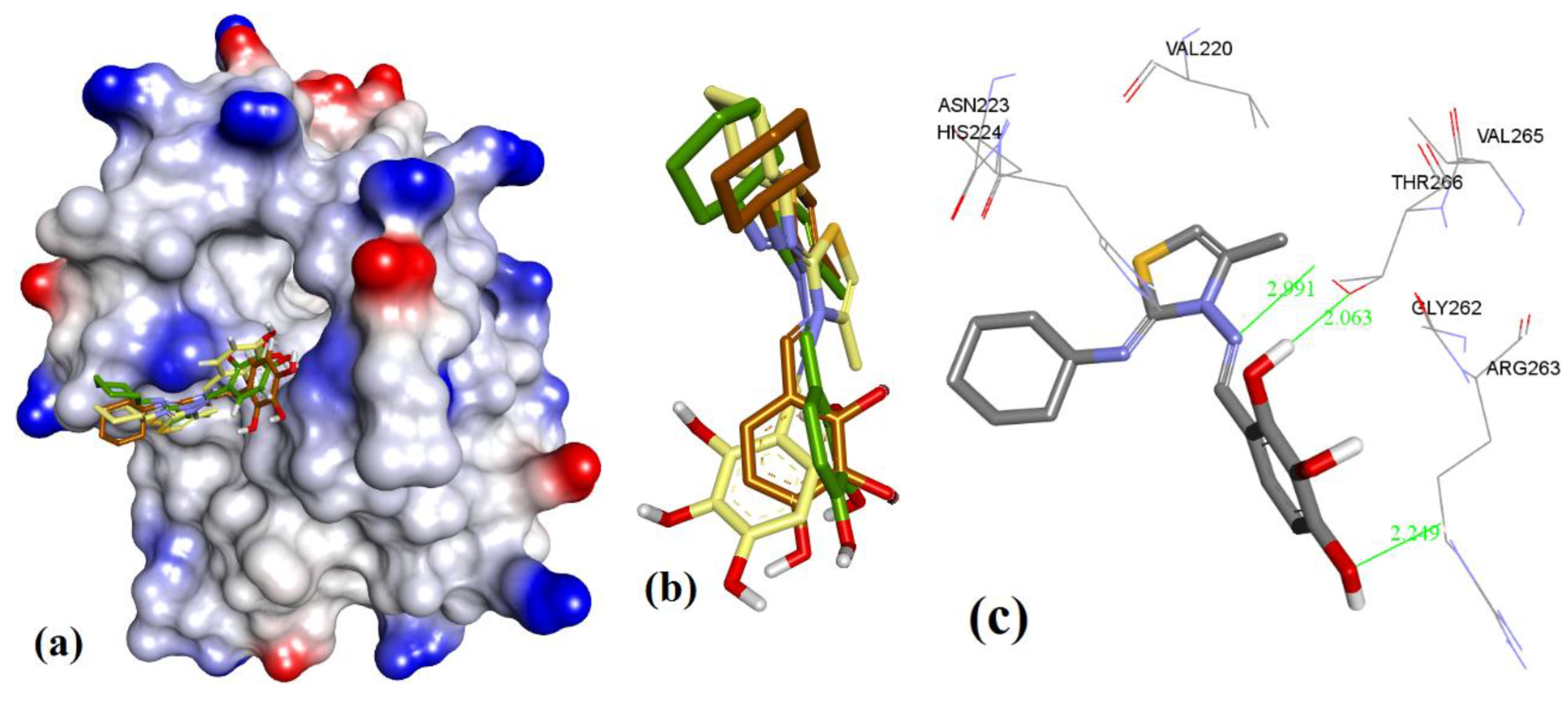
3. Results
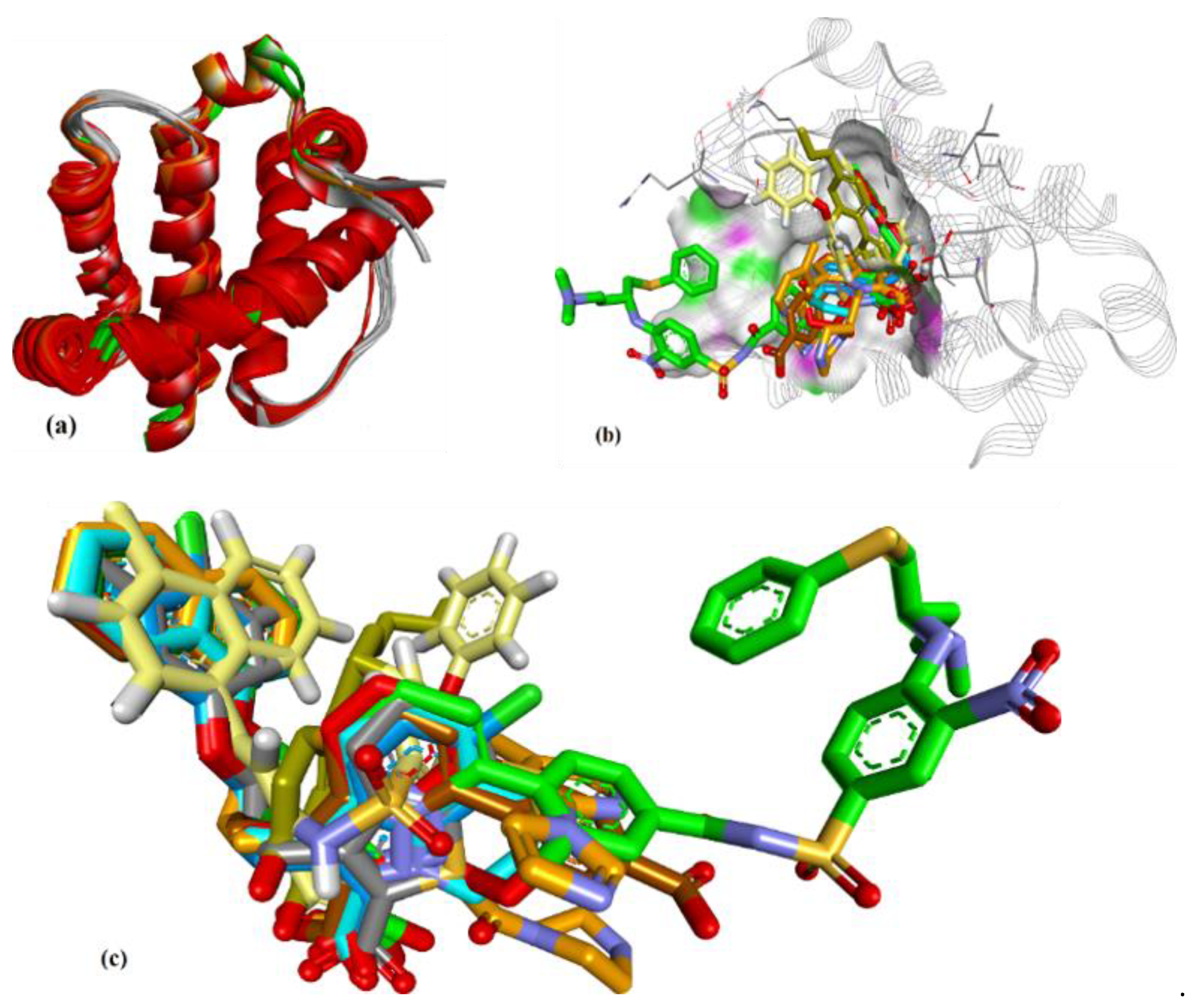
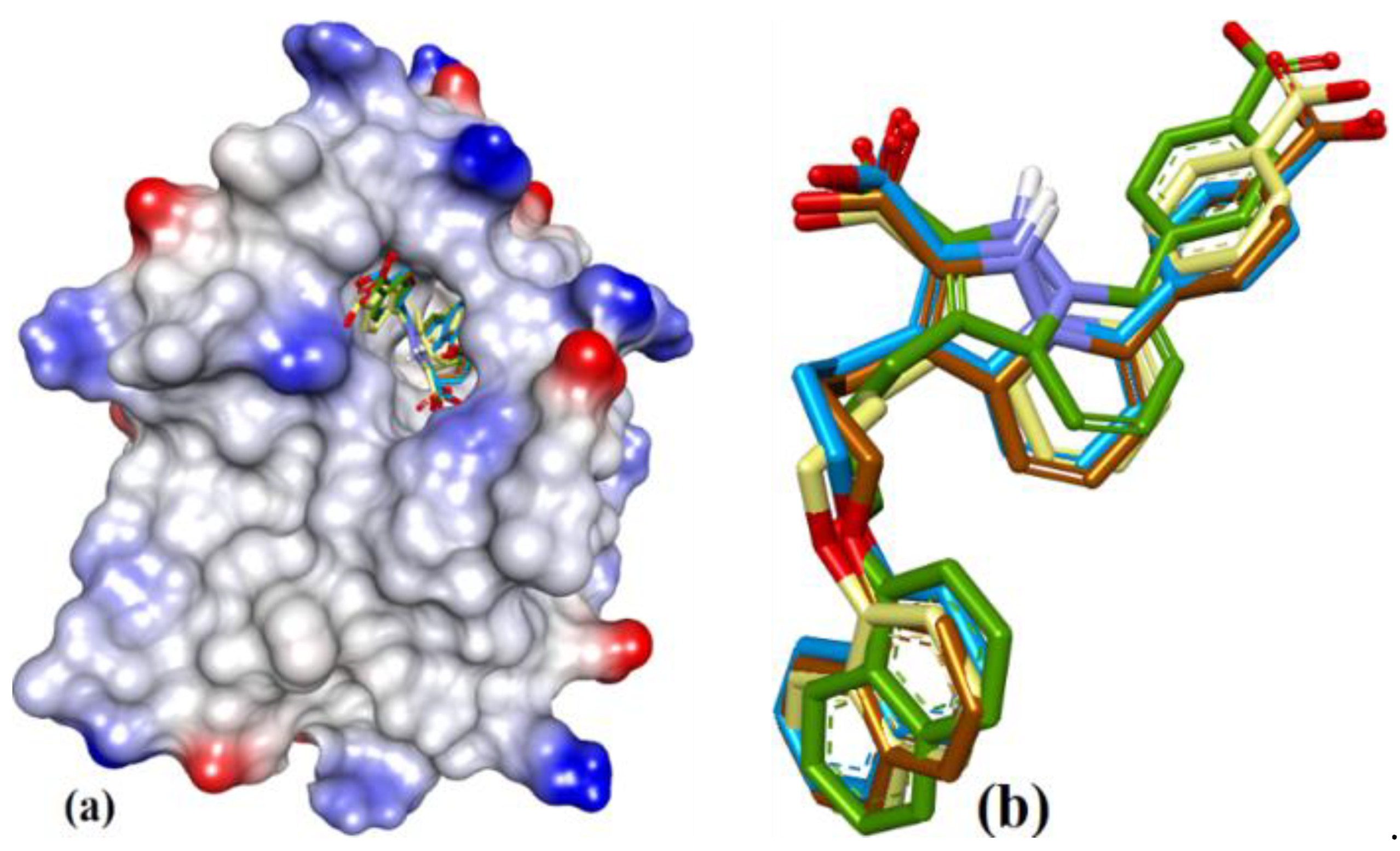
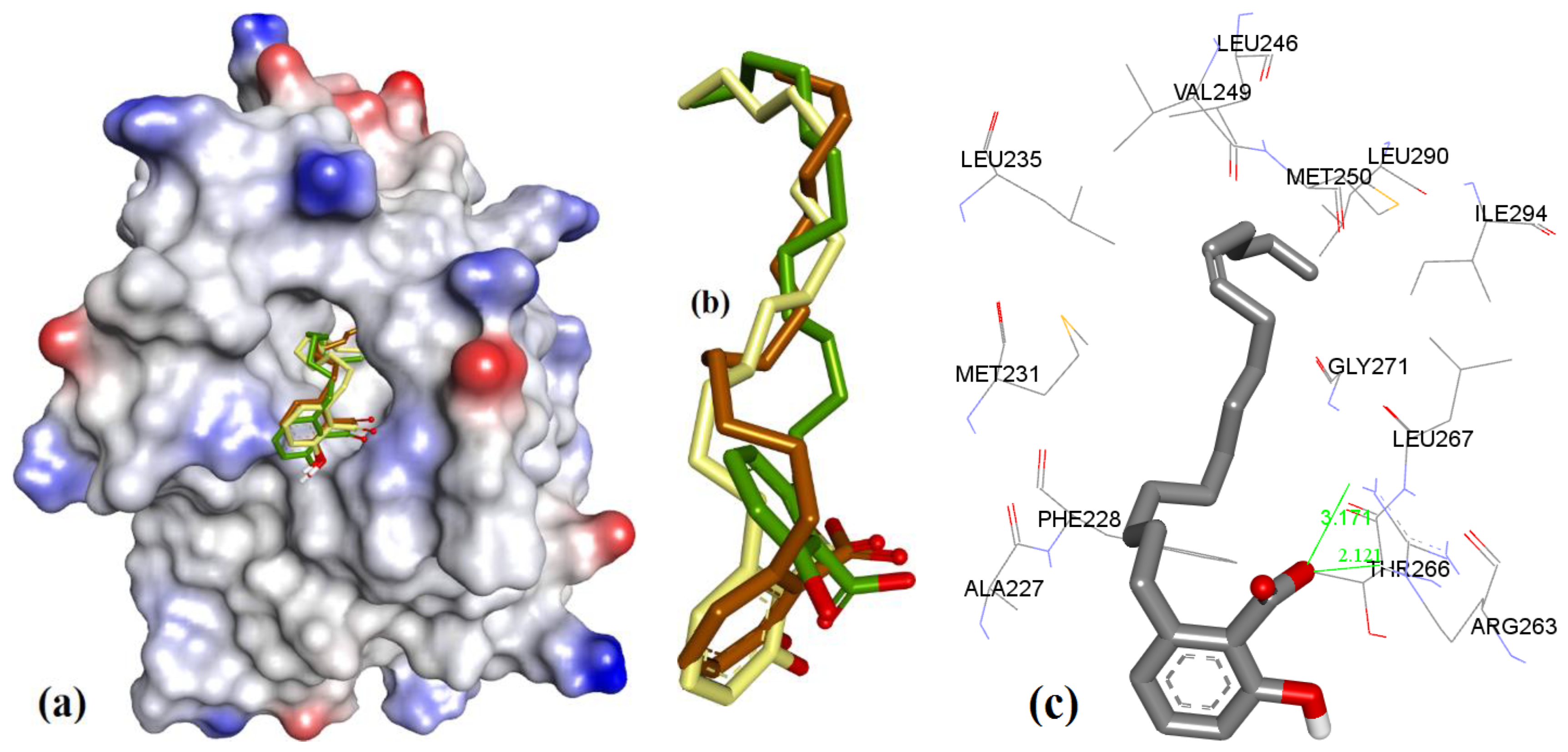
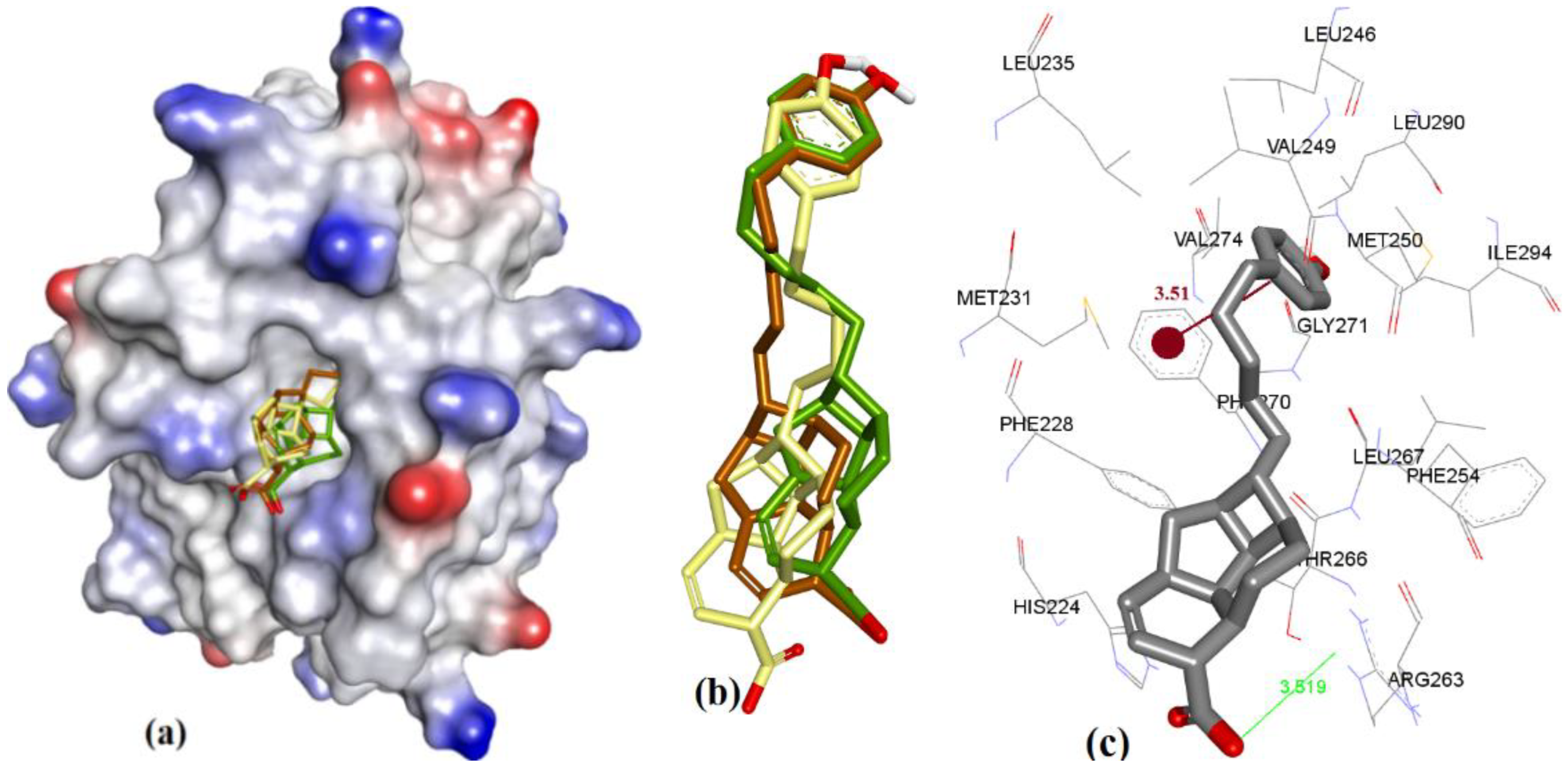
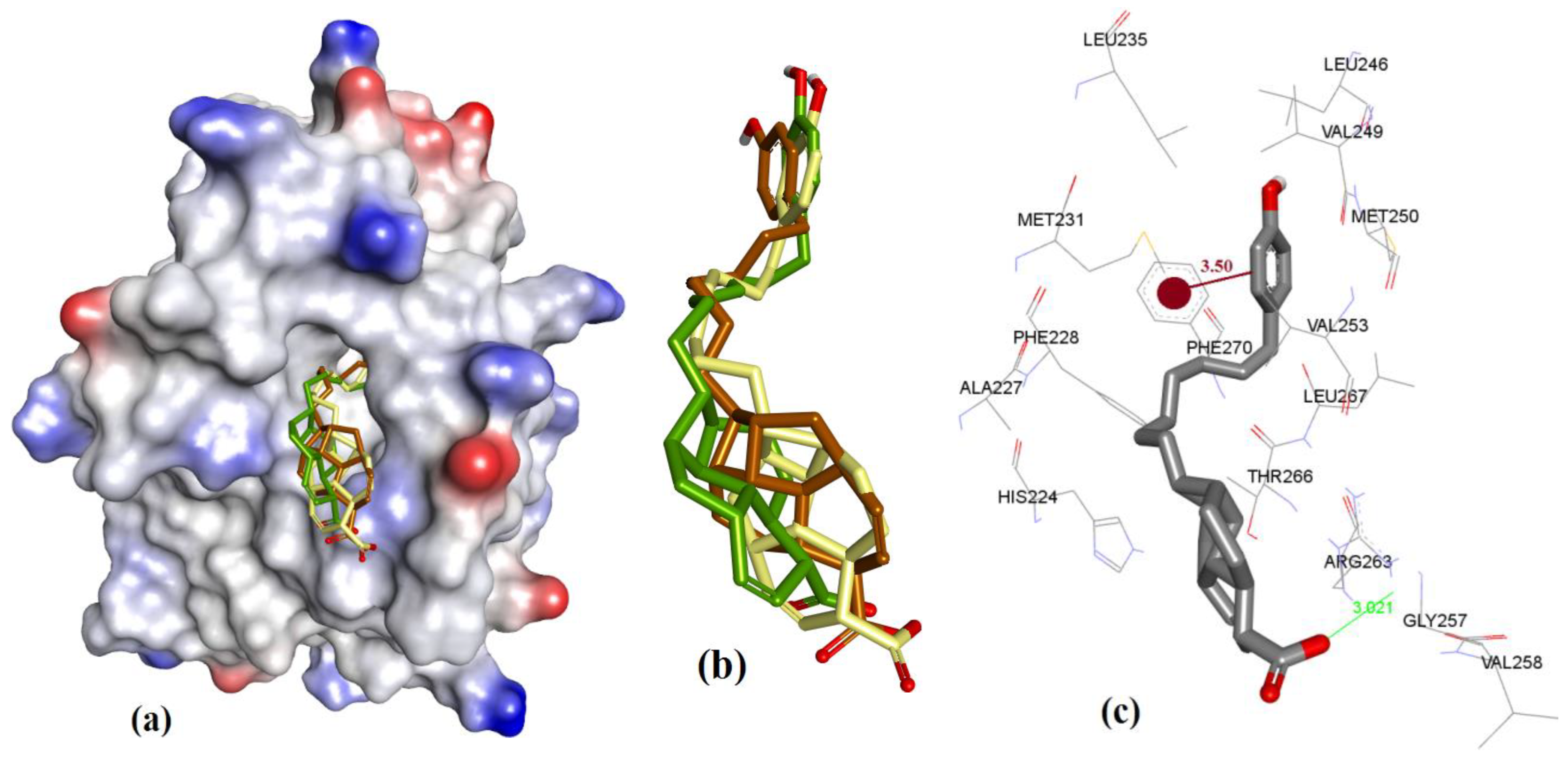
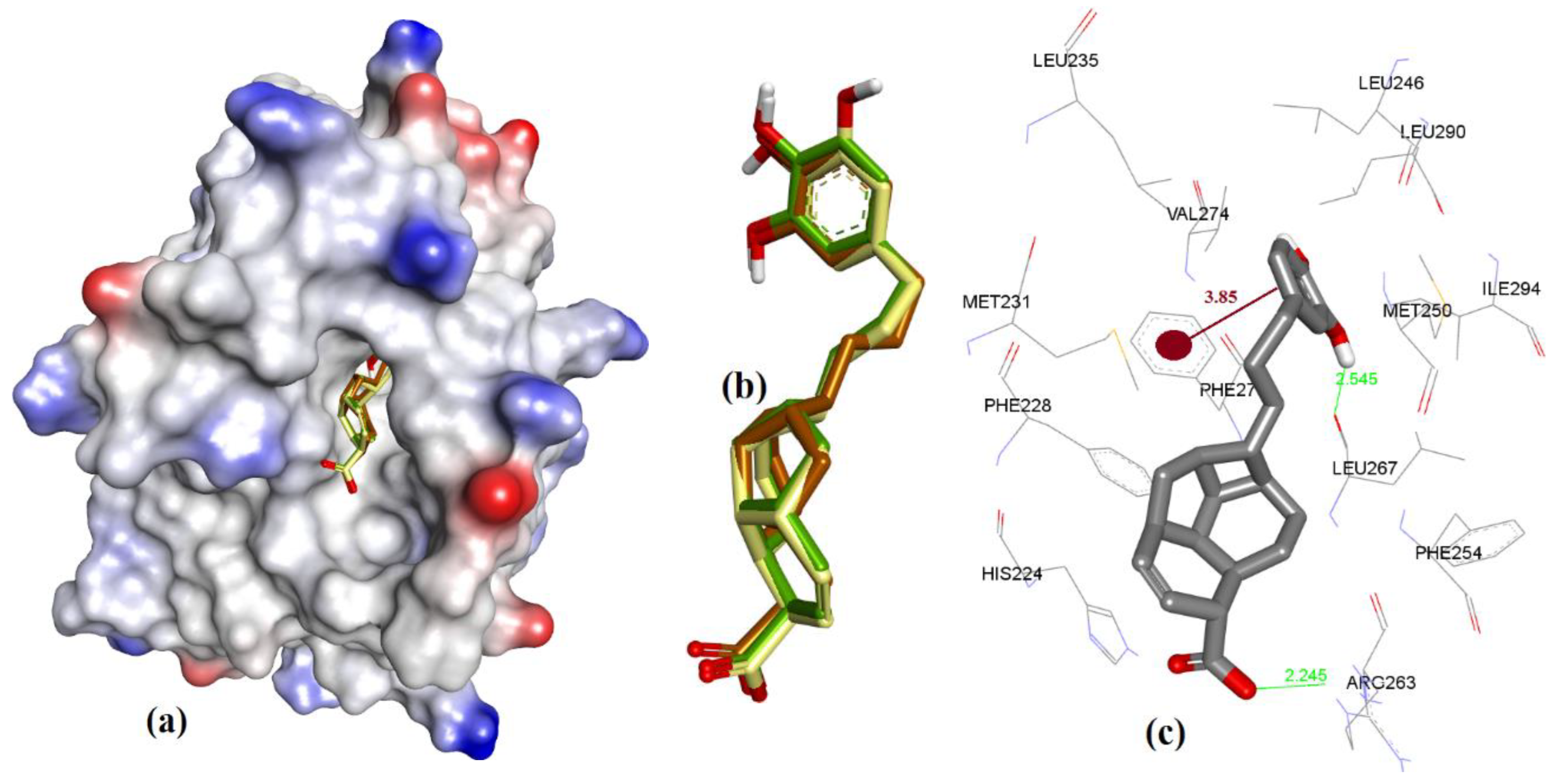
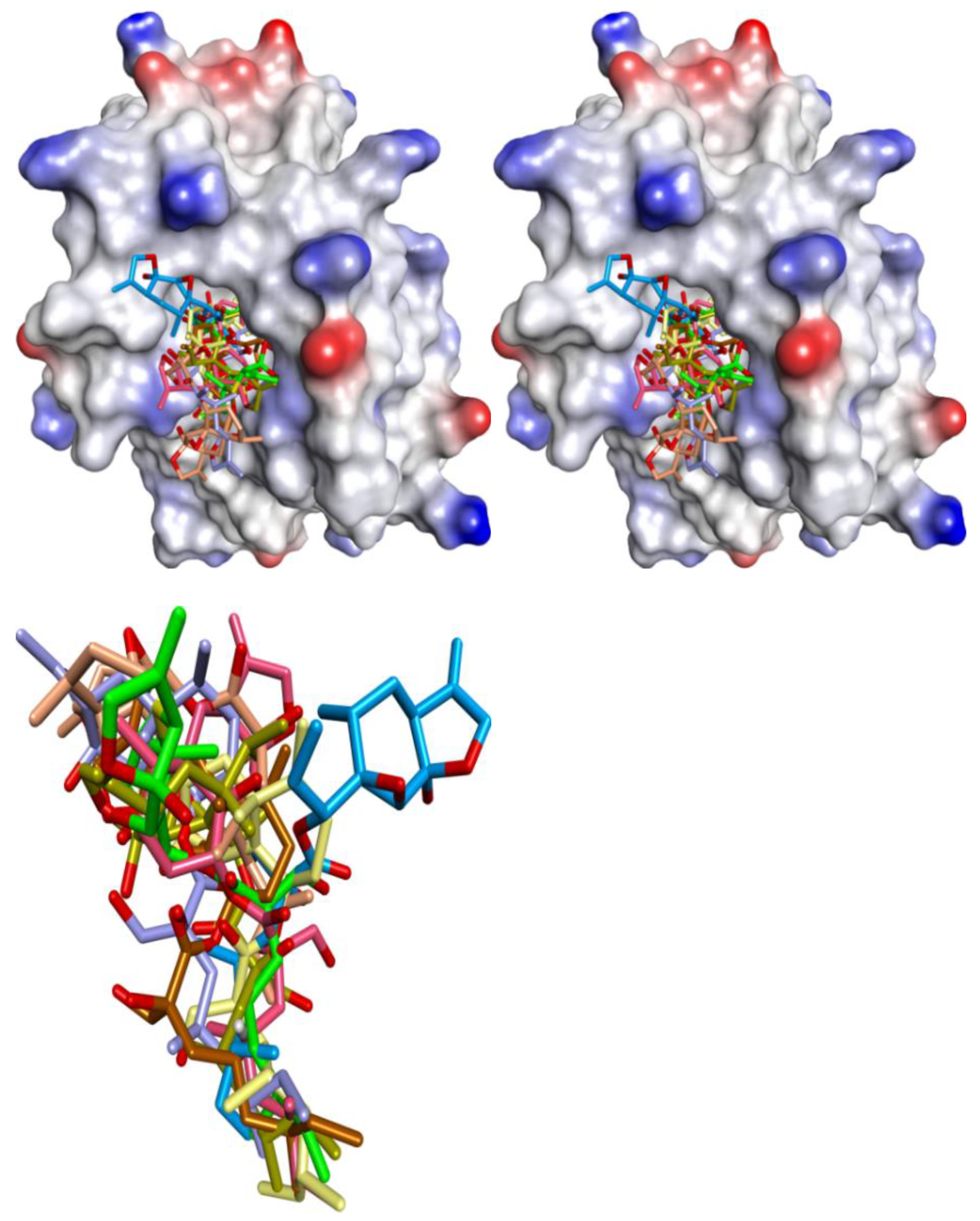
3.1. Validation of Molecular Modeling Protocols


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
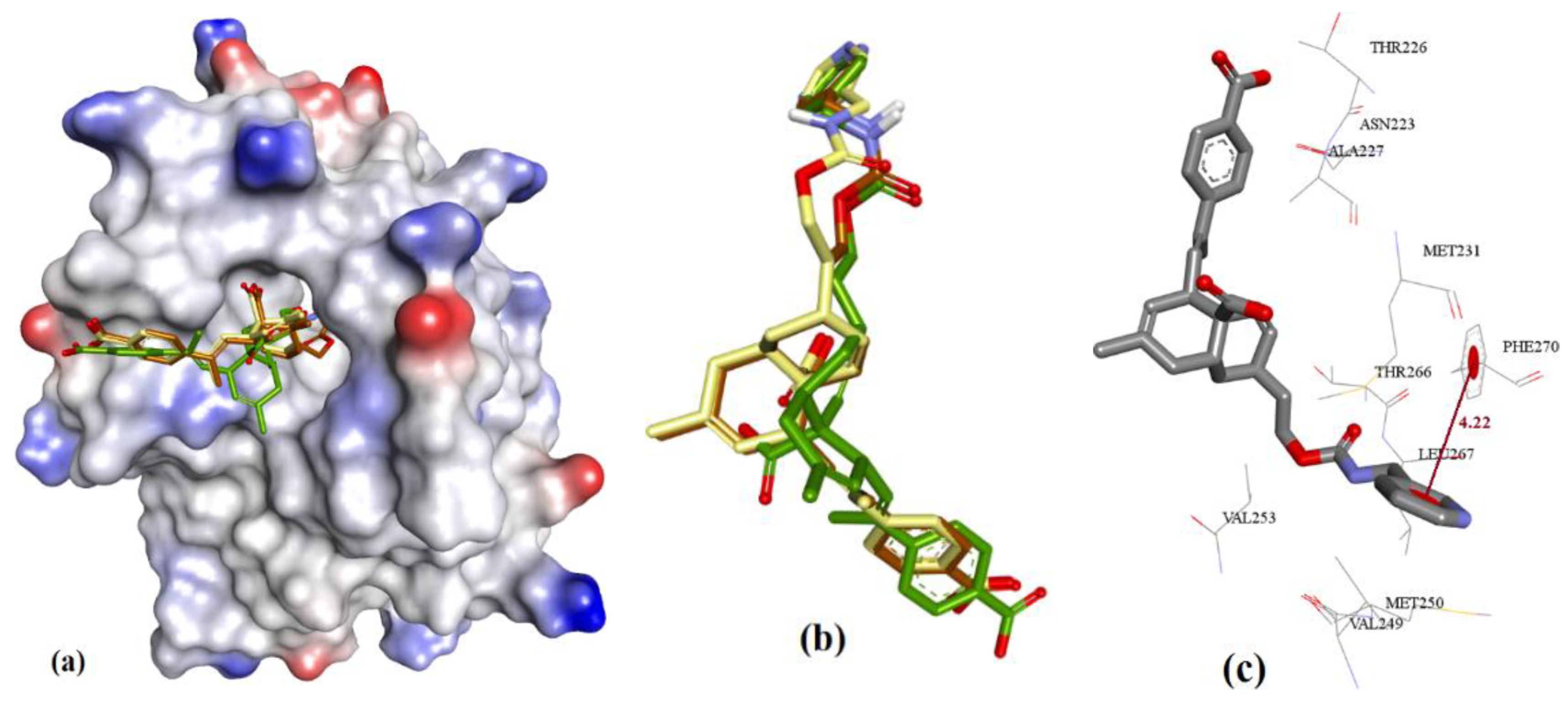{kind=link}
| Entry | PDB Code | Res. (Å) | Co-Crystallized Ligand Type | R-Value | R-Free | Clash Score | Ramachandran Outliers (%) | Side-Chain Outliers (%) | RSRZ Outliers (%) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 4WMR | 1.70 | SMI * | 0.171 | 0.206 | 1 | 0 | 0 | 4.0 |
| 2 | 4ZBF | 2.20 | SMI | 0.184 | 0.233 | 6 | 0.2 | 4.5 | 2.0 |
| 3 | 4ZBI | 2.50 | SMI | 0.183 | 0.242 | 7 | 0.6 | 8 | 1.7 |
| 4 | 4OQ5 | 2.86 | SMI | 0.200 | 0.235 | 9 | 2.1 | 11.3 | 8.1 |
| 5 | 4OQ6 | 1.81 | SMI | 0.203 | 0.205 | 11 | 0 | 7.3 | 1.4 |
| 6 | 3WIX | 1.90 | SMI | 0.246 | 0.291 | 4 | 0 | 3.3 | 3.2 |
| 7 | 3WIY | 2.15 | SMI | 0.214 | 0.283 | 3 | 0.7 | 4.0 | 2.5 |
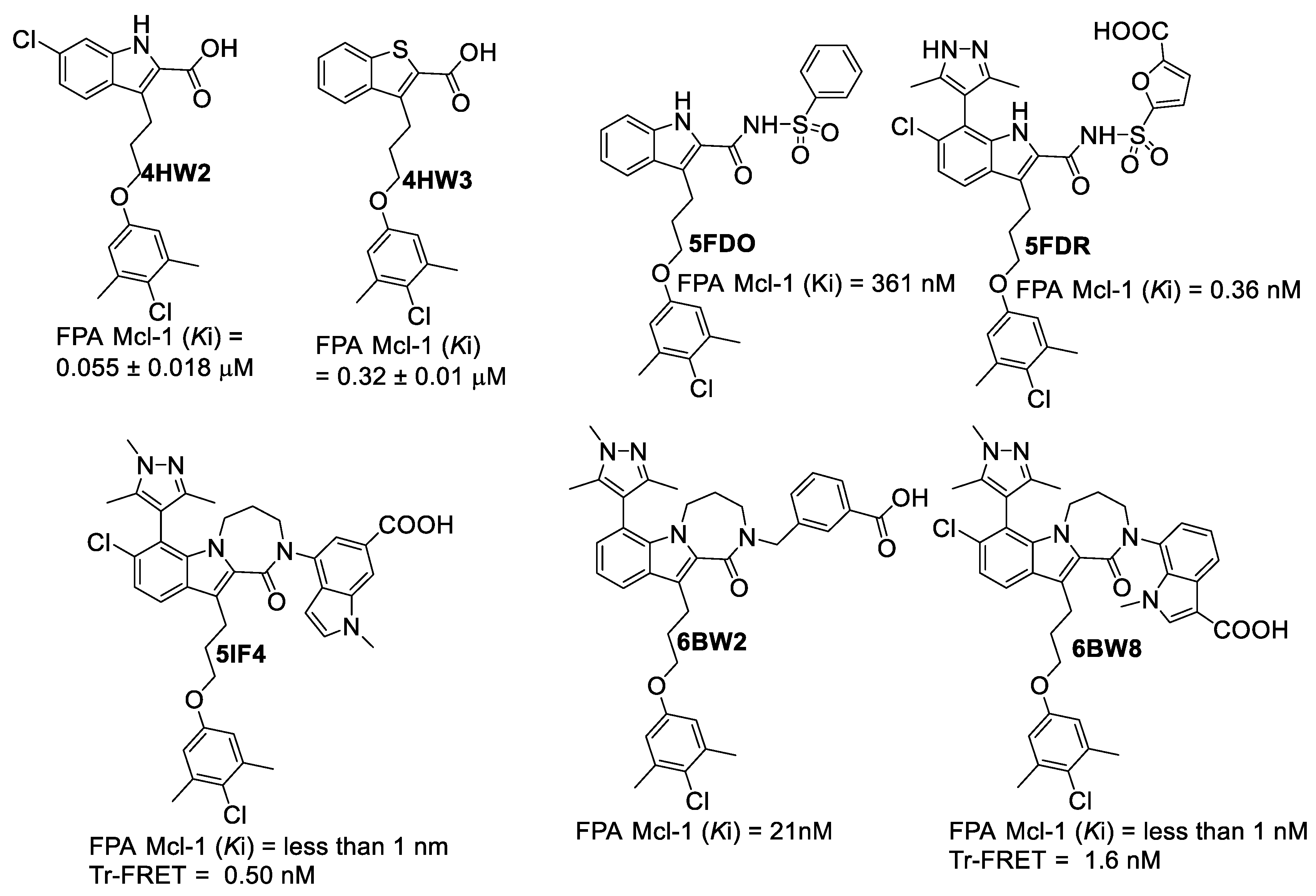
| 8 | 4HW2 | 2.80 | SMI | 0.217 | 0.251 | 32 | 1.7 | 17.6 | 1.8 |
| 9 | 4HW3 | 2.40 | SMI | 0.216 | 0.269 | 13 | 1 | 11.4 | 3.2 |
| 10 | 4HW4 | 1.53 | Peptide | 0.140 | 0.182 | 2 | 0 | 0 | 2.1 |
| 11 | 3TWU | 1.80 | Peptide | 0.184 | 0.223 | 2 | 0 | 0 | 2.3 |
| 12 | 3PK1 | 2.49 | Peptide | 0.213 | 0.245 | 3 | 0.9 | 8.1 | 0.9 |
| 13 | 3MK8 | 2.32 | Peptide | 0.233 | 0.275 | 9 | 0.7 | 0 | 1.9 |
| 14 | 3KZ0 | 2.35 | Peptide | 0.224 | 0.270 | 10 | 0 | 0 | 9.9 |
| 15 | 3KJ0 | 1.70 | Peptide | 0.187 | 0.223 | 8 | 0 | 0.6 | 4.4 |
| 16 | 3KJ1 | 1.95 | Peptide | 0.188 | 0.213 | 3 | 1.2 | 0.7 | 9.4 |
| 17 | 3KJ2 | 2.35 | Peptide | 0.210 | 0.233 | 2 | 0 | 1.3 | 6.7 |
| 18 | 3IO9 | 2.40 | Peptide | 0.211 | 0.269 | 7 | 0.6 | 4.9 | 4.1 |
| 19 | 2PQK | 2.00 | Peptide | 0.196 | 0.234 | 6 | 0 | 1.4 | 7 |
3.2. Evaluation of the Precision and Accuracy of Docking Methods
| Docking Phase Trials | RMSD (Å) | ||
|---|---|---|---|
| MOE | AutoDock | VLifeDock | |
| 1 | 0.689 | 1.007 | 0.902 |
| 2 | 0.681 | 0.989 | 0.884 |
| 3 | 0.682 | 0.996 | 0.879 |
| 4 | 0.681 | 0.996 | 0.857 |
| 5 | 0.681 | 0.994 | 0.871 |
| RMDSE (Δ = Å) | 0.008 | 0.018 | 0.025 |
| NRMSE | 85.35 | 55.36 | 36.08 |
3.3. Docking of Naturally Occurring hMcl-1 Inhibitors and Related Compounds
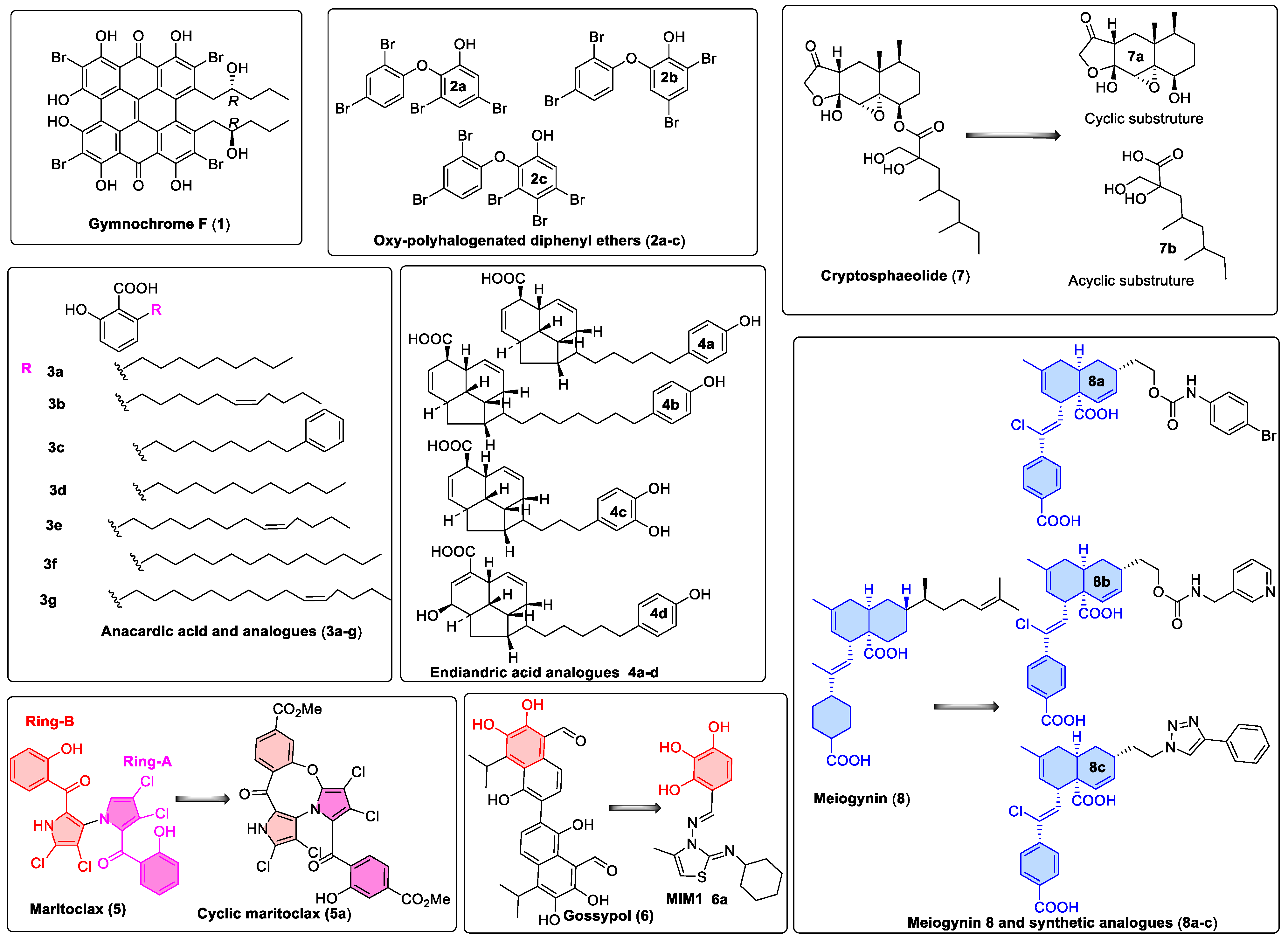
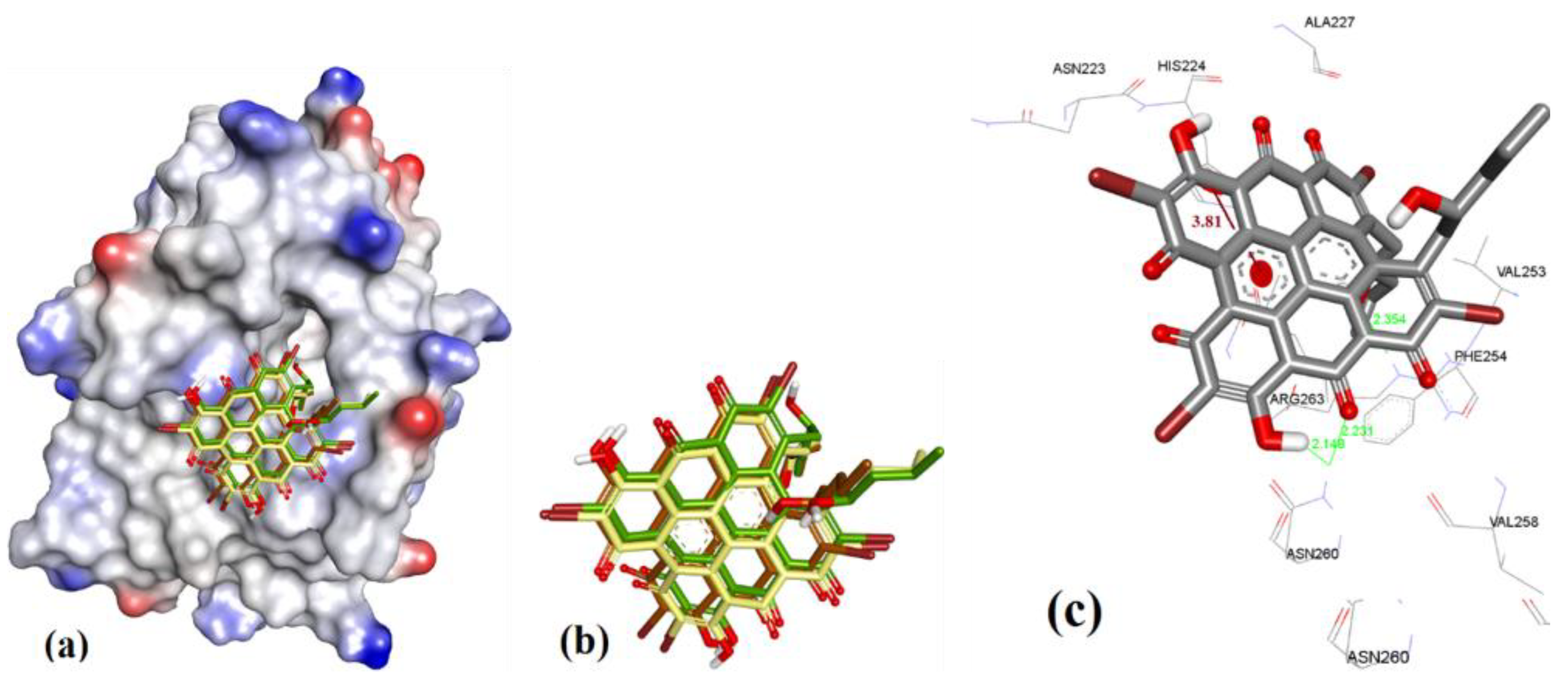
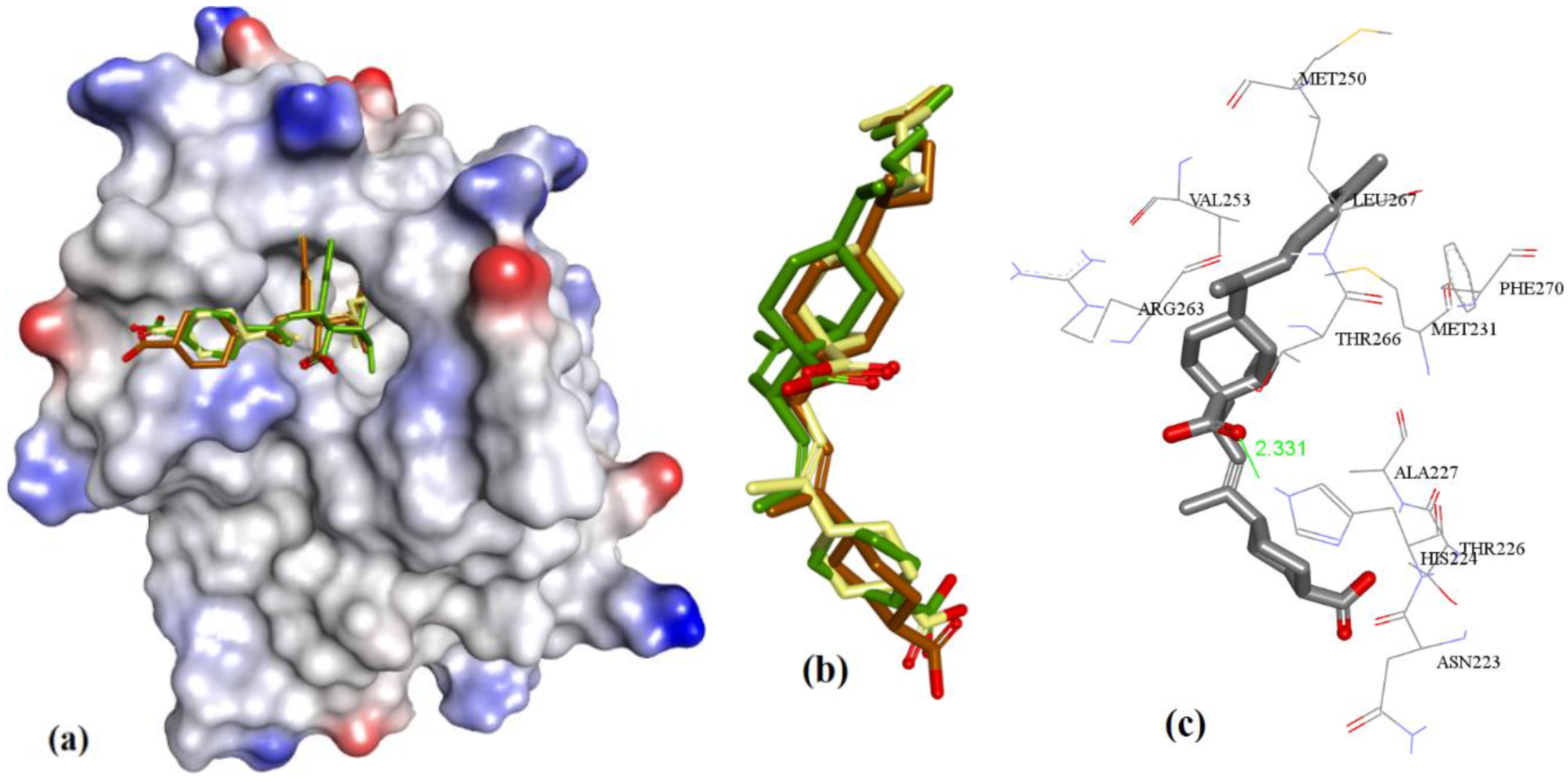
3.3.1. Gymnochrome-F
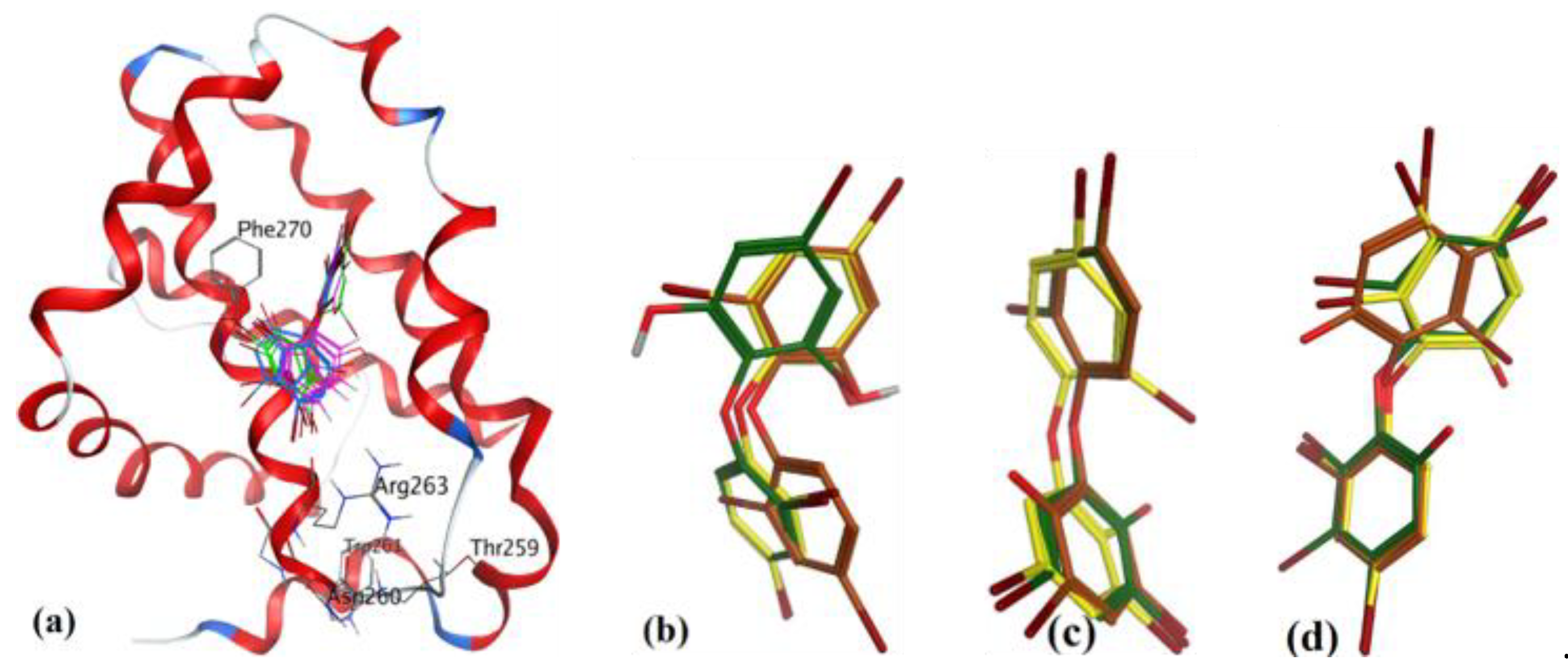
3.3.2. Sponge-Derived Oxy-Polyhalogenated Diphenyl Ethers 2
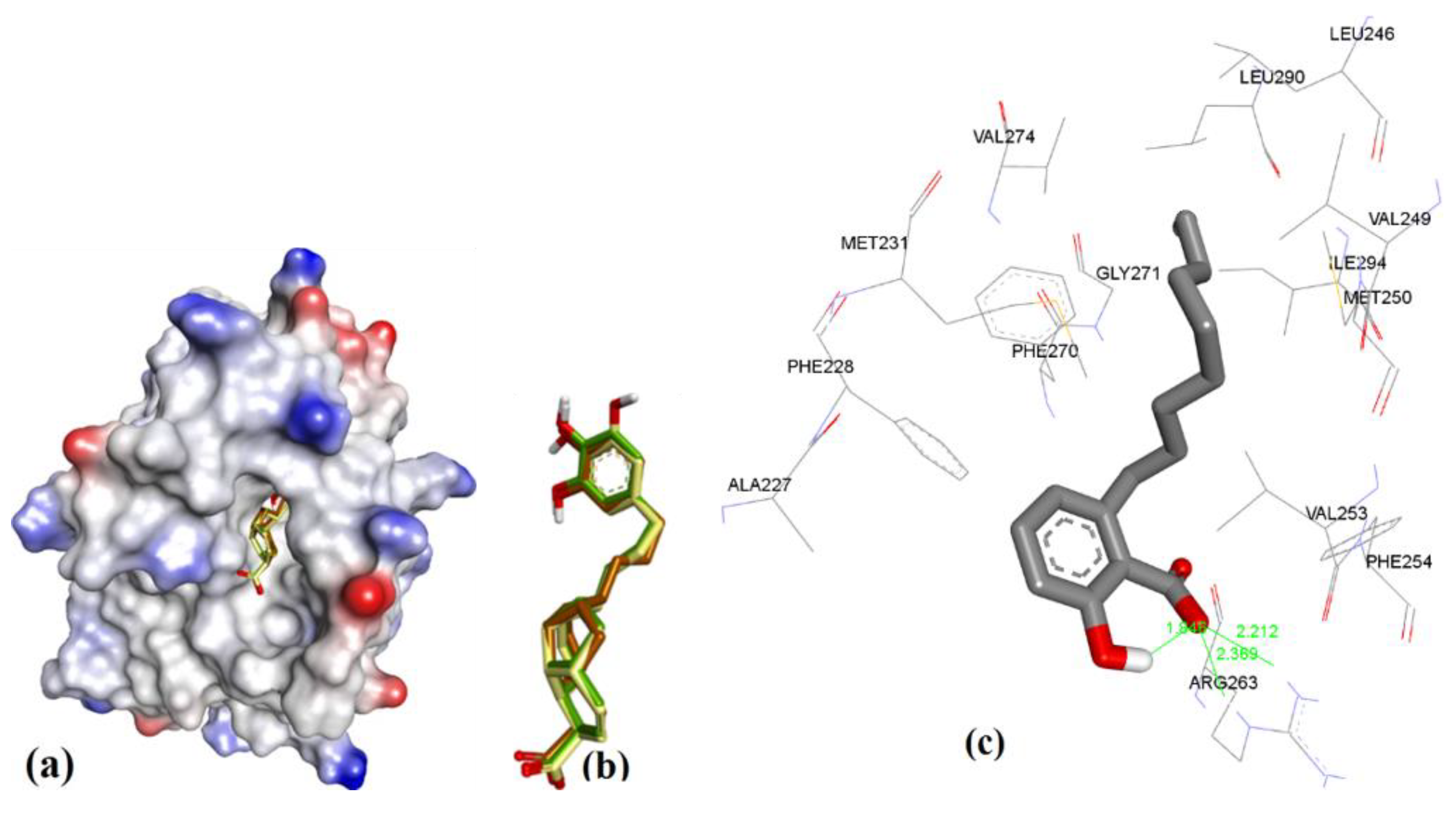
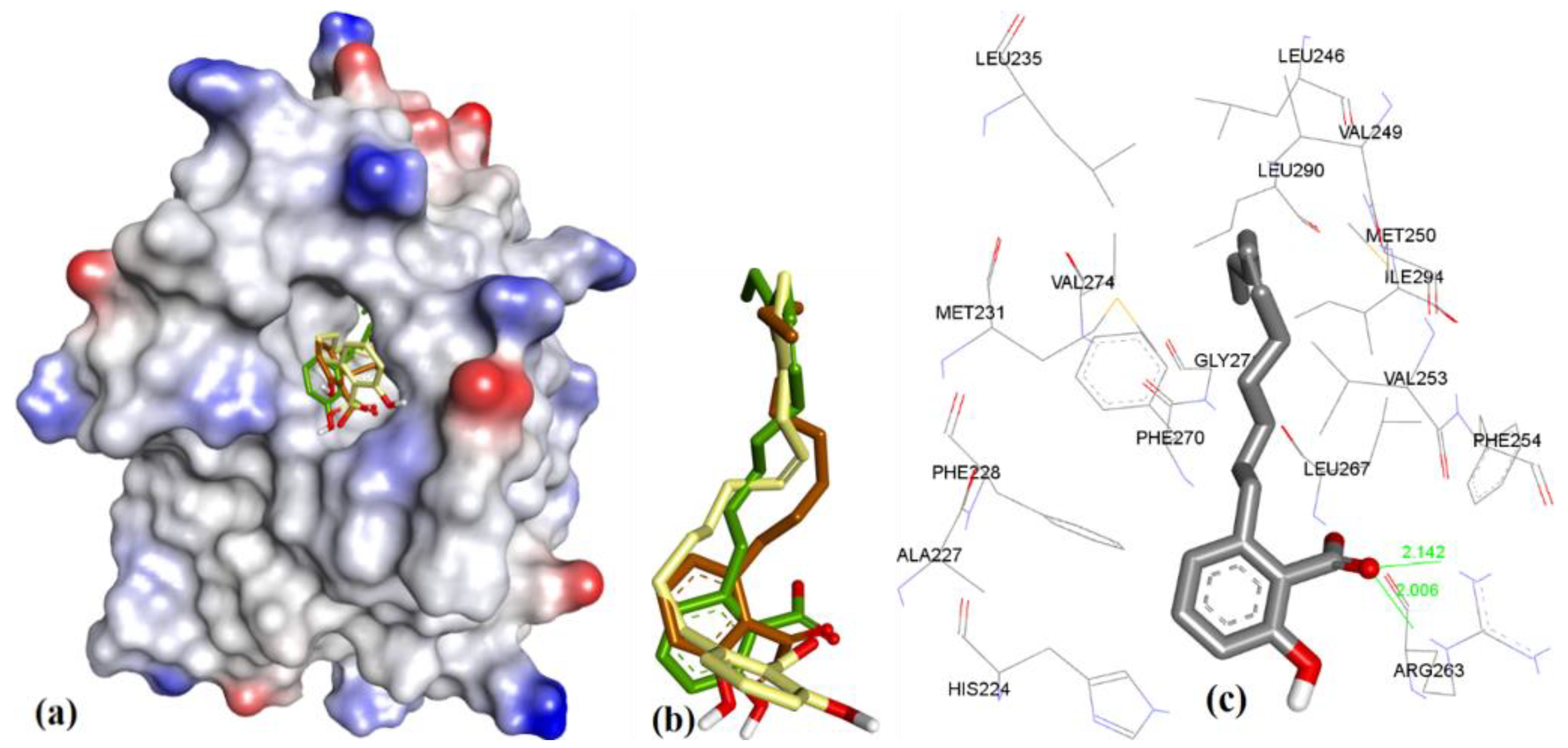
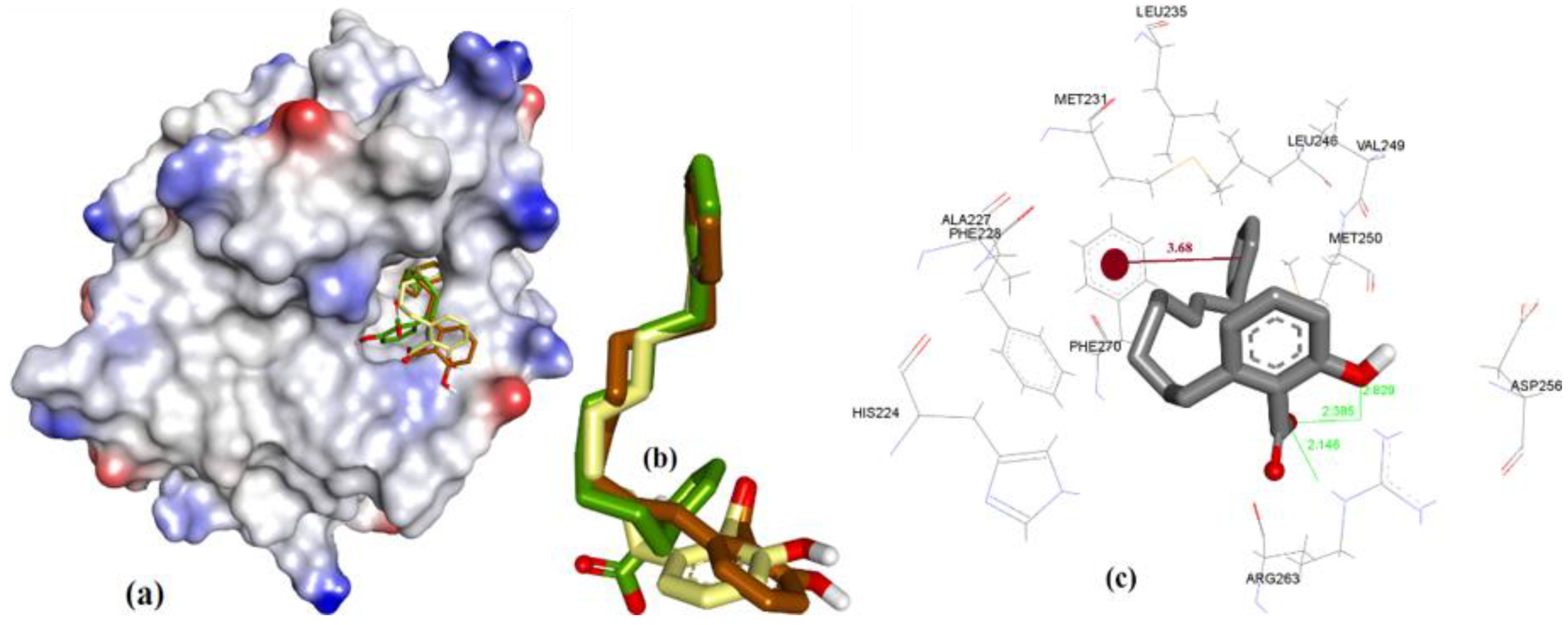
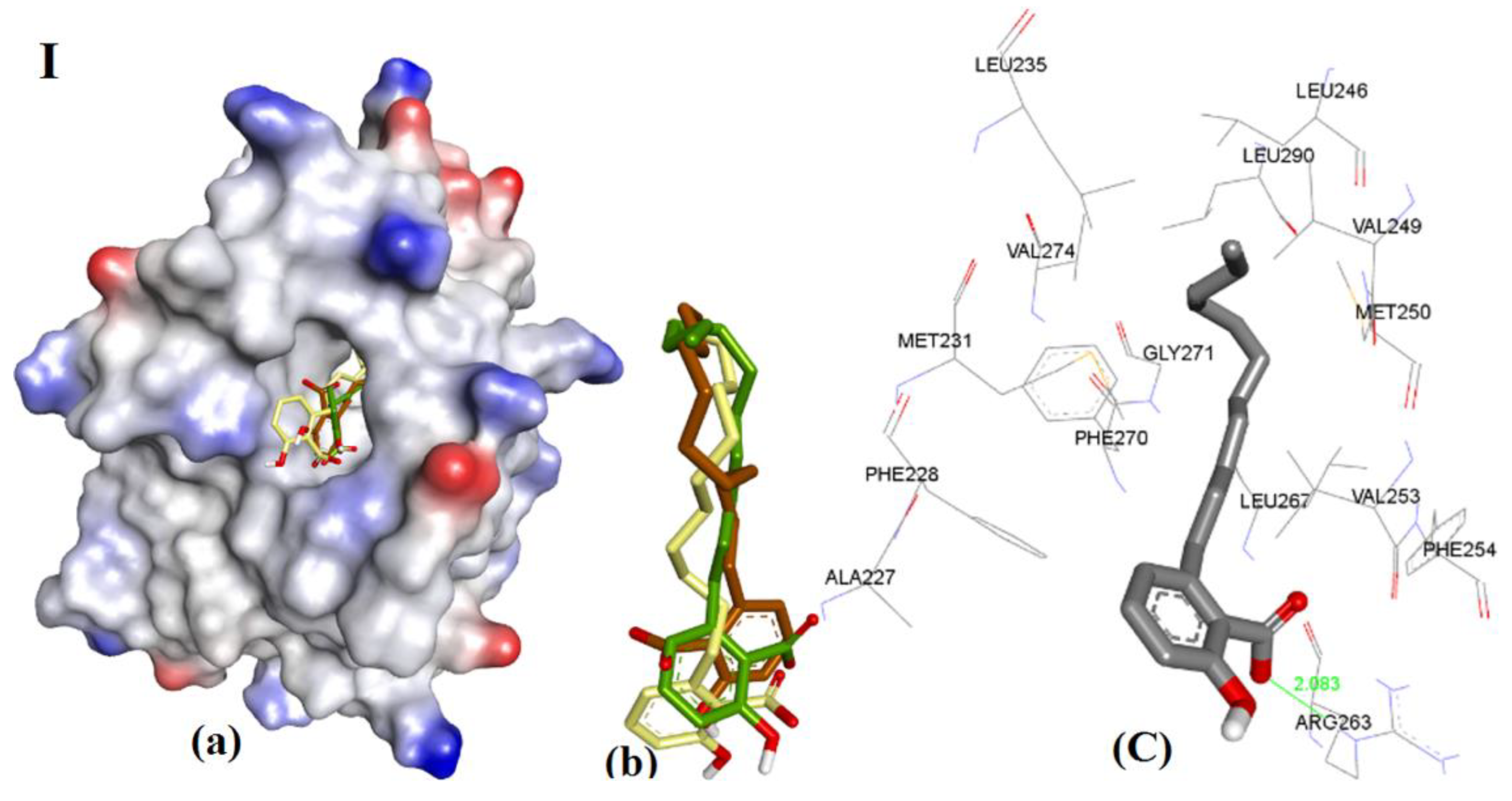
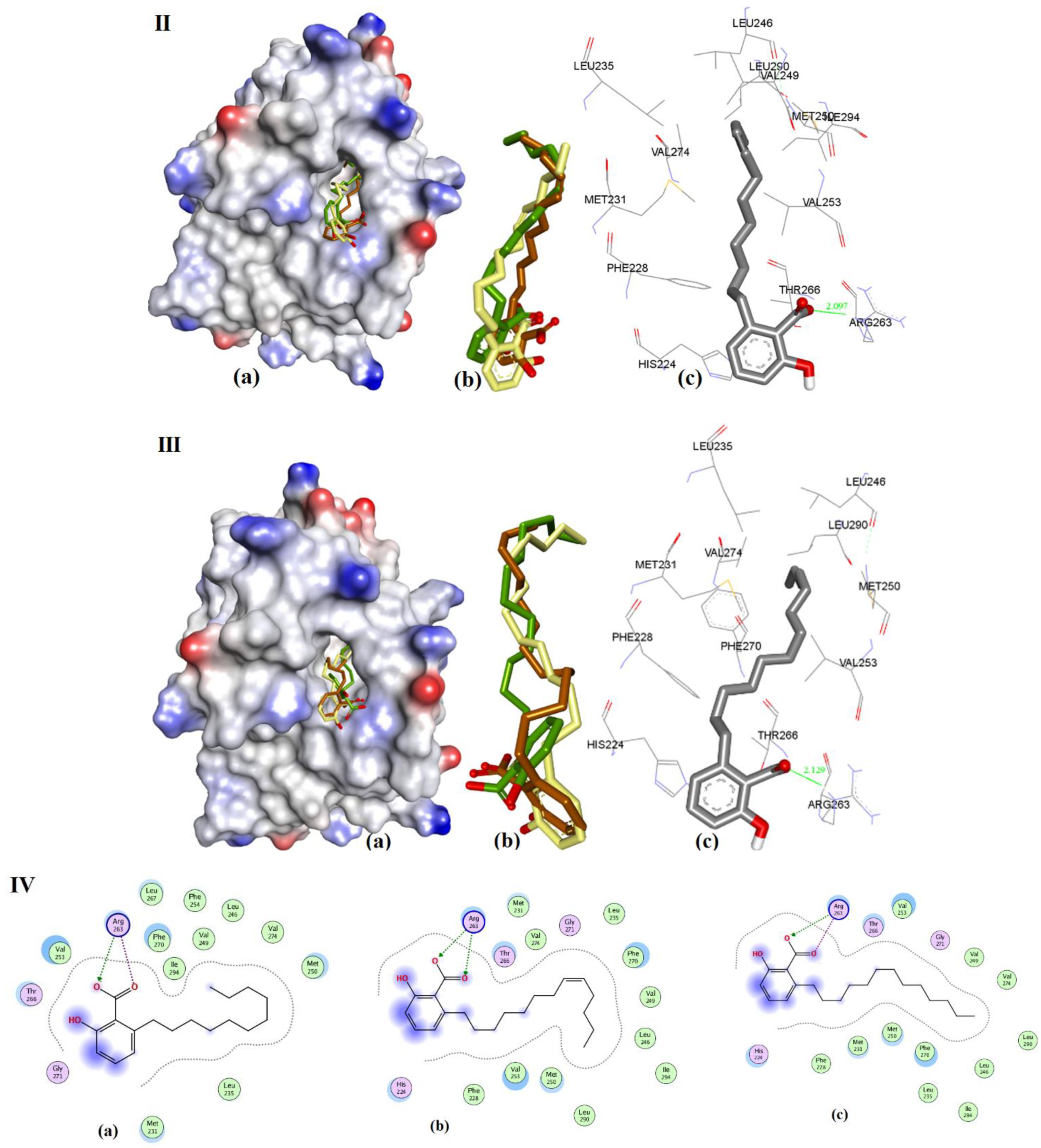
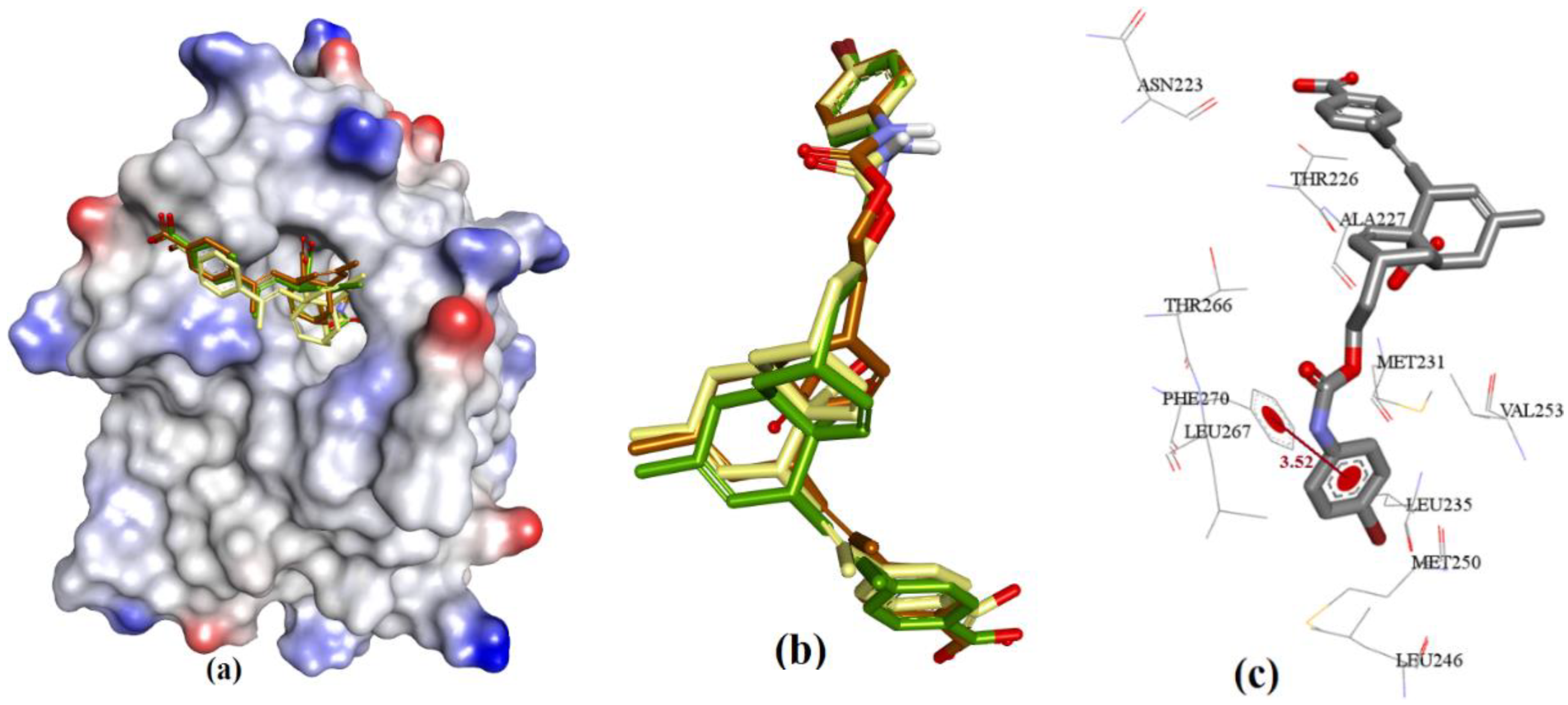
3.3.3. Anacardic Acid Derivatives (3a–g)
3.3.4. Endiandric Acid Analogues (4a–d)
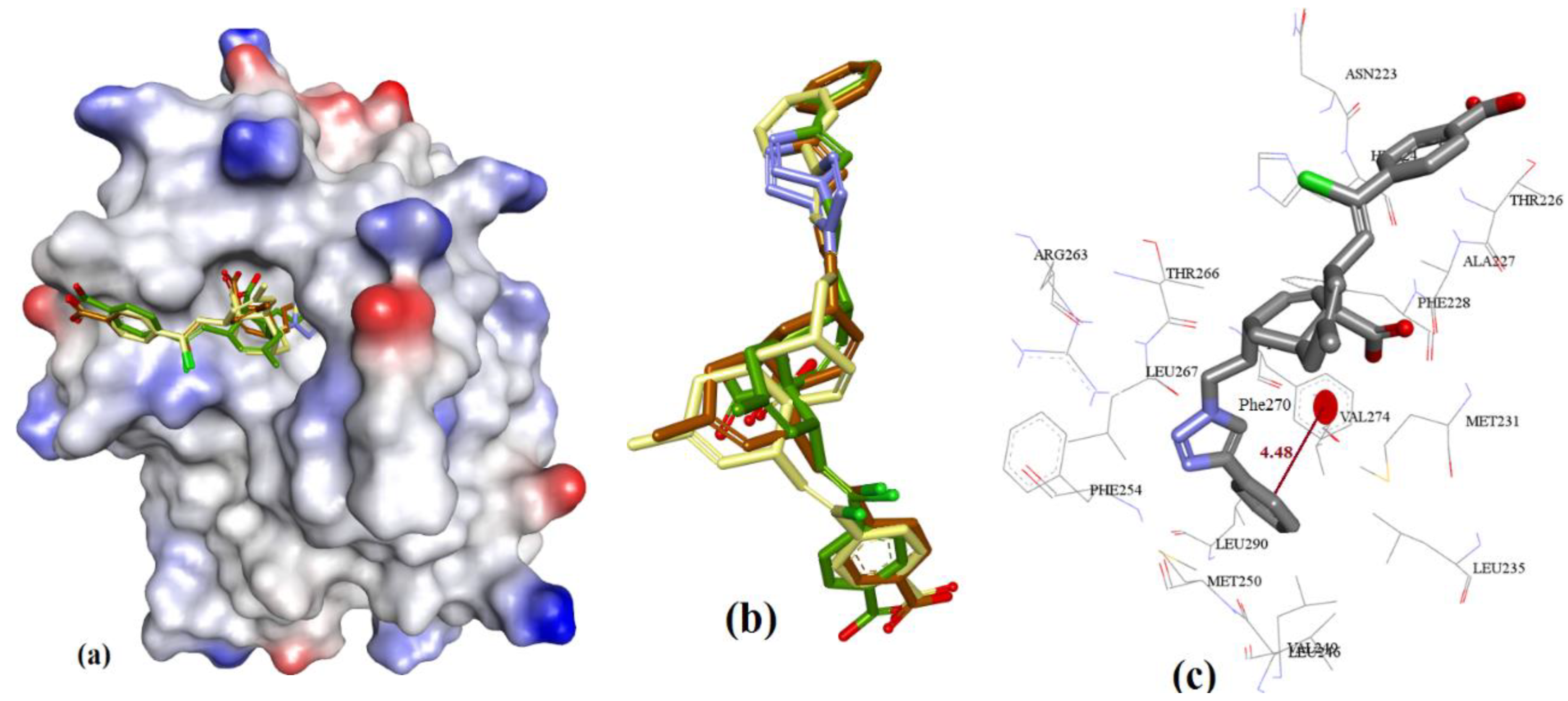
3.3.5. Marinopyrrole Analogs
3.3.6. MIM1 (Mcl-1 Inhibitor Molecule 1)
3.3.7. Cryptosphaerolide
3.3.8. Meiogynin-Derived hMcl-1 Inhibitors (8a–c)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| hMcl-1 | Human Mcl-1 protein |
| mMcl-1 | Mouse Mcl-1 protein |
| MRC | Multiple receptor conformations |
| MLC | Multiple ligand conformations |
| RMSD | Root-mean-square deviation |
| RMSE | Root-mean-square deviation error |
| NRMSE | Normalized root-mean-square deviation or error |
| MIM1 | Mcl-1 inhibitor molecule 1 |
| SMI | Small-molecule inhibitor |
References
- Denis, C.; Sopková-de Oliveira Santos, J.; Bureau, R.; Voisin-Chiret, A.S. Hot-Spots of Mcl-1 Protein: Miniperspective. J. Med. Chem. 2019, 63, 928–943. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Contractor, R.; Tsao, T.; Samudio, I.; Ruvolo, P.P.; Kitada, S.; Deng, X.; Zhai, D.; Shi, Y.-X.; Sneed, T. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 2006, 10, 375–388. [Google Scholar] [CrossRef] [PubMed]
- van Delft, M.F.; Wei, A.H.; Mason, K.D.; Vandenberg, C.J.; Chen, L.; Czabotar, P.E.; Willis, S.N.; Scott, C.L.; Day, C.L.; Cory, S. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 2006, 10, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Michels, J.; Obrist, F.; Vitale, I.; Lissa, D.; Garcia, P.; Behnam-Motlagh, P.; Kohno, K.; Wu, G.S.; Brenner, C.; Castedo, M. MCL-1 dependency of cisplatin-resistant cancer cells. Biochem. Pharmacol. 2014, 92, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, G.; Al-Harbi, S.; Mazumder, S.; Hill, B.; Smith, M.; Bodo, J.; Hsi, E.; Almasan, A. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis. 2015, 6, e1593. [Google Scholar] [CrossRef]
- Williams, M.M.; Cook, R.S. Bcl-2 family proteins in breast development and cancer: Could Mcl-1 targeting overcome therapeutic resistance? Oncotarget 2015, 6, 3519–3530. [Google Scholar] [CrossRef]
- Wei, A.; Roberts, A.W.; Spencer, A.; Rosenberg, A.S.; Siegel, D.; Walter, R.B.; Caenepeel, S.; Hughes, P.; McIver, Z.; Mezzi, K. Targeting MCL-1 in hematologic malignancies: Rationale and progress. Blood Rev. 2020, 44, 100672. [Google Scholar] [CrossRef]
- Keuling, A.M.; Felton, K.E.; Parker, A.A.; Akbari, M.; Andrew, S.E.; Tron, V.A. RNA silencing of Mcl-1 enhances ABT-737-mediated apoptosis in melanoma: Role for a caspase-8-dependent pathway. PLoS ONE 2009, 4, e6651. [Google Scholar] [CrossRef]
- Kang, M.H.; Wan, Z.; Kang, Y.H.; Sposto, R.; Reynolds, C.P. Mechanism of synergy of N-(4-hydroxyphenyl) retinamide and ABT-737 in acute lymphoblastic leukemia cell lines: Mcl-1 inactivation. J. Natl. Cancer Inst. 2008, 100, 580–595. [Google Scholar] [CrossRef]
- Negi, A.; Voisin-Chiret, A.S. Strategies to Reduce the On-Target Platelet Toxicity of Bcl-xL Inhibitors: PROTACs, SNIPERs and Prodrug-Based Approaches. ChemBioChem 2022, 23, e202100689. [Google Scholar] [CrossRef]
- Negi, A.; Reilly, C.O.; Jarikote, D.V.; Zhou, J.; Murphy, P.V. Multi-targeting protein-protein interaction inhibitors: Evolution of macrocyclic ligands with embedded carbohydrates (MECs) to improve selectivity. Eur. J. Med. Chem. 2019, 176, 292–309. [Google Scholar] [CrossRef] [PubMed]
- Negi, A.; Murphy, P.V. Development of Mcl-1 inhibitors for cancer therapy. Eur. J. Med. Chem. 2020, 210, 113038. [Google Scholar] [CrossRef] [PubMed]
- Kairys, V.; Baranauskiene, L.; Kazlauskiene, M.; Matulis, D.; Kazlauskas, E. Binding affinity in drug design: Experimental and computational techniques. Expert Opin. Drug Discov. 2019, 14, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Negi, A.; Mirallai, S.I.; Warta, R.; Herold-Mende, C.; Carty, M.P.; Ye, X.-S.; Murphy, P.V. N-Alkyl-1, 5-dideoxy-1, 5-imino-L-fucitols as fucosidase inhibitors: Synthesis, molecular modelling and activity against cancer cell lines. Bioorganic Chem. 2019, 84, 418–433. [Google Scholar] [CrossRef]
- Negi, A.; Zhou, J.; Sweeney, S.; Murphy, P.V. Ligand design for somatostatin receptor isoforms 4 and 5. Eur. J. Med. Chem. 2019, 163, 148–159. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Negi, A.; Bhandari, N.; Shyamlal, B.R.K.; Chaudhary, S. Inverse docking based screening and identification of protein targets for Cassiarin alkaloids against Plasmodium falciparum. Saudi Pharm. J. 2018, 26, 546–567. [Google Scholar] [CrossRef]
- Negi, A.; & Kesari, K.K. Chitosan Nanoparticle Encapsulation of Antibacterial Essential Oils. Micromachines 2022, 13, 1265. [Google Scholar] [CrossRef]
- Kemami Wangun, H.V.; Wood, A.; Fiorilla, C.; Reed, J.K.; McCarthy, P.J.; Wright, A.E. Gymnochromes E and F, cytotoxic phenanthroperylenequinones from a deep-water crinoid, Holopus rangii. J. Nat. Prod. 2010, 73, 712–715. [Google Scholar] [CrossRef]
- Calcul, L.; Chow, R.; Oliver, A.G.; Tenney, K.; White, K.N.; Wood, A.W.; Fiorilla, C.; Crews, P. NMR strategy for unraveling structures of bioactive sponge-derived oxy-polyhalogenated diphenyl ethers. J. Nat. Prod. 2009, 72, 443–449. [Google Scholar] [CrossRef] [Green Version]
- Gény, C.; Rivière, G.; Bignon, J.r.; Birlirakis, N.; Guittet, E.; Awang, K.; Litaudon, M.; Roussi, F.; Dumontet, V. Anacardic acids from Knema hookeriana as modulators of Bcl-xL/Bak and Mcl-1/Bid interactions. J. Nat. Prod. 2016, 79, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Apel, C.c.; Gény, C.; Dumontet, V.; Birlirakis, N.; Roussi, F.; Pham, V.C.; Doan Thi Mai, H.; Nguyen, V.H.; Chau, V.M.; Litaudon, M. Endiandric acid analogues from Beilschmiedia ferruginea as dual inhibitors of Bcl-xL/Bak and Mcl-1/Bid interactions. J. Nat. Prod. 2014, 77, 1430–1437. [Google Scholar] [CrossRef] [PubMed]
- Doi, K.; Li, R.; Sung, S.-S.; Wu, H.; Liu, Y.; Manieri, W.; Krishnegowda, G.; Awwad, A.; Dewey, A.; Liu, X. Discovery of marinopyrrole A (maritoclax) as a selective Mcl-1 antagonist that overcomes ABT-737 resistance by binding to and targeting Mcl-1 for proteasomal degradation. J. Biol. Chem. 2012, 287, 10224–10235. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.C.; Prieto-Davo, A.; Jensen, P.R.; Fenical, W. The marinopyrroles, antibiotics of an unprecedented structure class from a marine Streptomyces sp. Org. Lett. 2008, 10, 629–631. [Google Scholar] [CrossRef]
- Etxebarria, A.; Landeta, O.; Antonsson, B.; Basañez, G. Regulation of antiapoptotic MCL-1 function by gossypol: Mechanistic insights from in vitro reconstituted systems. Biochem. Pharmacol. 2008, 76, 1563–1576. [Google Scholar] [CrossRef]
- Hron, R.; Kuk, M.; Abraham, G.; Wan, P. Ethanol extraction of oil, gossypol and aflatoxin from cottonseed. J. Am. Oil Chem. Soc. 1994, 71, 417–421. [Google Scholar] [CrossRef]
- Cohen, N.A.; Stewart, M.L.; Gavathiotis, E.; Tepper, J.L.; Bruekner, S.R.; Koss, B.; Opferman, J.T.; Walensky, L.D. A competitive stapled peptide screen identifies a selective small molecule that overcomes MCL-1-dependent leukemia cell survival. Chem. Biol. 2012, 19, 1175–1186. [Google Scholar] [CrossRef]
- Oh, H.; Jensen, P.R.; Murphy, B.T.; Fiorilla, C.; Sullivan, J.F.; Ramsey, T.; Fenical, W. Cryptosphaerolide, a cytotoxic Mcl-1 inhibitor from a marine-derived ascomycete related to the genus Cryptosphaeria. J. Nat. Prod. 2010, 73, 998–1001. [Google Scholar] [CrossRef]
- Samra, A.A.; Robert, A.; Gov, C.; Favre, L.; Eloy, L.; Jacquet, E.; Bignon, J.; Wiels, J.; Desrat, S.; Roussi, F. Dual inhibitors of the pro-survival proteins Bcl-2 and Mcl-1 derived from natural compound meiogynin A. Eur. J. Med. Chem. 2018, 148, 26–38. [Google Scholar] [CrossRef]
- Litaudon, M.; Bousserouel, H.; Awang, K.; Nosjean, O.; Martin, M.-T.; Dau, M.E.T.H.; Hadi, H.A.; Boutin, J.A.; Sevenet, T.; Gueritte, F. A dimeric sesquiterpenoid from a Malaysian Meiogyne as a new inhibitor of Bcl-xL/BakBH3 domain peptide interaction. J. Nat. Prod. 2009, 72, 480–483. [Google Scholar] [CrossRef]
- Gore, S.; García, E.S.; Hendrickx, P.M.; Gutmanas, A.; Westbrook, J.D.; Yang, H.; Feng, Z.; Baskaran, K.; Berrisford, J.M.; Hudson, B.P. Validation of structures in the Protein Data Bank. Structure 2017, 25, 1916–1927. [Google Scholar] [CrossRef] [PubMed]
- Touw, W.G.; Joosten, R.P.; Vriend, G. New biological insights from better structure models. J. Mol. Biol. 2016, 428, 1375–1393. [Google Scholar] [CrossRef]
- Kleywegt, G.J.; Harris, M.R.; Zou, J.y.; Taylor, T.C.; Wählby, A.; Jones, T.A. The Uppsala electron-density server. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2240–2249. [Google Scholar] [CrossRef]
- Rueda, M.; Bottegoni, G.; Abagyan, R. Consistent improvement of cross-docking results using binding site ensembles generated with elastic network normal modes. J. Chem. Inf. Modeling 2009, 49, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Corbeil, C.R.; Williams, C.I.; Labute, P. Variability in docking success rates due to dataset preparation. J. Comput. -Aided Mol. Des. 2012, 26, 775–786. [Google Scholar] [CrossRef]
- Molecular Operating Environment; Software; Chemical Computing Group: Montreal, QC, Canada, 2014.
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905. [Google Scholar] [CrossRef] [PubMed]
- Molecular Design Suite; Software; VLife Sciences Technologies Pvt. Ltd.: Pune, India, 2008.
- Friberg, A.; Vigil, D.; Zhao, B.; Daniels, R.N.; Burke, J.P.; Garcia-Barrantes, P.M.; Camper, D.; Chauder, B.A.; Lee, T.; Olejniczak, E.T. Discovery of potent myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods and structure-based design. J. Med. Chem. 2013, 56, 15–30. [Google Scholar] [CrossRef]
- Pelz, N.F.; Bian, Z.; Zhao, B.; Shaw, S.; Tarr, J.C.; Belmar, J.; Gregg, C.; Camper, D.V.; Goodwin, C.M.; Arnold, A.L. Discovery of 2-indole-acylsulfonamide myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods. J. Med. Chem. 2016, 59, 2054–2066. [Google Scholar] [CrossRef]
- Lee, T.; Bian, Z.; Zhao, B.; Hogdal, L.J.; Sensintaffar, J.L.; Goodwin, C.M.; Belmar, J.; Shaw, S.; Tarr, J.C.; Veerasamy, N. Discovery and biological characterization of potent myeloid cell leukemia-1 inhibitors. FEBS Lett. 2017, 591, 240–251. [Google Scholar] [CrossRef]
- Shaw, S.; Bian, Z.; Zhao, B.; Tarr, J.C.; Veerasamy, N.; Jeon, K.O.; Belmar, J.; Arnold, A.L.; Fogarty, S.A.; Perry, E. Optimization of Potent and Selective Tricyclic Indole Diazepinone Myeloid Cell Leukemia-1 Inhibitors Using Structure-Based Design. J. Med. Chem. 2018, 61, 2410–2421. [Google Scholar] [CrossRef]
- Svensson, F.; Norinder, U.; Bender, A. Improving screening efficiency through iterative screening using docking and conformal prediction. J. Chem. Inf. Modeling 2017, 57, 439–444. [Google Scholar] [CrossRef]
- Ferreira, L.; dos Santos, R.; Oliva, G.; Andricopulo, A. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.A. Conformational adaptation in drug-target interactions and residence time. Future Med. Chem. 2011, 3, 1491–1501. [Google Scholar] [CrossRef] [PubMed]
- Ashtawy, H.M.; Mahapatra, N.R. Task-specific scoring functions for predicting ligand binding poses and affinity and for screening enrichment. J. Chem. Inf. Modeling 2017, 58, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Pons, J.-L.; Bourguet, W.; Labesse, G. Towards accurate high-throughput ligand affinity prediction by exploiting structural ensembles, docking metrics and ligand similarity. bioRxiv 2019. [Google Scholar] [CrossRef]
- Fukunishi, Y.; Yamashita, Y.; Mashimo, T.; Nakamura, H. Prediction of Protein−compound Binding Energies from Known Activity Data: Docking-score-based Method and its Applications. Mol. Inform. 2018, 37, 1700120. [Google Scholar] [CrossRef]
- Greenidge, P.; Kramer, C.; Mozziconacci, J.-C.; Sherman, W. Improving docking results via reranking of ensembles of ligand poses in multiple X-ray protein conformations with MM-GBSA. J. Chem. Inf. Modeling 2014, 54, 2697–2717. [Google Scholar] [CrossRef]
- Hawkins, P.C.; Skillman, A.G.; Nicholls, A. Comparison of shape-matching and docking as virtual screening tools. J. Med. Chem. 2007, 50, 74–82. [Google Scholar] [CrossRef]
- Makeneni, S.; Thieker, D.F.; Woods, R.J. Applying pose clustering and MD simulations to eliminate false positives in molecular docking. J. Chem. Inf. Modeling 2018, 58, 605–614. [Google Scholar] [CrossRef]
- Negi, A.; Mirallai, S.I.; Konda, S.; & Murphy, P.V. An improved method for synthesis of non-symmetric triarylpyridines. Tetrahedron 2022, 121, 132930. [Google Scholar] [CrossRef]
- Cheng, C.; Liu, Y.; Balasis, M.E.; Simmons, N.L.; Li, J.; Song, H.; Pan, L.; Qin, Y.; Nicolaou, K.; Sebti, S.M. Cyclic marinopyrrole derivatives as disruptors of Mcl-1 and Bcl-xL binding to Bim. Mar. Drugs 2014, 12, 1335–1348. [Google Scholar] [CrossRef] [PubMed]
- Desrat, S.; Remeur, C.; Geny, C.; Riviere, G.; Colas, C.; Dumontet, V.; Birlirakis, N.; Iorga, B.; Roussi, F. From meiogynin A to the synthesis of dual inhibitors of Bcl-xL and Mcl-1 anti-apoptotic proteins. Chem. Commun. 2014, 50, 8593–8596. [Google Scholar] [CrossRef] [PubMed]
- Colas, C.; Roussi, F.; Iorga, B. Focused ligand libraries as tools for in silico design of anti-apoptotic proteins inhibitors. Chem. Life Sci. 2011, 41–46. [Google Scholar]
- Zhang, T.; Walensky, L.D.; Saghatelian, A. A nonapoptotic role for BAX and BAK in eicosanoid metabolism. ACS Chem. Biol. 2015, 10, 1398–1403. [Google Scholar] [CrossRef] [Green Version]






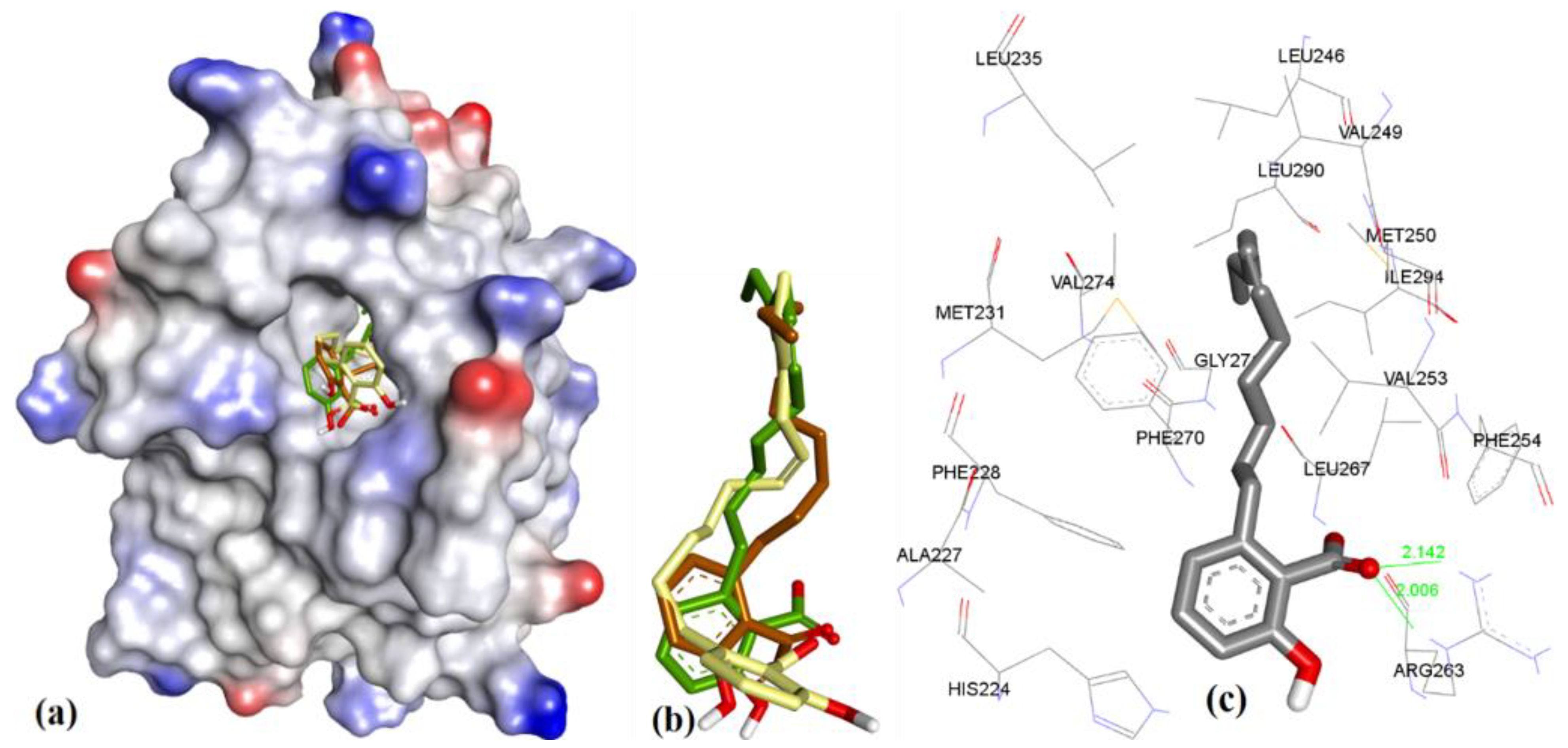



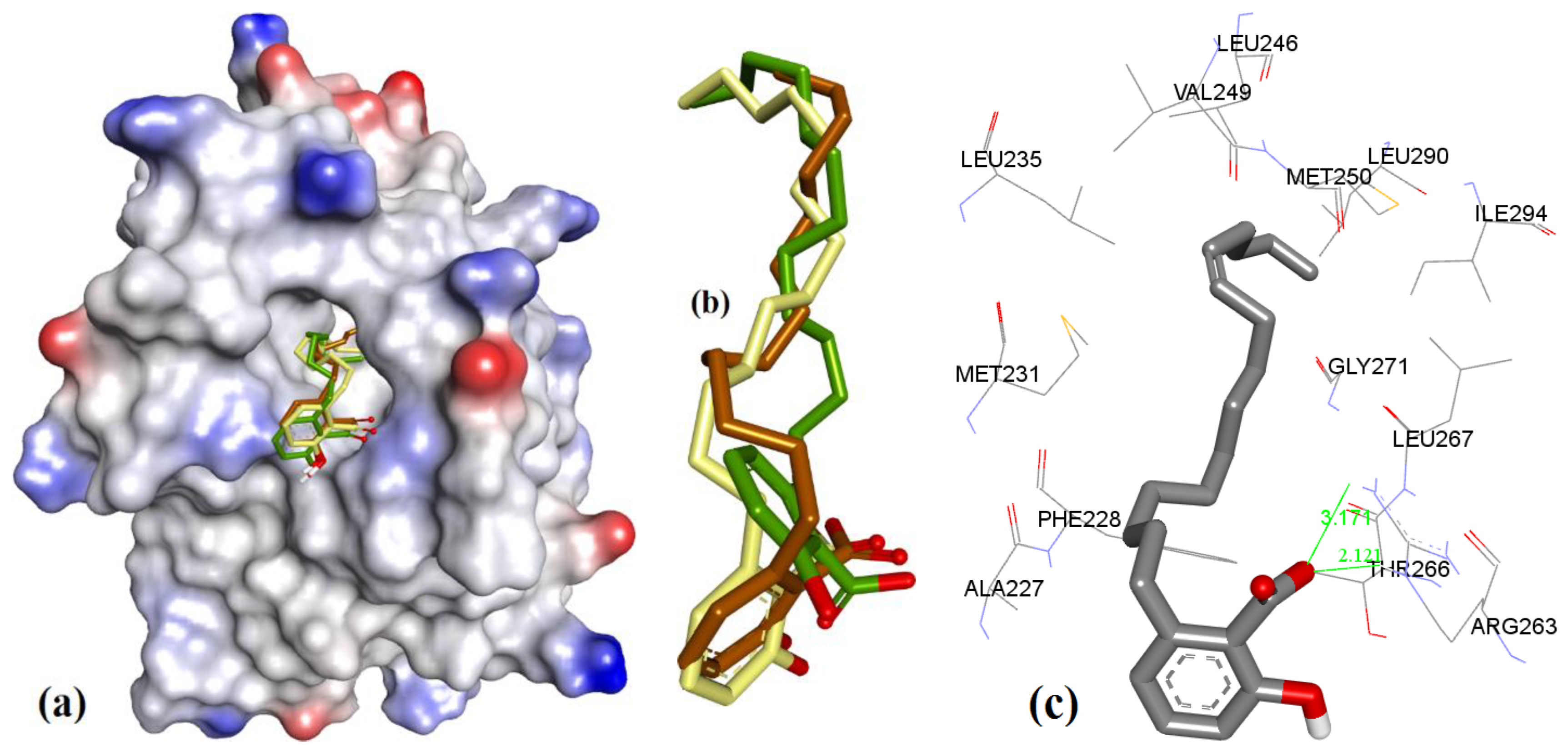
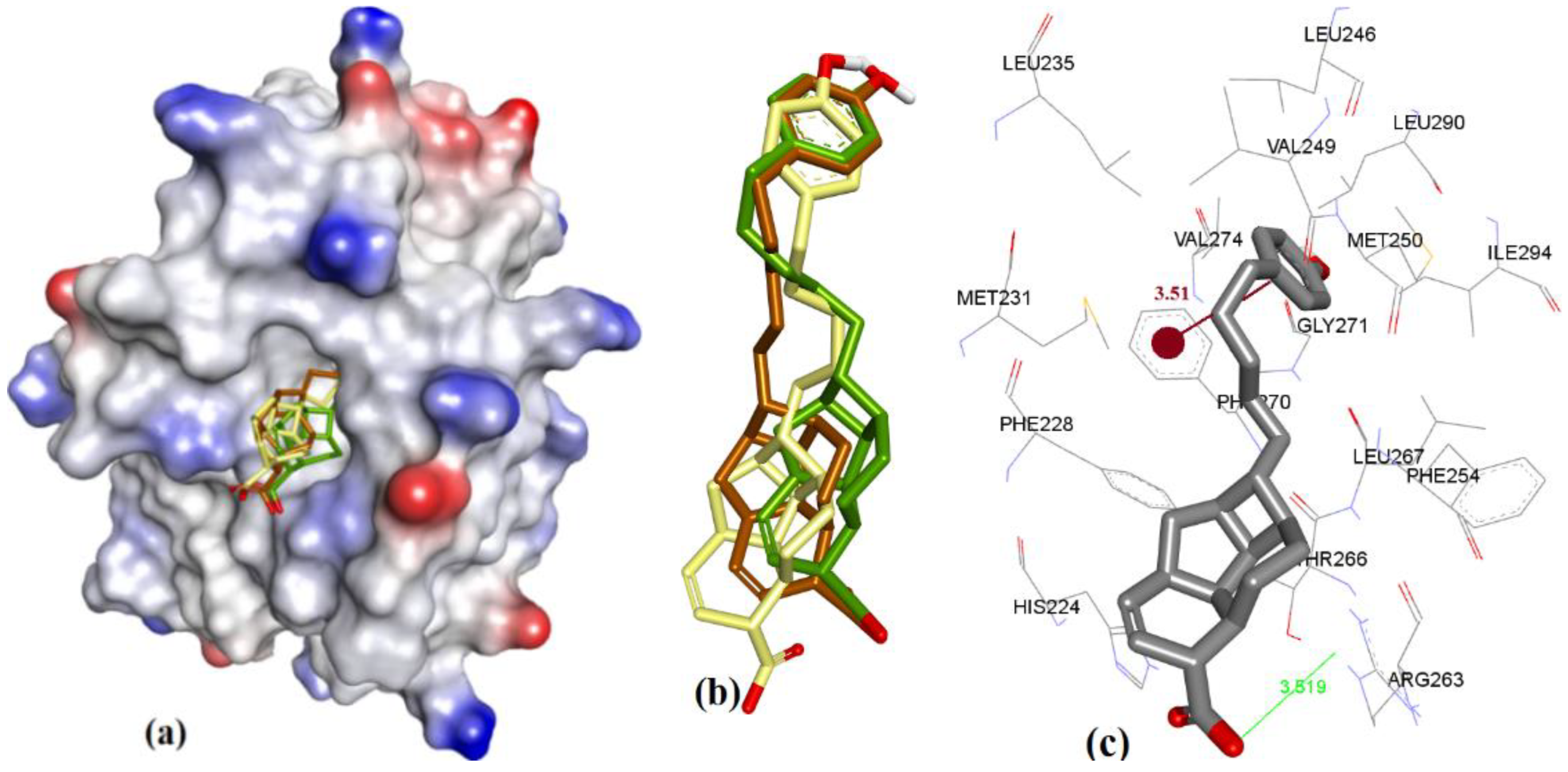

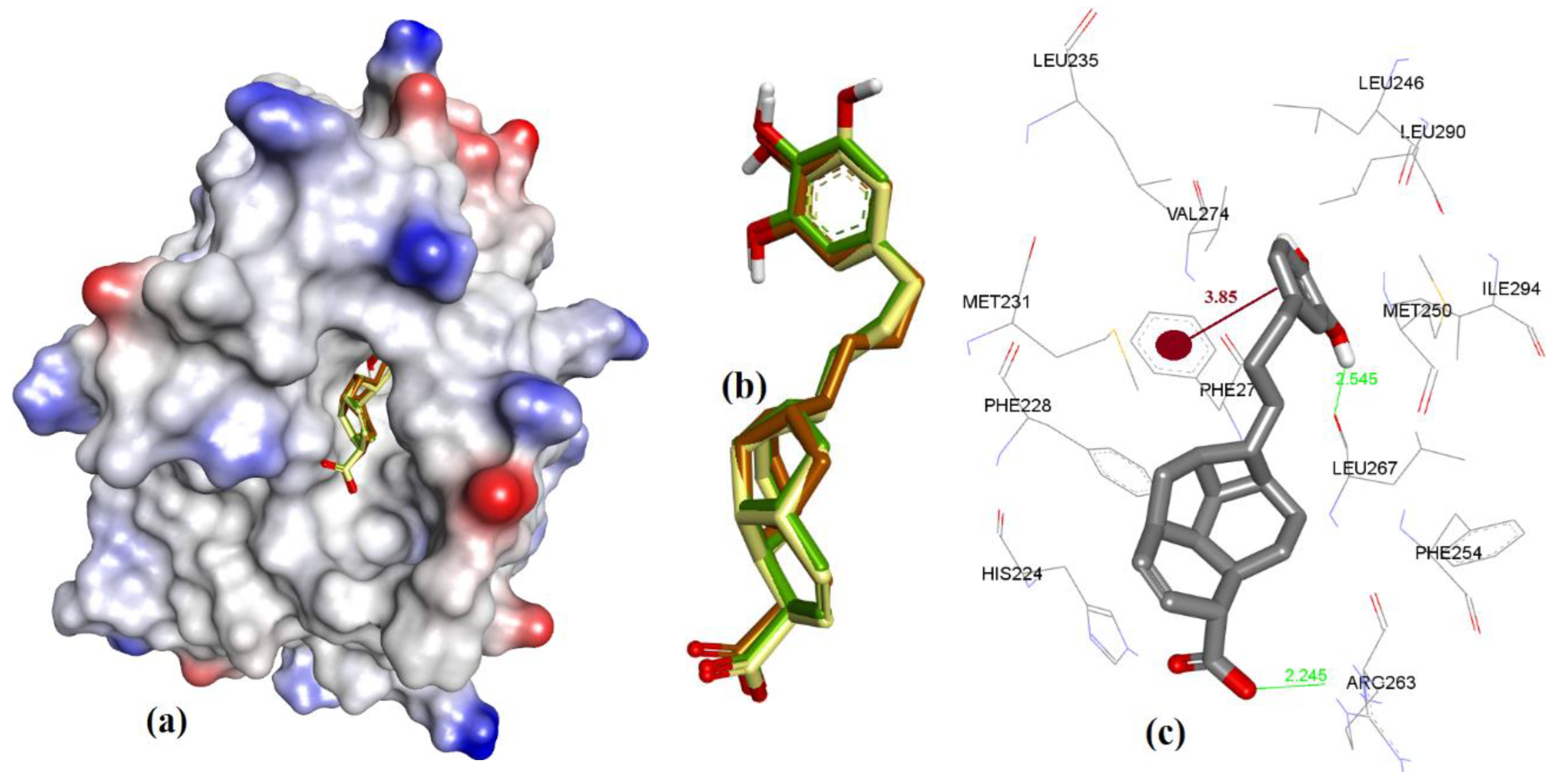
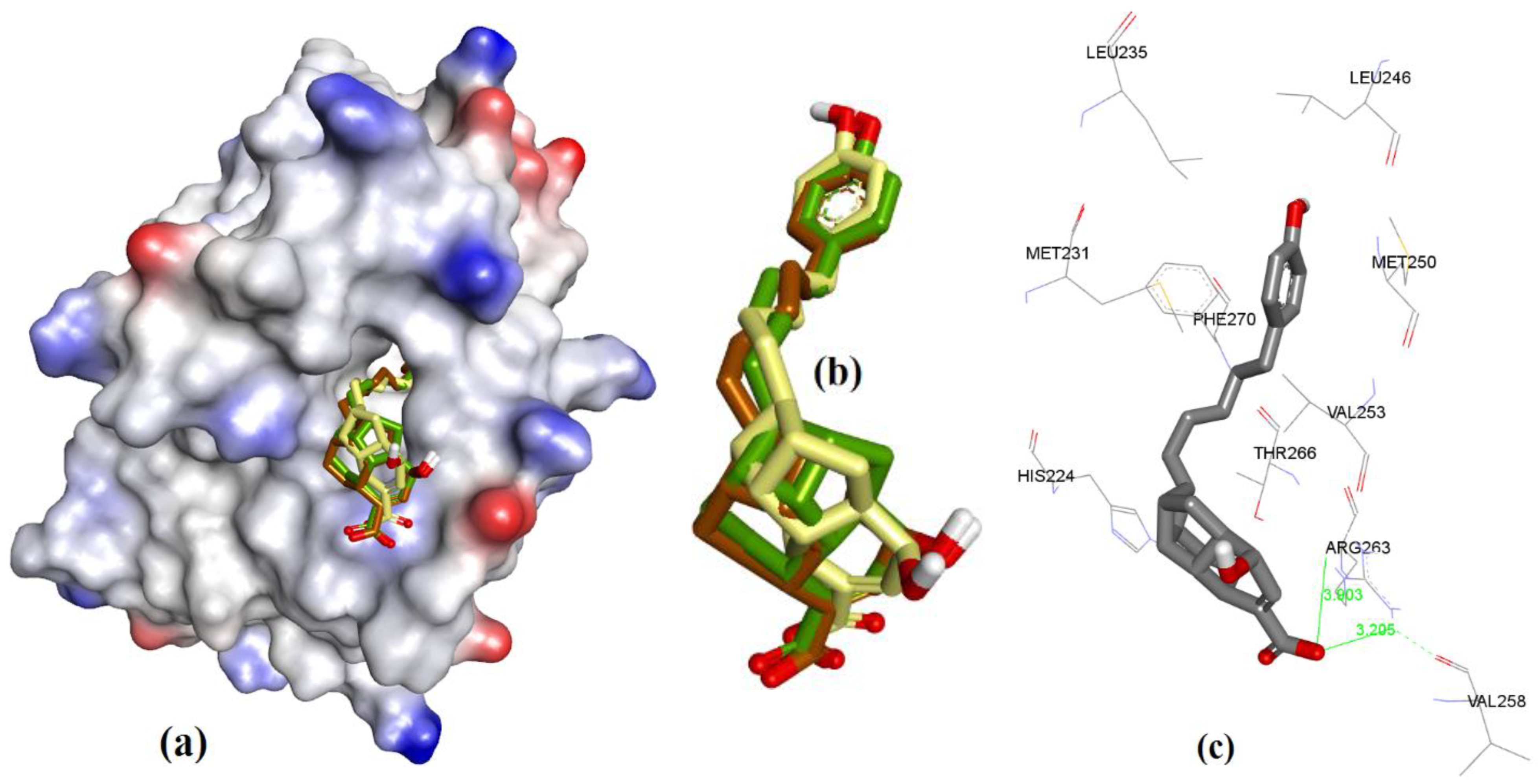
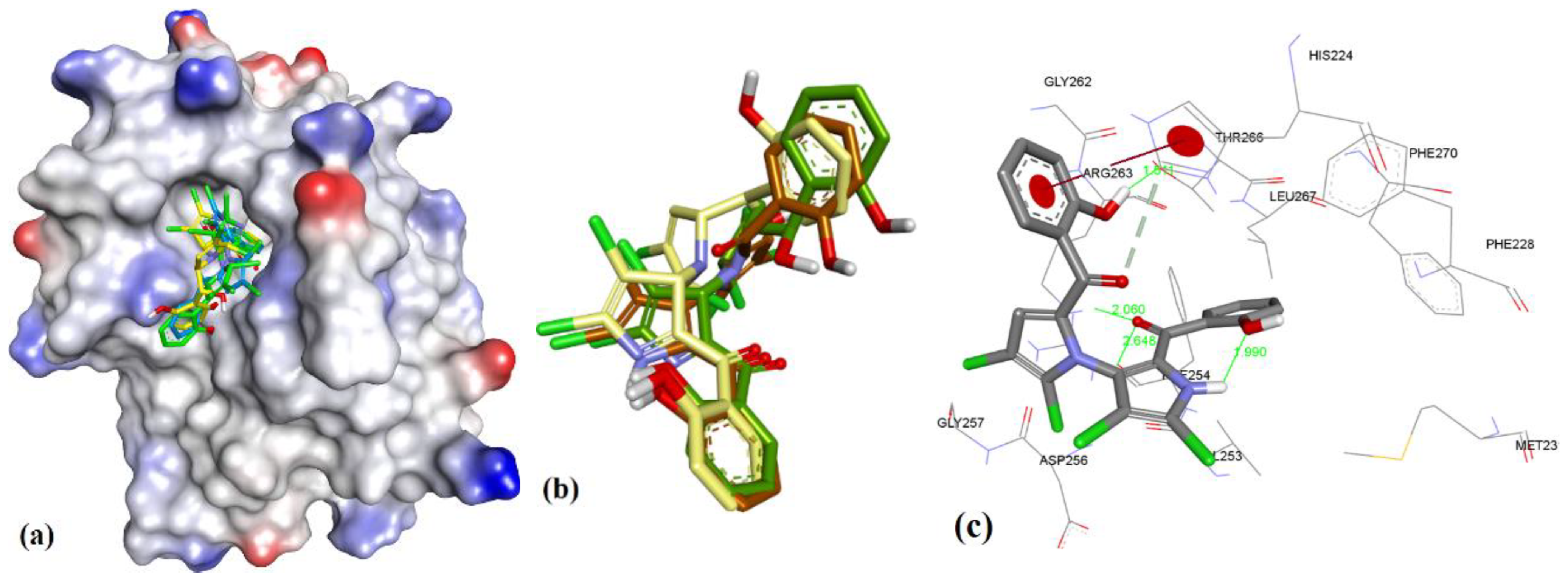
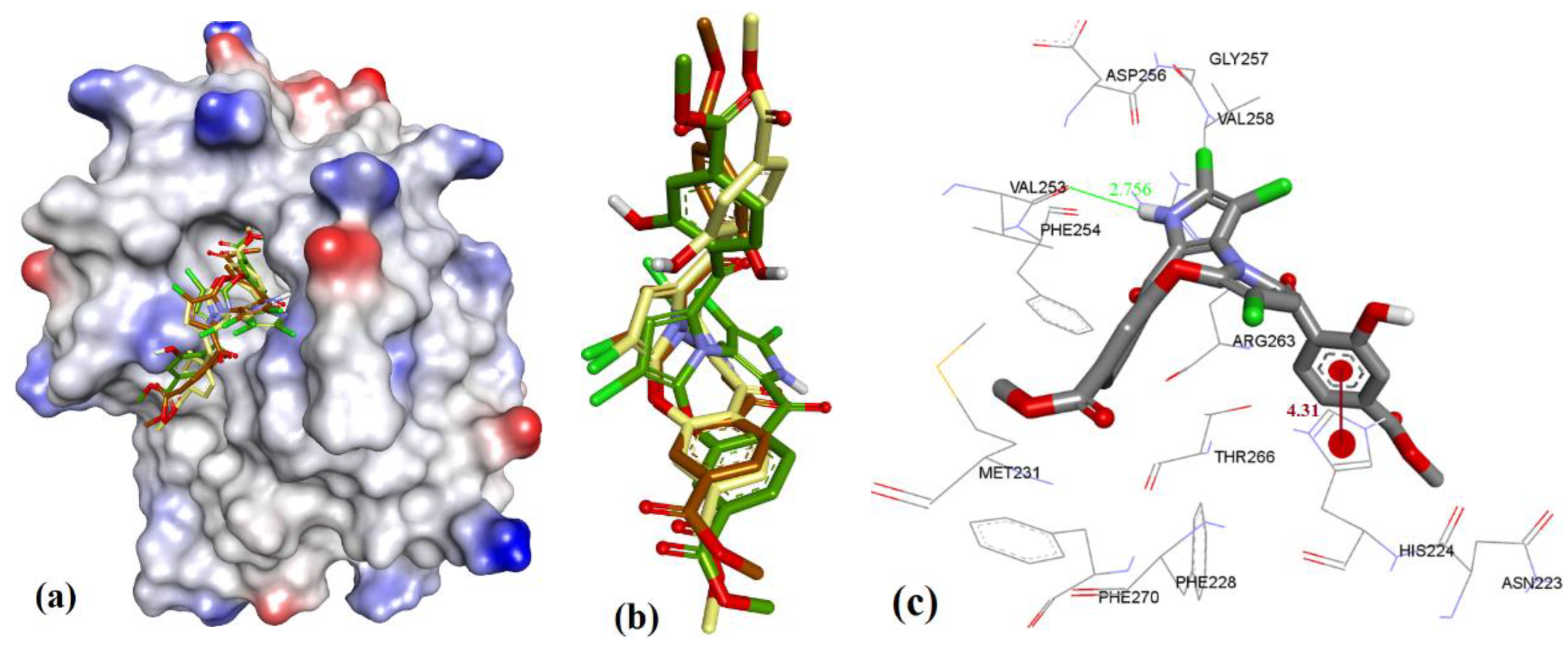
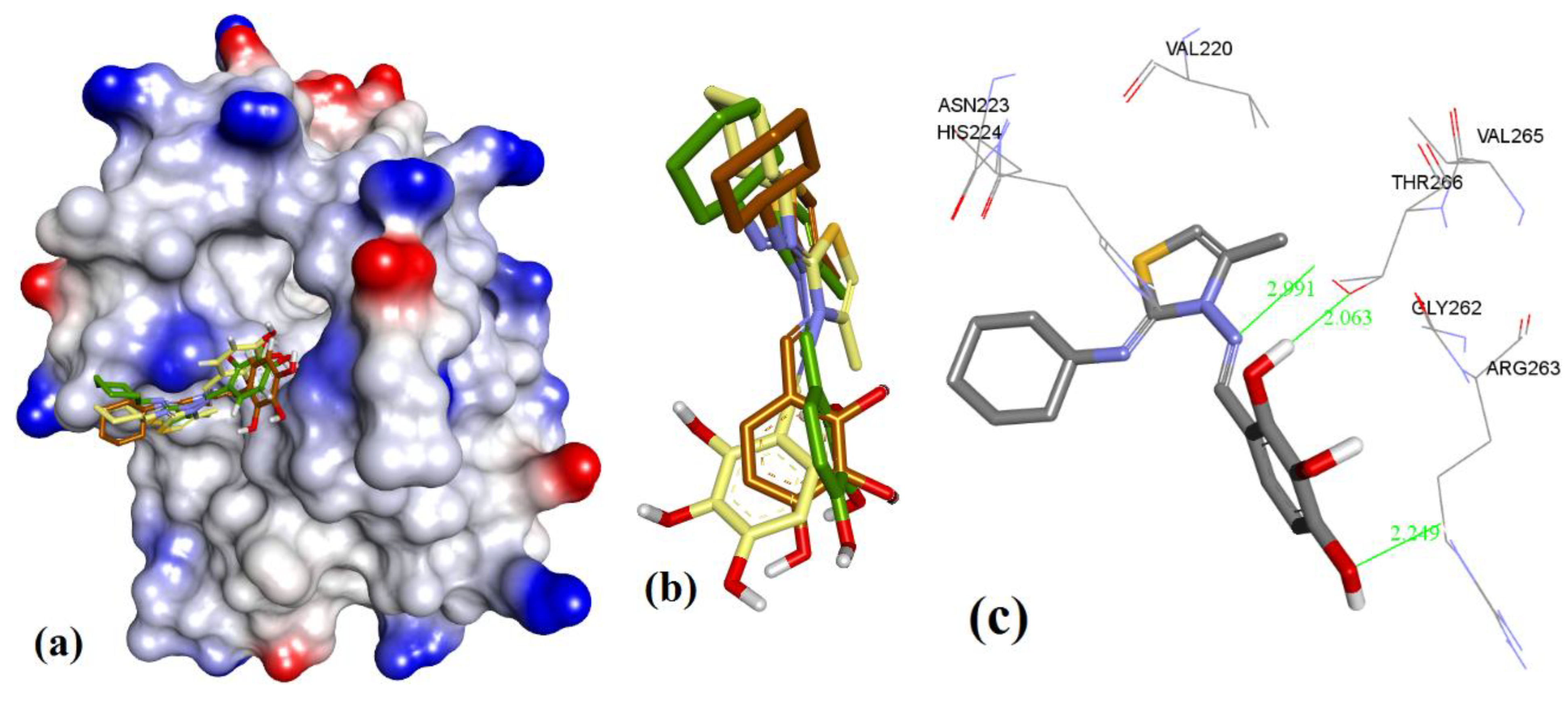
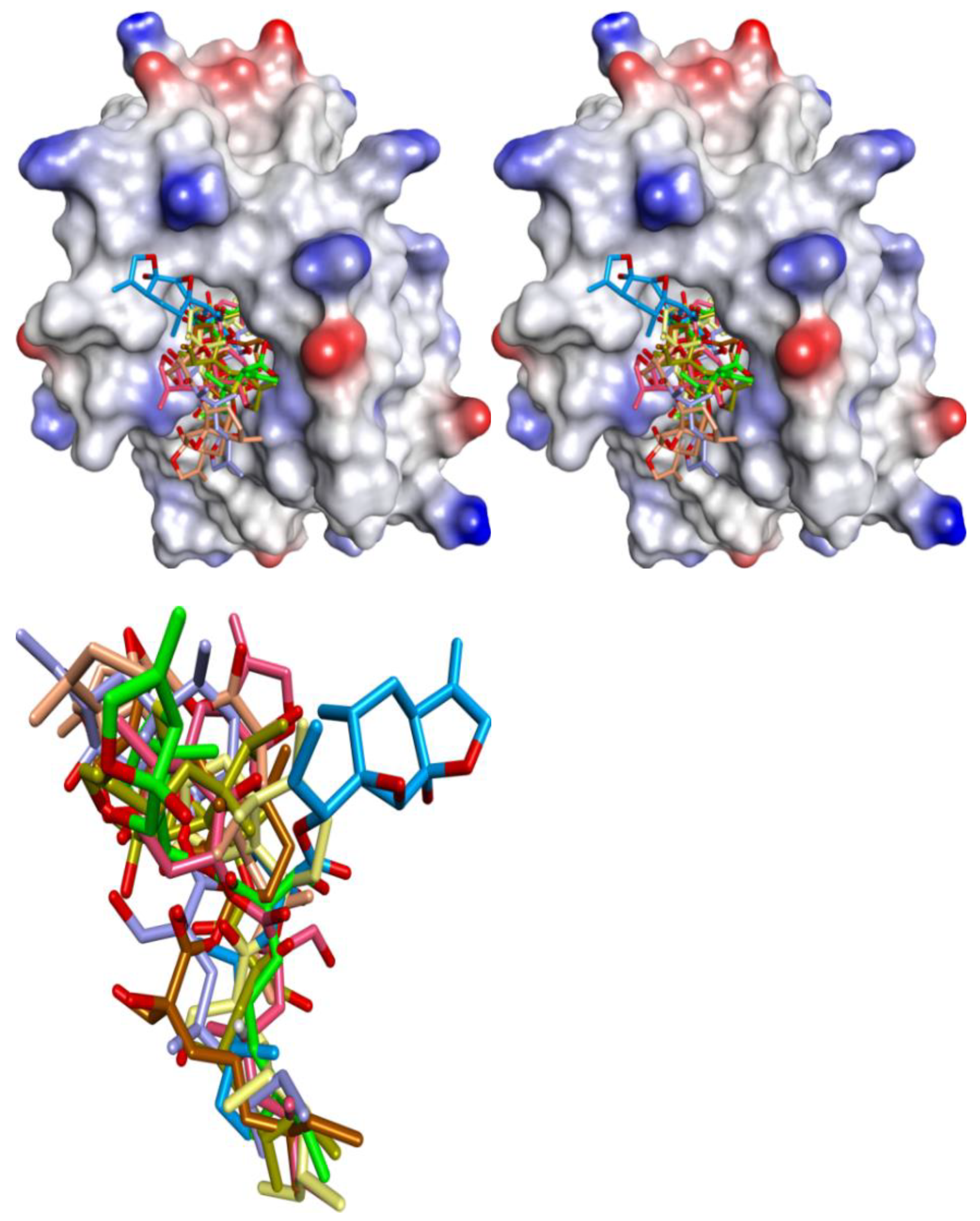




| Proteins | 4WMR | 4ZBF | 4ZBI | 4OQ5 | 4OQ6 | 3WIX | 3WIY | 4HW2 | Average | SD |
|---|---|---|---|---|---|---|---|---|---|---|
| Proteins | ||||||||||
| 4WMR | 0.888 | 0.174 | ||||||||
| 4ZBF | 0.86 | 0.644 | 0.156 | |||||||
| 4ZBI | 0.68 | 0.43 | 0.638 | 0.134 | ||||||
| 4OQ5 | 0.66 | 0.83 | 0.73 | 0.858 | 0.143 | |||||
| 4OQ6 | 1.06 | 0.78 | 0.85 | 0.90 | 0.873 | 0.102 | ||||
| 3WIX | 1.05 | 0.65 | 0.67 | 1.07 | 0.96 | 0.829 | 0.161 | |||
| 3WIY | 0.72 | 0.46 | 0.46 | 0.71 | 0.70 | 0.74 | 0.658 | 0.117 | ||
| 4HW2 | 1.13 | 0.62 | 0.72 | 1.05 | 0.88 | 0.70 | 0.78 | 0.808 | 0.185 | |
| 4HW3 | 0.94 | 0.52 | 0.56 | 0.91 | 0.85 | 0.79 | 0.69 | 0.58 | 0.730 | 0.155 |
| Proteins | 4WMR | 4ZBF | 4ZBI | 4OQ5 | 4OQ6 | 3WIX | 3WIY | 4HW2 | 4HW3 |
|---|---|---|---|---|---|---|---|---|---|
| Ligands | |||||||||
| 4WMR | 0.73 | 1.12 | 1.23 | 1.61 | 1.45 | 0.81 | 0.89 | 1.26 | 1.32 |
| 4ZBF | 1.27 | 0.75 | 1.19 | 1.32 | 1.09 | 0.90 | 1.03 | 1.31 | 1.42 |
| 4ZBI | 1.19 | 1.12 | 0.96 | 1.12 | 1.11 | 0.86 | 1.12 | 1.21 | 1.36 |
| 4OQ5 | 1.70 | 0.95 | 1.09 | 0.81 | 1.18 | 0.93 | 0.82 | 1.44 | 1.24 |
| 4OQ6 | 1.57 | 0.83 | 1.12 | 0.95 | 1.02 | 1.09 | 1.11 | 1.38 | 1.42 |
| 3WIX | 1.20 | 0.98 | 1.03 | 0.99 | 1.08 | 0.69 | 0.78 | 1.25 | 1.07 |
| 3WIY | 1.33 | 1.26 | 0.92 | 1.04 | 0.80 | 0.74 | 0.79 | 1.18 | 1.04 |
| 4HW2 | 1.87 | 1.14 | 1.30 | 1.61 | 1.49 | 0.98 | 0.86 | 1.12 | 1.13 |
| 4HW3 | 1.71 | 1.32 | 1.21 | 1.53 | 1.34 | 0.82 | 0.78 | 1.20 | 0.97 |
| Average | 1.397 | 1.052 | 1.117 | 1.220 | 1.173 | 0.869 | 0.909 | 1.261 | 1.219 |
| SD | 0.331 | 0.179 | 0.121 | 0.288 | 0.207 | 0.116 | 0.132 | 0.095 | 0.162 |
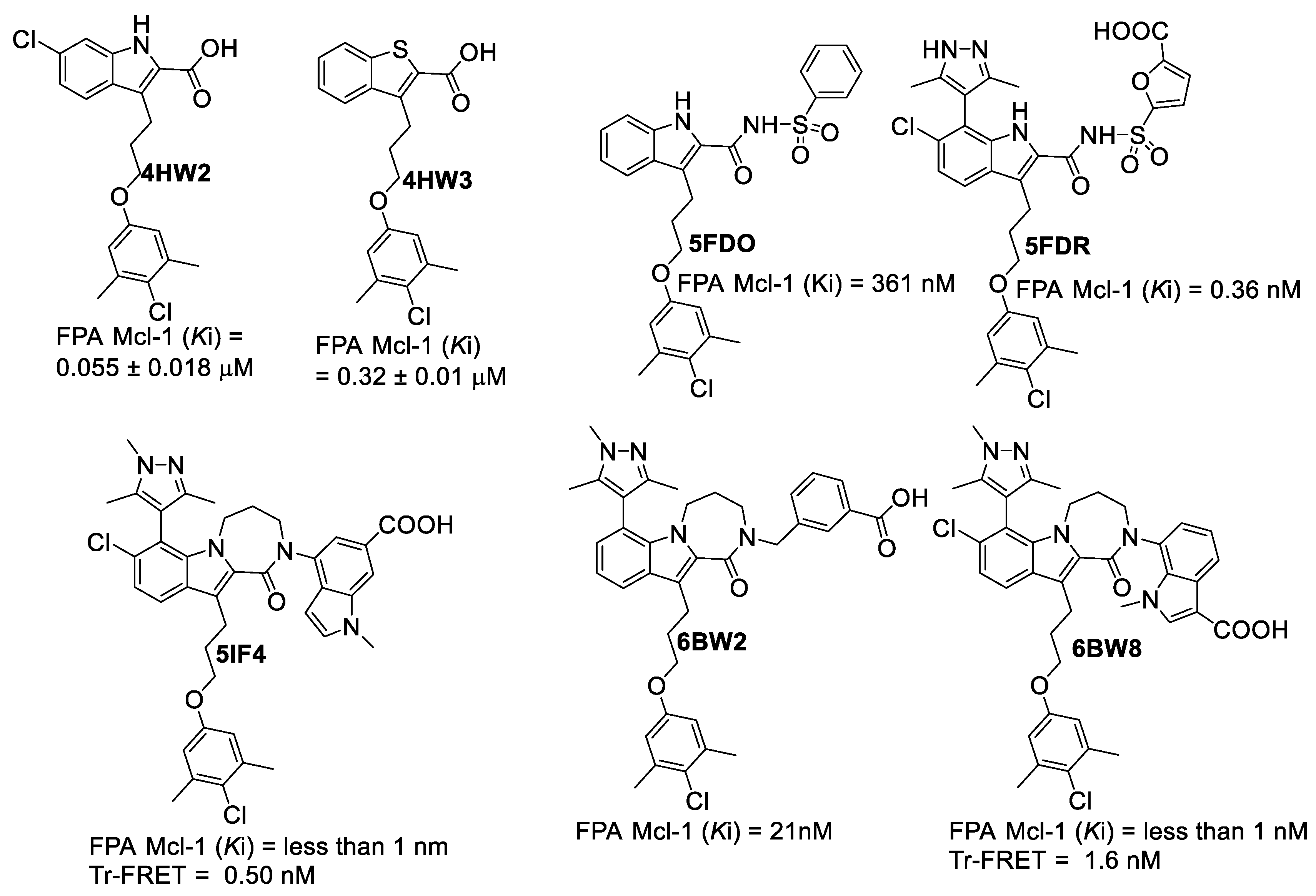
| PDB File | Ki Value (nM) | MOE | AutoDock | VLifeDock | |||
|---|---|---|---|---|---|---|---|
| Score a | RMSD b | Score a | RMSD b | Score a | RMSD b | ||
| 5FDO | 361 | −7.29 | 1.81 | −8.30 | 1.94 | −7.02 | 3.49 |
| 5FDR | 0.94 | −8.59 | 2.82 | −7.22 | 3.07 | −7.48 | 3.14 |
| 6BW2 | 21.0 | −10.52 | 1.86 | −9.94 | 2.30 | −9.11 | 2.33 |
| 6BW8 | <1.00 | −10.34 | 0.81 | −9.84 | 1.36 | −9.17 | 1.45 |
| 5IF4 | <1.00 | −10.76 | 1.24 | −10.14 | 1.36 | −10.87 | 1.51 |
| 4HW2 | 55 ± 18 | −8.41 | 1.12 | −7.99 | 1.27 | −8.02 | 1.30 |
| 4HW3 | 320 ± 10 | −6.46 | 0.97 | −6.47 | 1.19 | −6.10 | 1.43 |
| Corresponding Amino Acids | |||||
|---|---|---|---|---|---|
| mMcl-1 | hMcl-1 | mMcl-1 | hMcl-1 | mMcl-1 | hMcl-1 |
| G150 | - | N241 | N260 | R229 | R248 |
| I163 | I182 | G243 | G262 | F235 | F254 |
| T172 | T191 | I245 | I264 | D237 | D256 |
| G184 | G203 | S250 | S269 | R244 | R263 |
| G200 | G219 | F251 | F270 | V246 | V265 |
| Q202 | Q221 | V255 | V274 | ||
| R203 | R222 | V262 | I281 | ||
| N204 | N223 | V278 | V297 | ||
| R214 | R233 | L279 | L298 | ||
| L216 | L235 | F300 | F319 | ||
| N220 | N239 | Q306 | |||
| G222 | D241 | G307 | |||
| S228 | S247 | ||||
| Entry | Compound | hMcl-1 Ki/IC50 in µM | Bcl-2 a/Bcl-xL b Ki/IC50 = µM | Ref. | Triangle Matcher f | AutoDock f | GRIP Docking f | RMSD (Å) |
|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 3.3 d | NR | [19] | −7.36 | −7.71 | −6.17 | 1.08 |
| 2 | 2a | 2.4 ± 0.1 | NR | [20] | −8.33 | −8.29 | −8.76 | 0.81 |
| 3 | 2b | 8.9 | NR | [20] | −8.42 | −8.22 | −8.67 | 0.72 |
| 4 | 2c | 7.3 | NR | [20] | −8.27 | −8.16 | −8.64 | 1.04 |
| 5 | 3a | 17.7 ± 3.1 d | >23 b,d | [21] | −7.14 | −6.82 | −7.17 | 1.22 |
| 6 | 3b | 5.8 ± 0.3 d | 3.2 ± 0.1 b,d | [21] | −7.83 | −7.14 | −7.67 | 2.09 |
| 7 | 3c | 3.7 ± 2.0 d | 16.3 ± 0.5 b,d | [21] | −7.98 | −7.61 | −8.13 | 1.58 |
| 8 | 3d | 0.7 ± 0.1 d | 1.2 ± 0.1 b,d | [21] | −8.34 | −7.42 | −8.91 | 2.18 |
| 9 | 3e | 0.2 ± 0.1 d | 0.3 ± 0.1 b,d | [21] | −8.47 | −8.22 | −9.09 | 1.97 |
| 10 | 3f | 0.2 ± 0.1 d | 0.2 ± 0.1 b,d | [21] | −8.22 | −7.53 | −8.76 | 2.05 |
| 11 | 3g | 1.2 ± 0.9 d | 5.7 ± 0.6 b,d | [21] | −8.00 | −7.68 | −8.27 | 2.31 |
| 12 | 4aa | 14 ± 3.3 e | 19.2 ± 1.6 b,e | [22] | −7.15 | −6.71 | −7.19 | 1.86 |
| 13 | 4b | 13 ± 5.0 e | 12.6 ± 0.2 b,e | [22] | −7.42 | −6.32 | −7.53 | 1.59 |
| 14 | 4c | 5.2 ± 0.2 e | >100 b,e | [22] | −8.02 | −7.60 | −8.11 | 0.88 |
| 15 | 4d | 5.9 ± 0.5 e | 19.4 ± 3.0 b,e | [22] | −7.89 | −7.14 | −8.02 | 1.03 |
| 17 | 5 | 8.9 ± 1.0 d | 16.4 ± 3.3 b,d | [53] | −7.51 | −6.67 | −7.92 | 1.23 |
| 18 | 5a | 4.3 ± 1.5 d | 3.4 ± 0.9 b,d | [53] | −7.06 | −6.39 | −7.84 | 1.70 |
| 19 | 6a | 4.72 d | NR | [27] | −8.77 | −7.88 | −8.90 | 2.13 |
| 20 | 7 (R, R, R) | NAc | NR | [28] | −6.88 | −5.29 | −6.16 | 2.64 |
| 21 | 7 (R, R, S) | NAc | NR | [28] | −7.17 | −6.02 | −6.31 | 2.37 |
| 22 | 7 (R, S, S) | NAc | NR | [28] | −7.04 | −5.89 | −6.20 | 3.14 |
| 23 | 7 (S, S, S) | NAc | NR | [28] | −7.22 | −6.17 | −6.47 | 1.76 |
| 24 | 7 (S, R, R) | NAc | NR | [28] | −6.53 | −5.56 | −5.90 | 2.22 |
| 25 | 7 (S, S, R) | NAc | NR | [28] | −6.70 | −5.82 | −6.13 | 2.90 |
| 26 | 7 (R, S, R) | NAc | NR | [28] | −7.34 | −6.31 | −6.41 | 2.46 |
| 27 | 7 (S, R, S) | NAc | NR | [28] | −7.12 | −6.12 | −6.28 | 1.94 |
| 29 | 8 | 5.2 ± 1.2 e | 1.46 ± 0.12 a,e/8.30 ± 1.20 b,e | [29] | −6.66 | −6.42 | −6.24 | 1.48 |
| 30 | 8a | 0.46 ± 0.06 e | 0.83 ± 0.16 a,e/2.19 ± 0.09 b,e | [29] | −7.22 | −7.14 | −6.94 | 1.14 |
| 31 | 8b | 5.92 ± 0.47 e | >23 a,e/8.48 ± 0.40 b,e | [29] | −7.33 | −7.25 | −7.22 | 1.85 |
| 32 | 8c | 0.56 ± 0.04 e | 1.54 ± 0.44 a,e/2.44 ± 0.02 b,e | [29] | −6.97 | −6.89 | −6.77 | 1.57 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Negi, A.; Murphy, P.V. Natural Products as Mcl-1 Inhibitors: A Comparative Study of Experimental and Computational Modelling Data. Chemistry 2022, 4, 983-1009. https://doi.org/10.3390/chemistry4030067
Negi A, Murphy PV. Natural Products as Mcl-1 Inhibitors: A Comparative Study of Experimental and Computational Modelling Data. Chemistry. 2022; 4(3):983-1009. https://doi.org/10.3390/chemistry4030067
Chicago/Turabian StyleNegi, Arvind, and Paul V. Murphy. 2022. "Natural Products as Mcl-1 Inhibitors: A Comparative Study of Experimental and Computational Modelling Data" Chemistry 4, no. 3: 983-1009. https://doi.org/10.3390/chemistry4030067
APA StyleNegi, A., & Murphy, P. V. (2022). Natural Products as Mcl-1 Inhibitors: A Comparative Study of Experimental and Computational Modelling Data. Chemistry, 4(3), 983-1009. https://doi.org/10.3390/chemistry4030067