
Metal–Organic Cages: Applications in Organic Reactions



Abstract
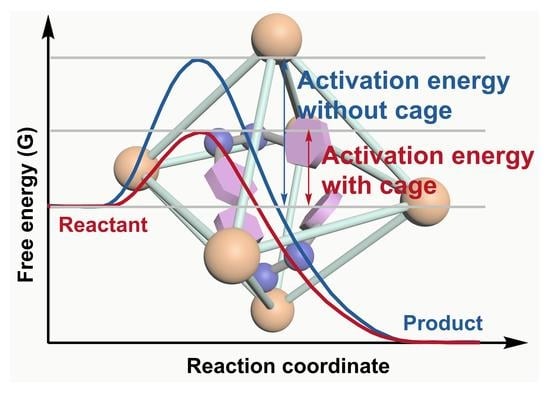
:
1. Introduction
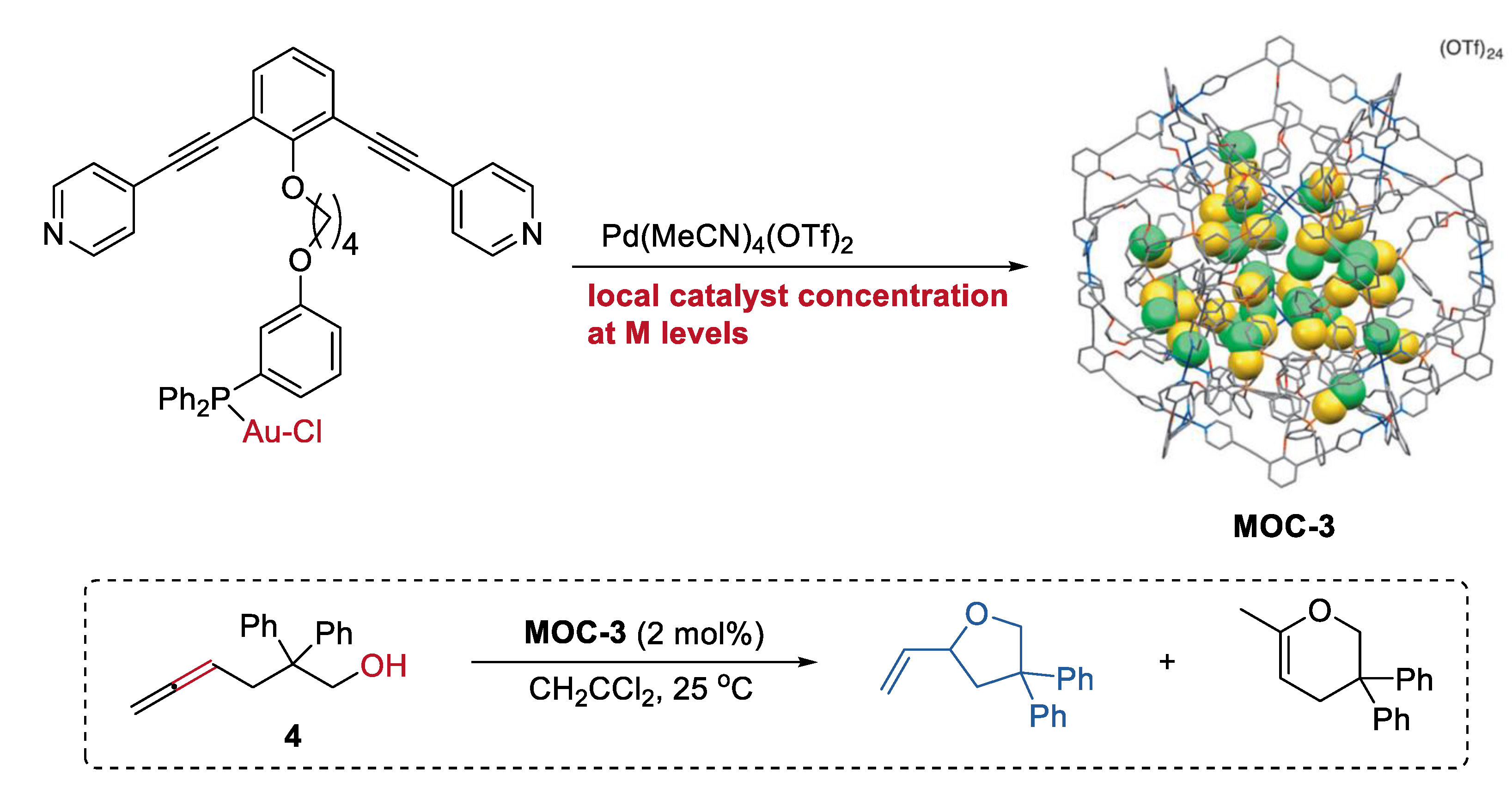
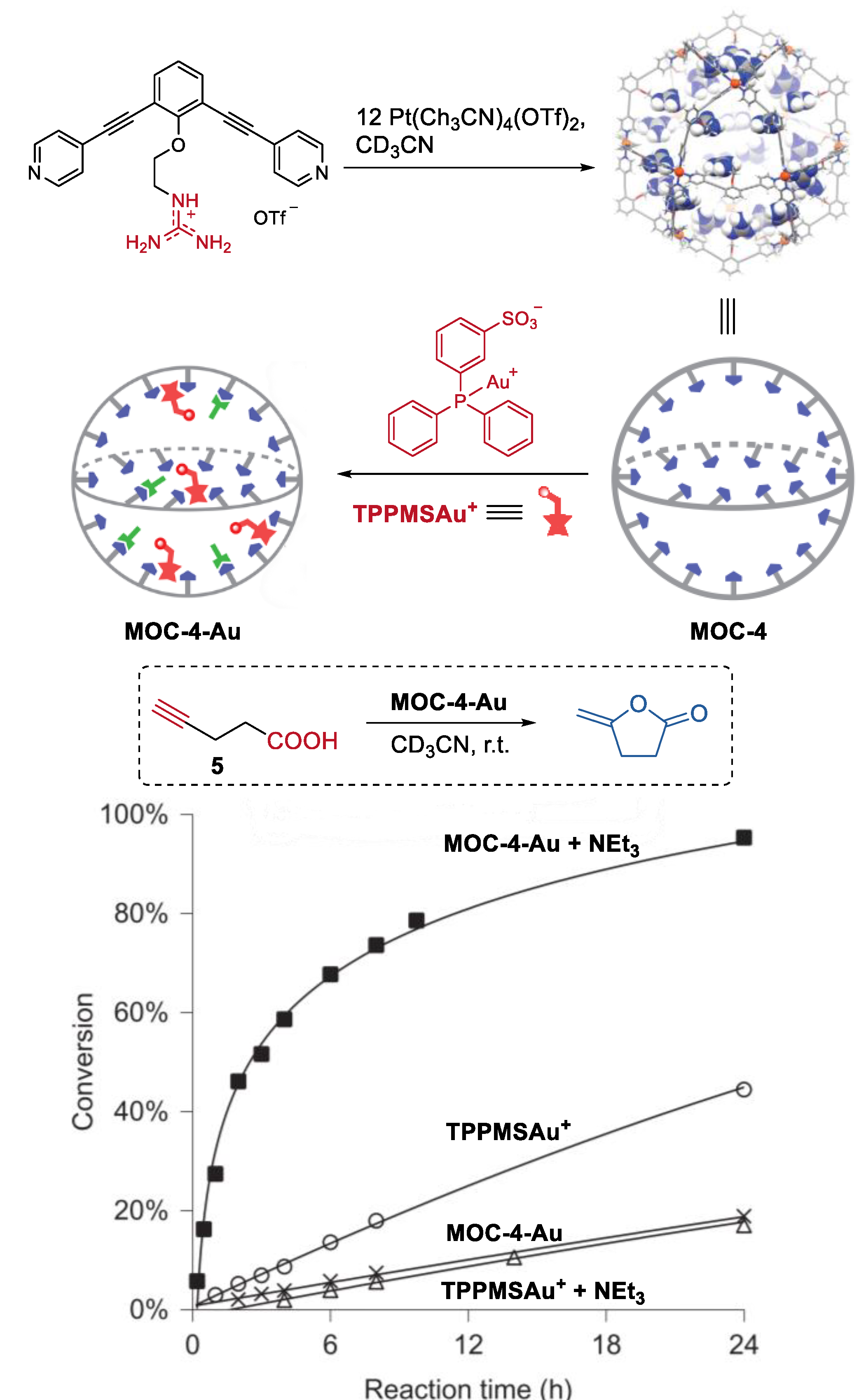
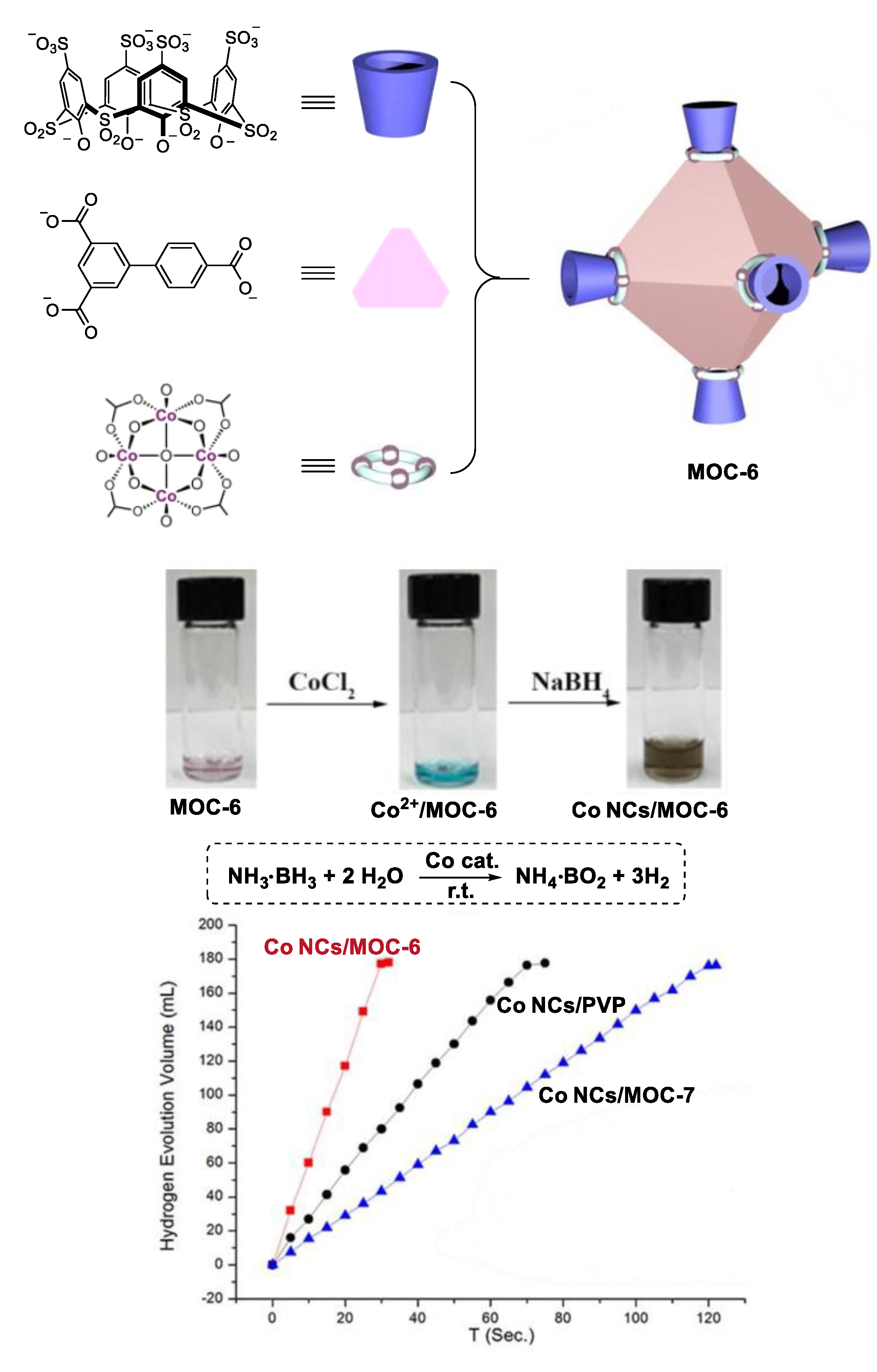
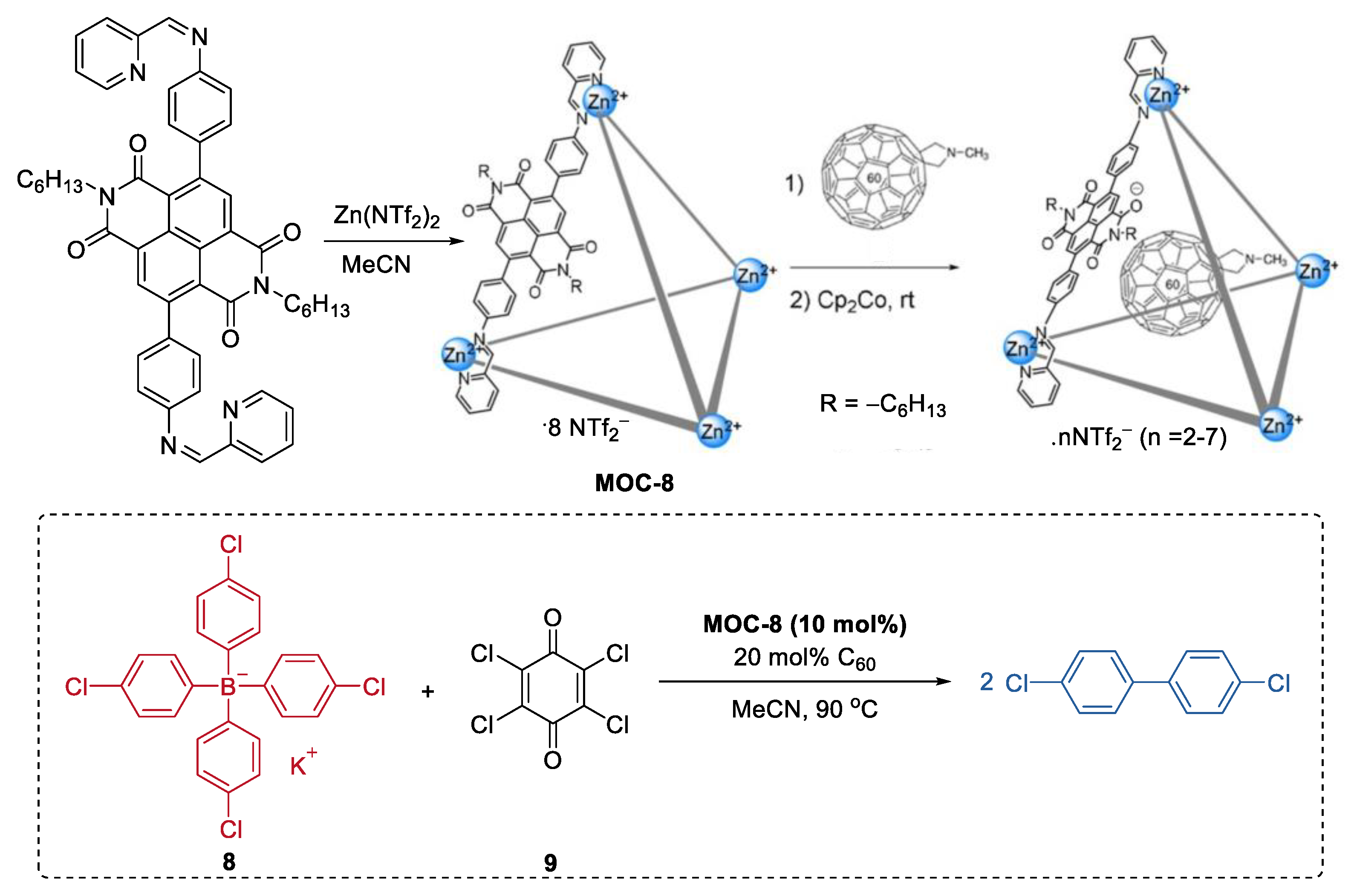
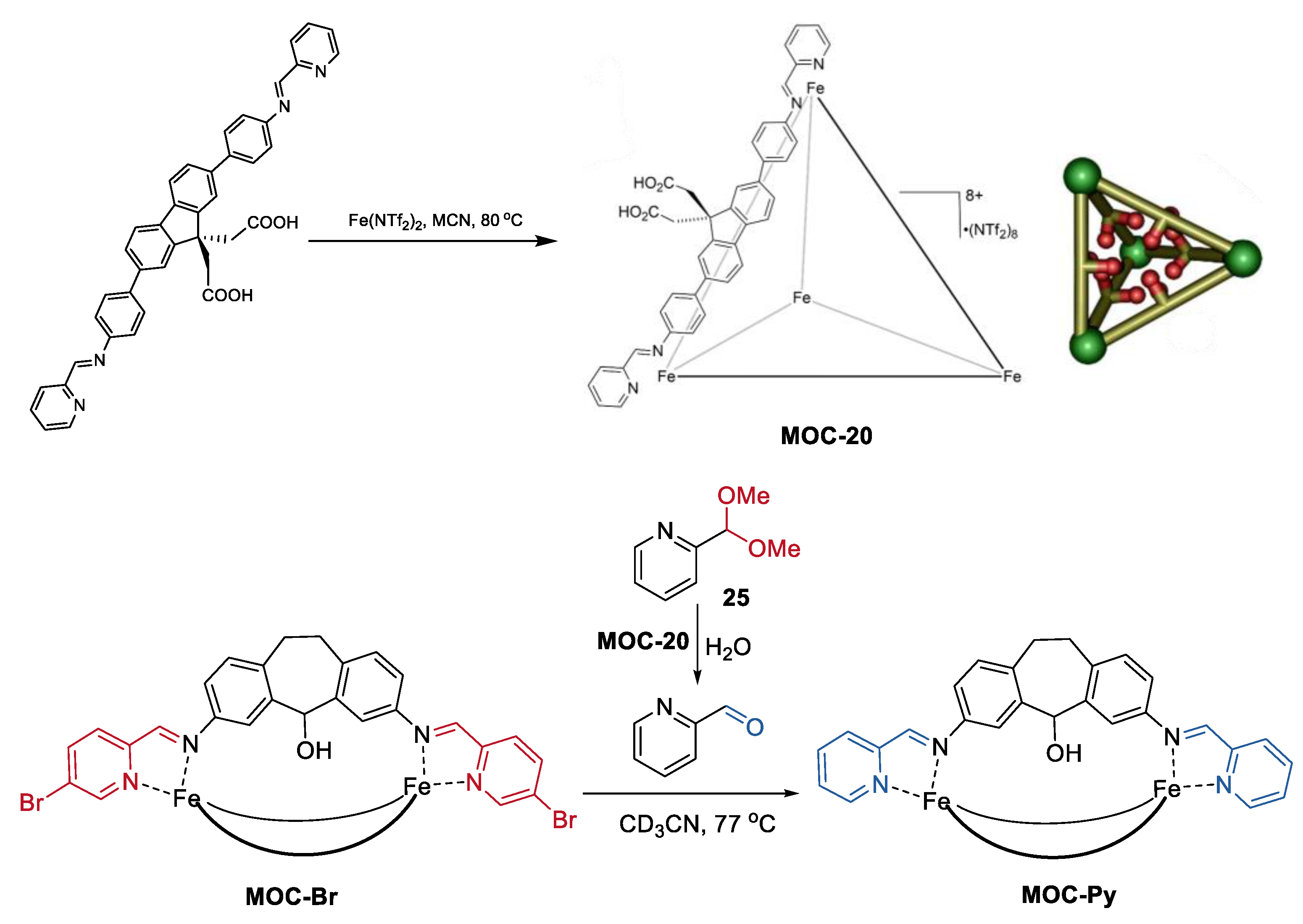
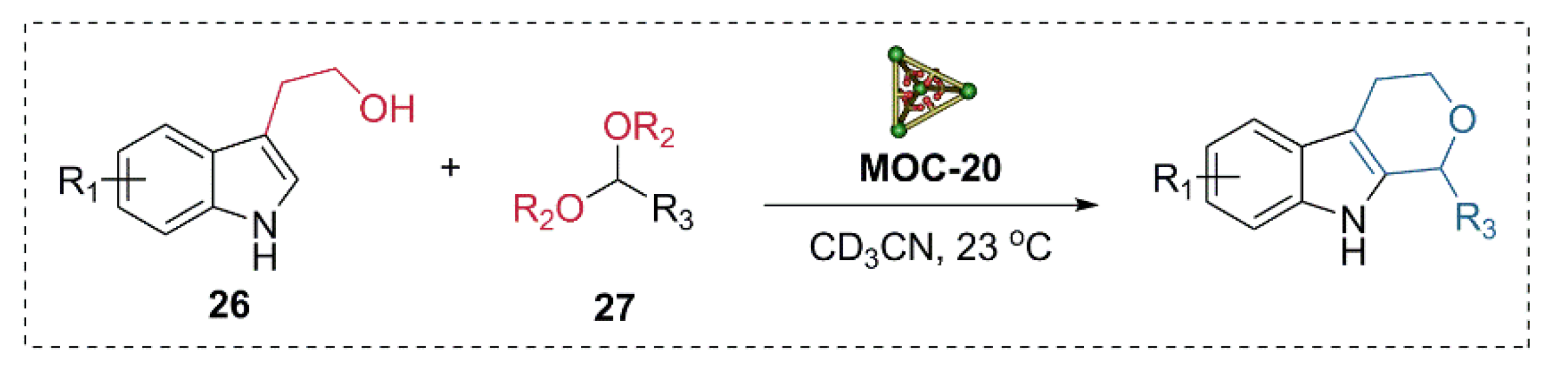
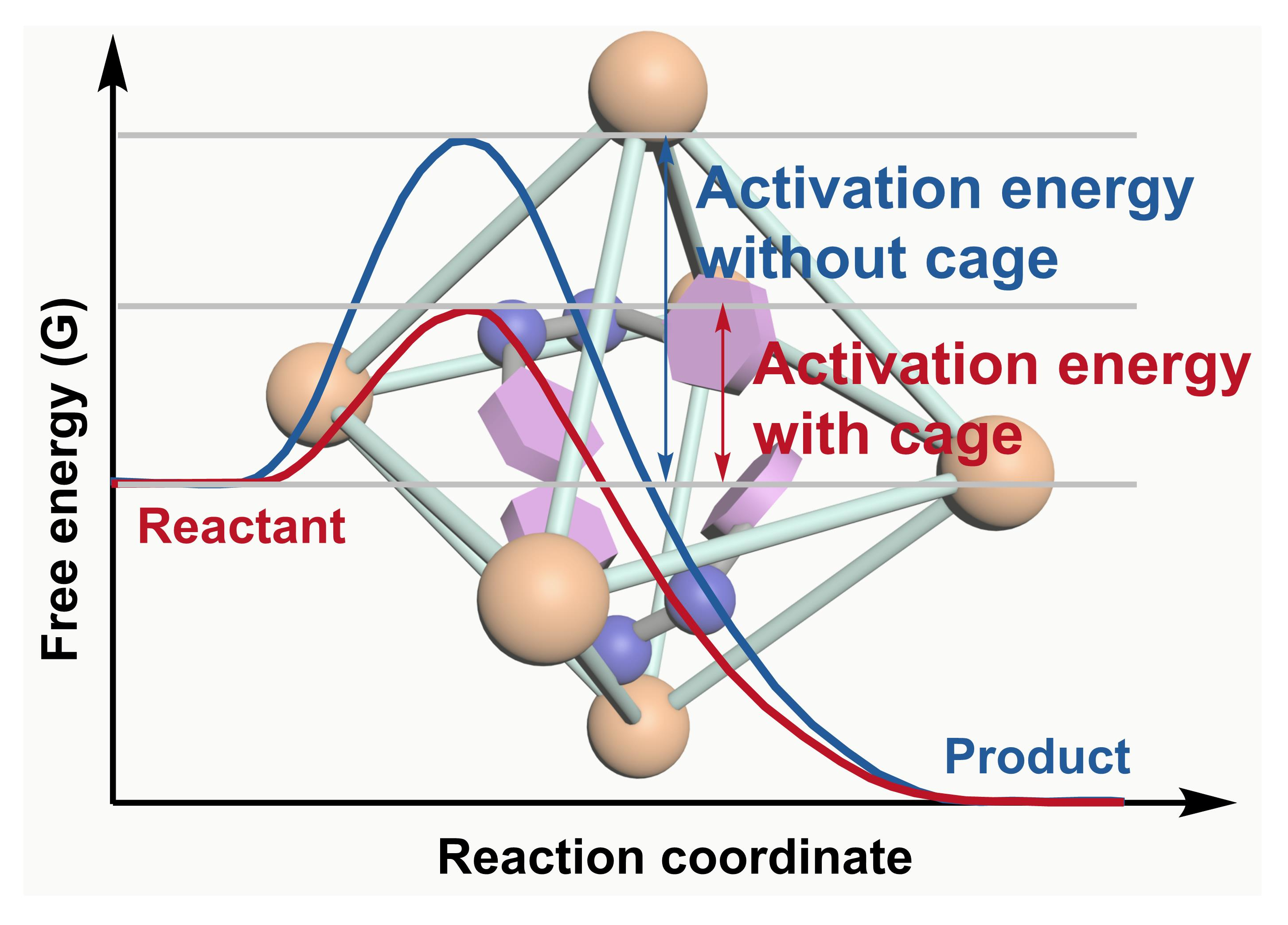
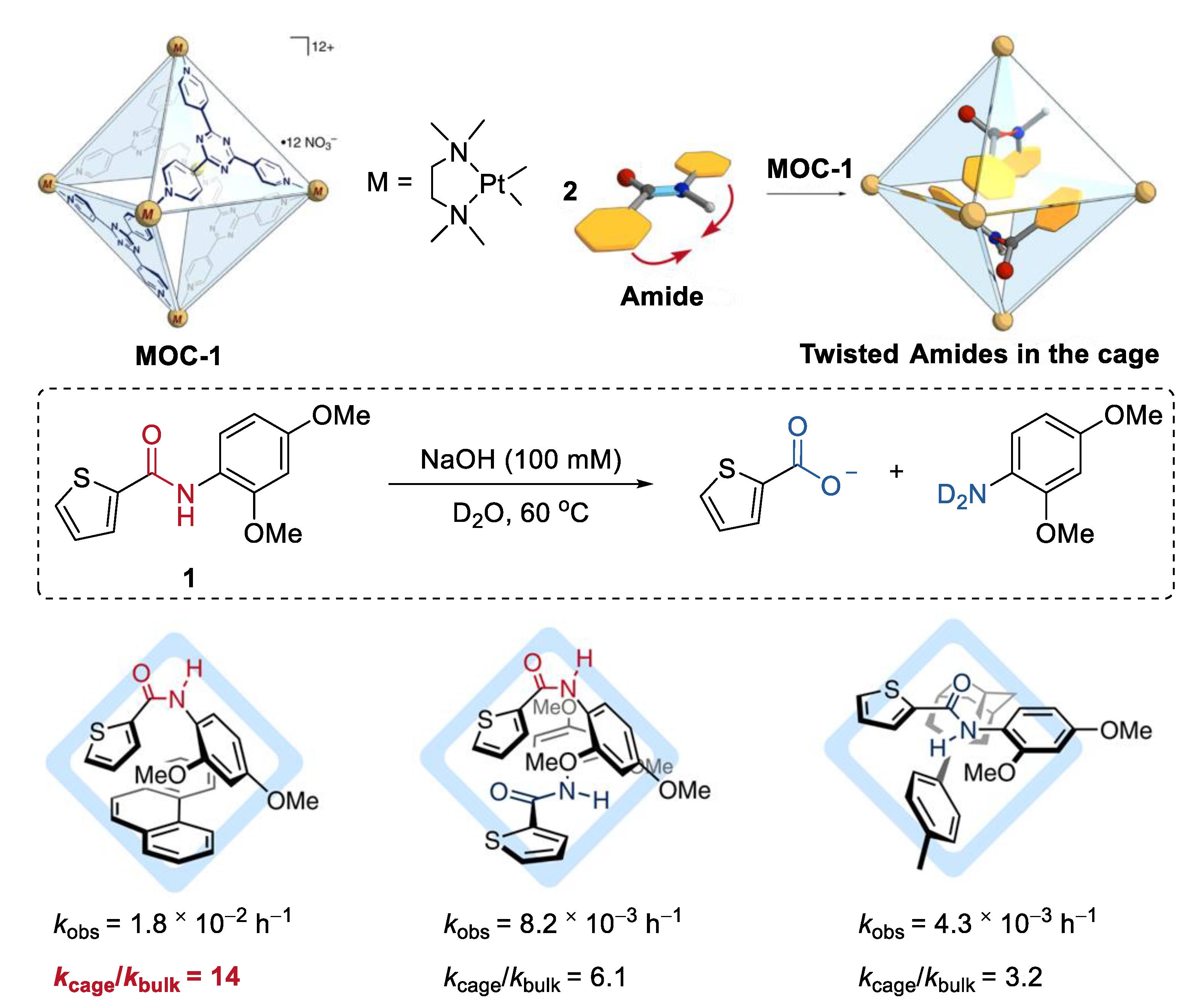
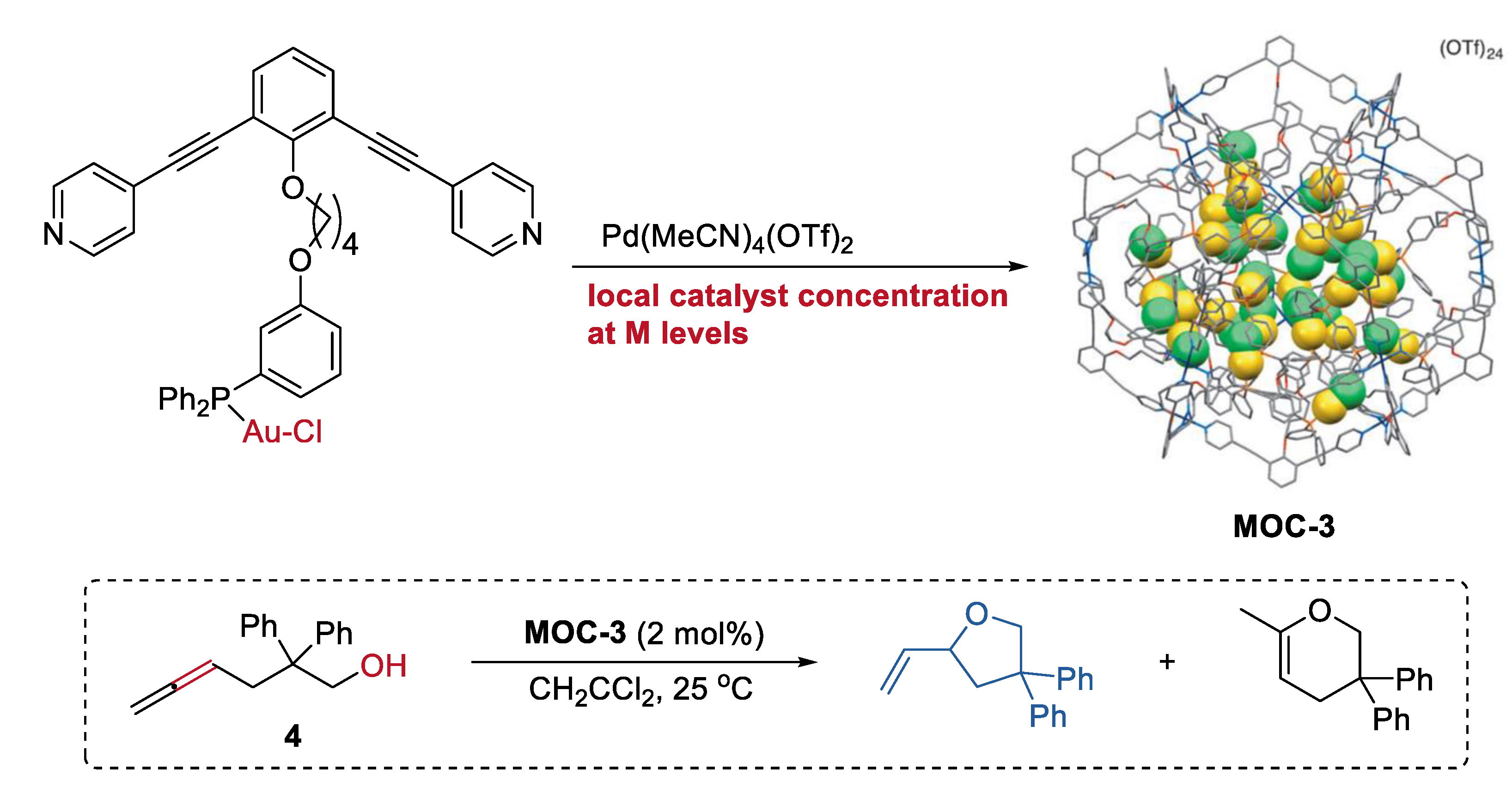
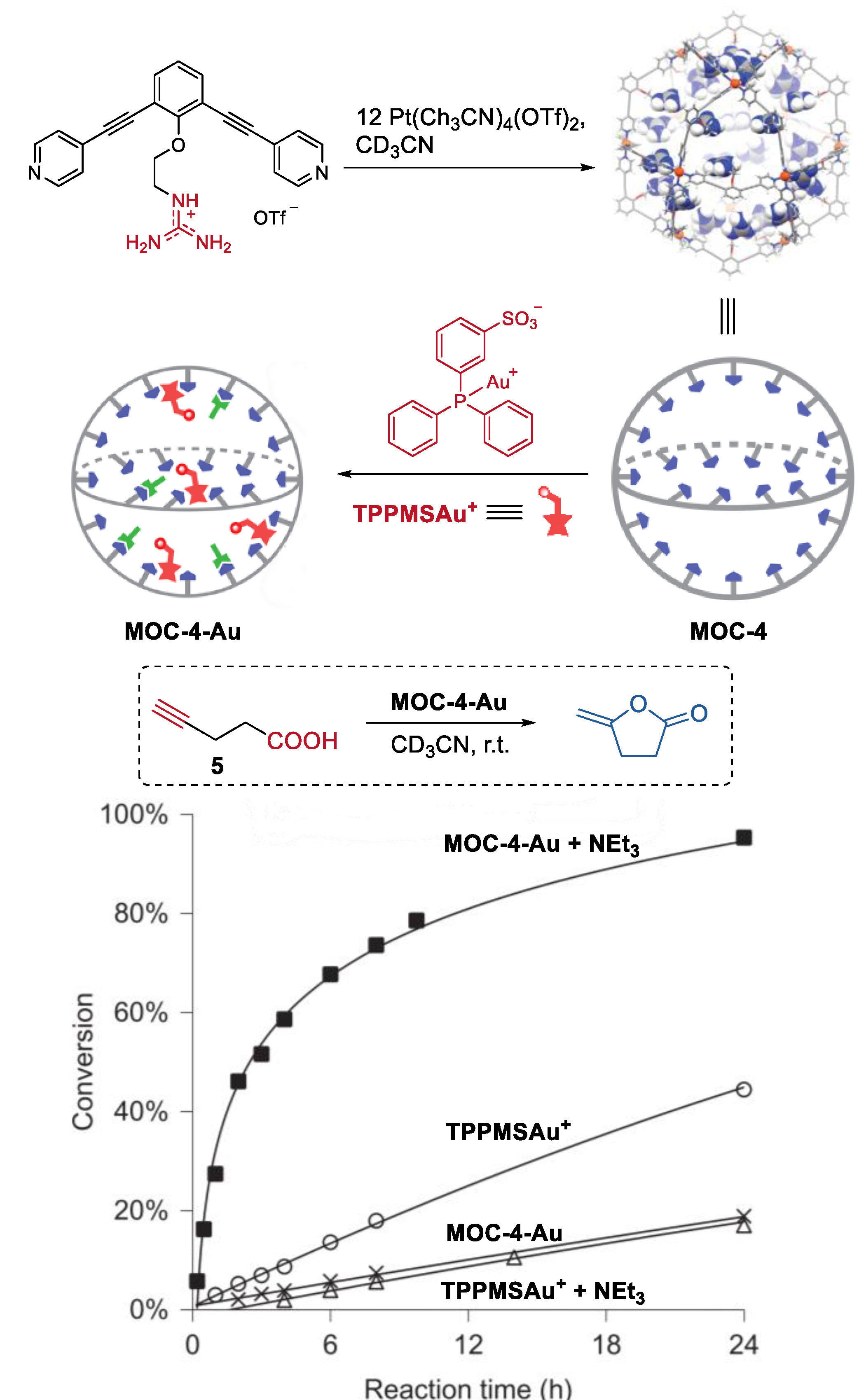
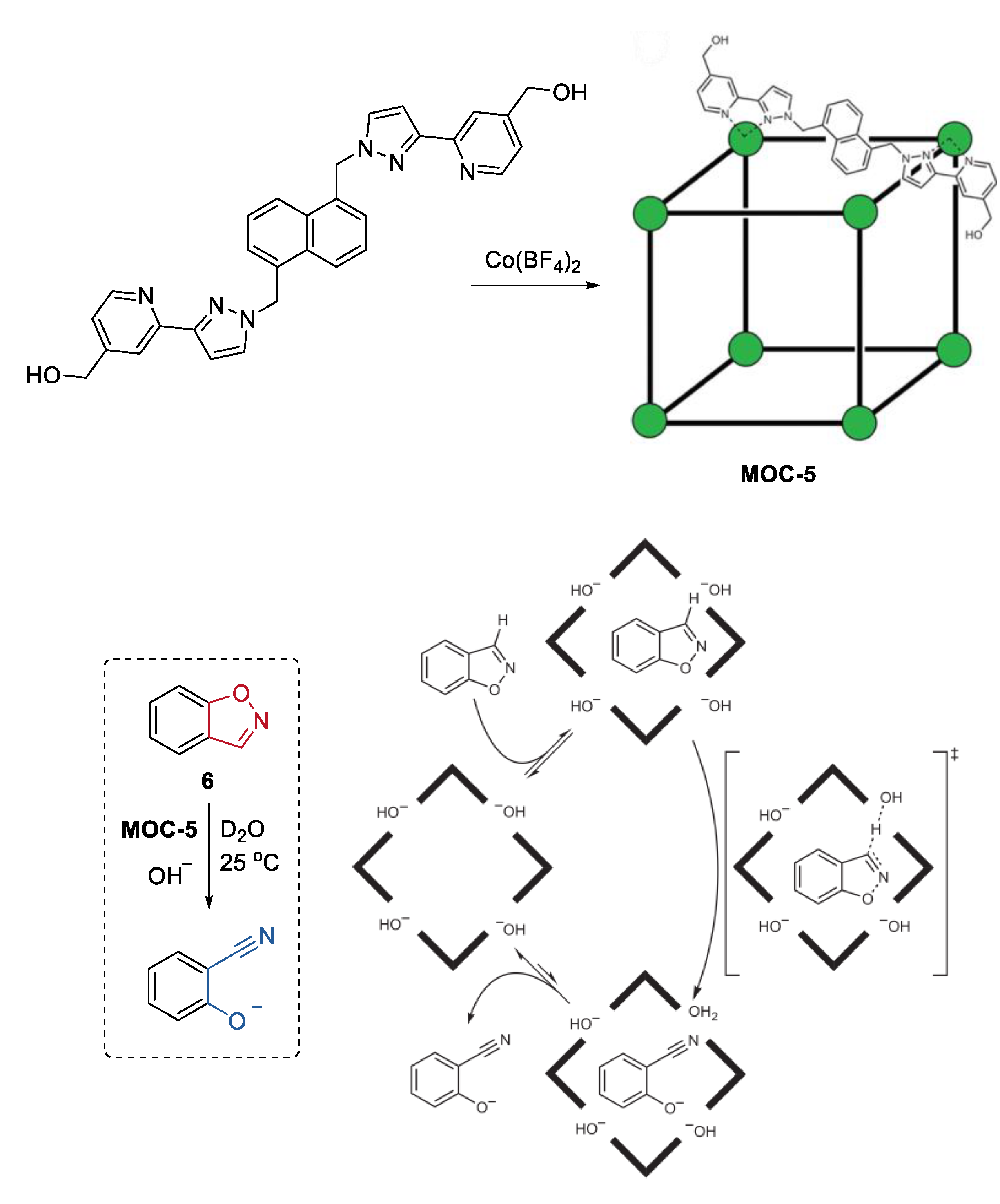
2. Acceleration of Catalytic Activity by Metal–Organic Cages
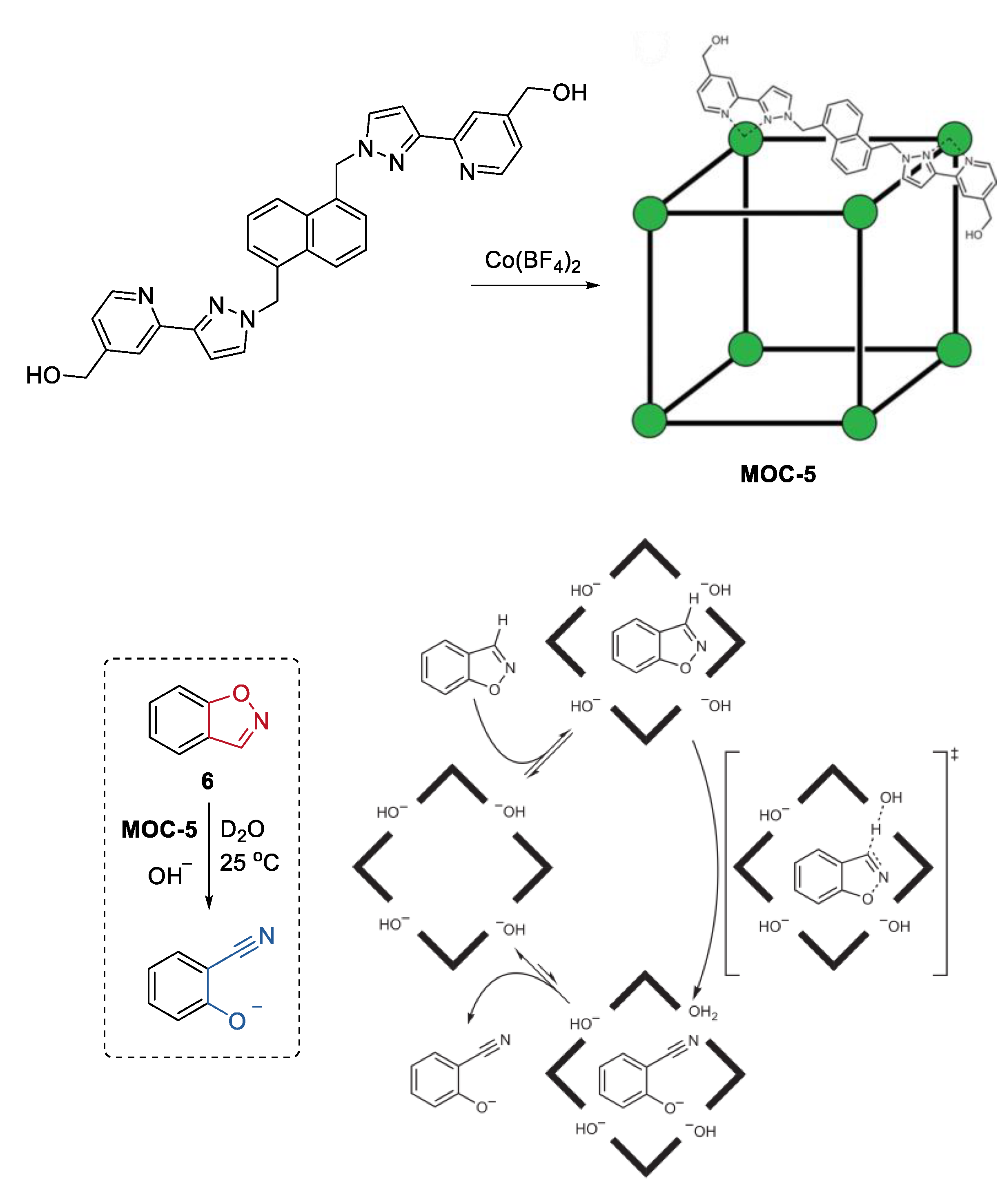
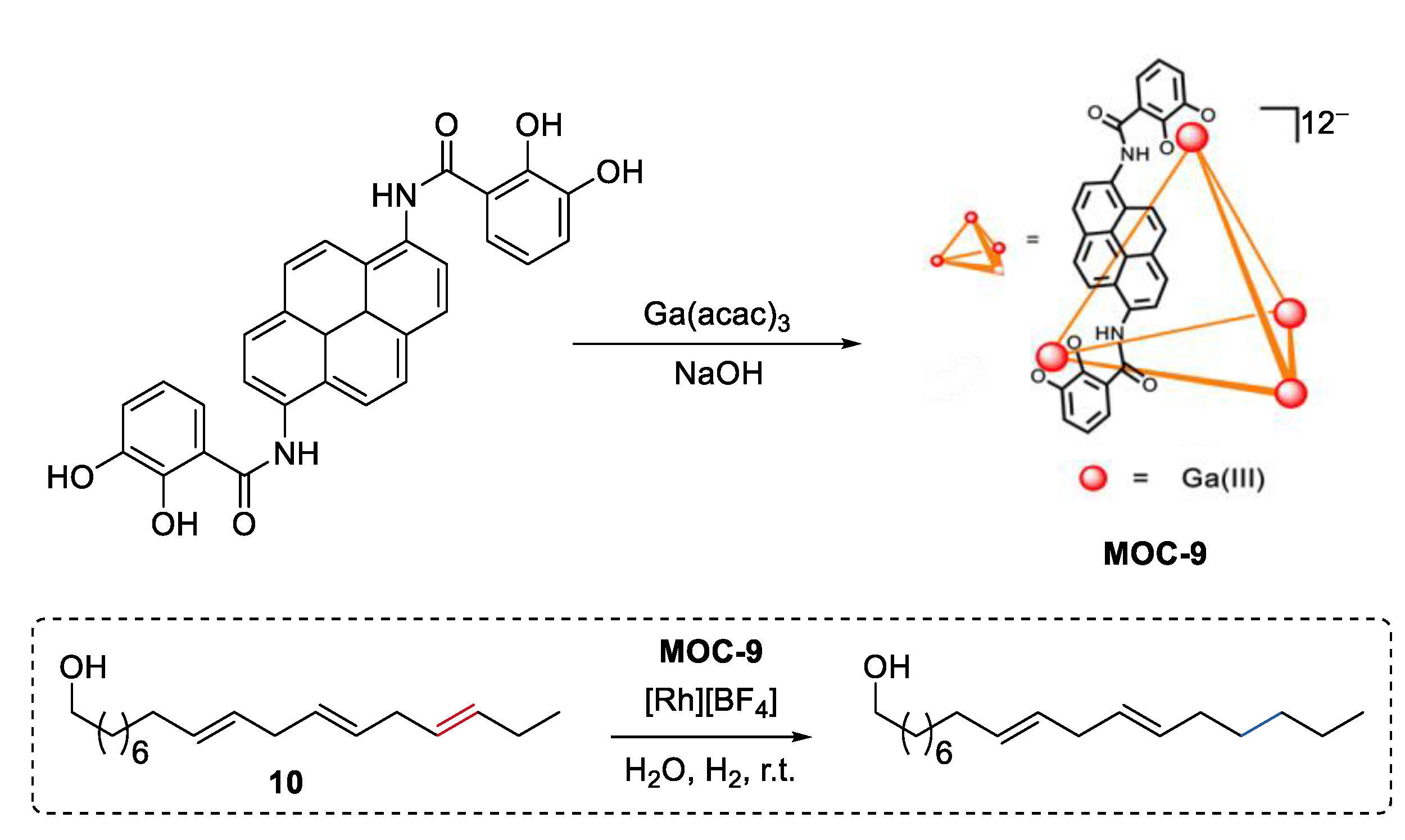
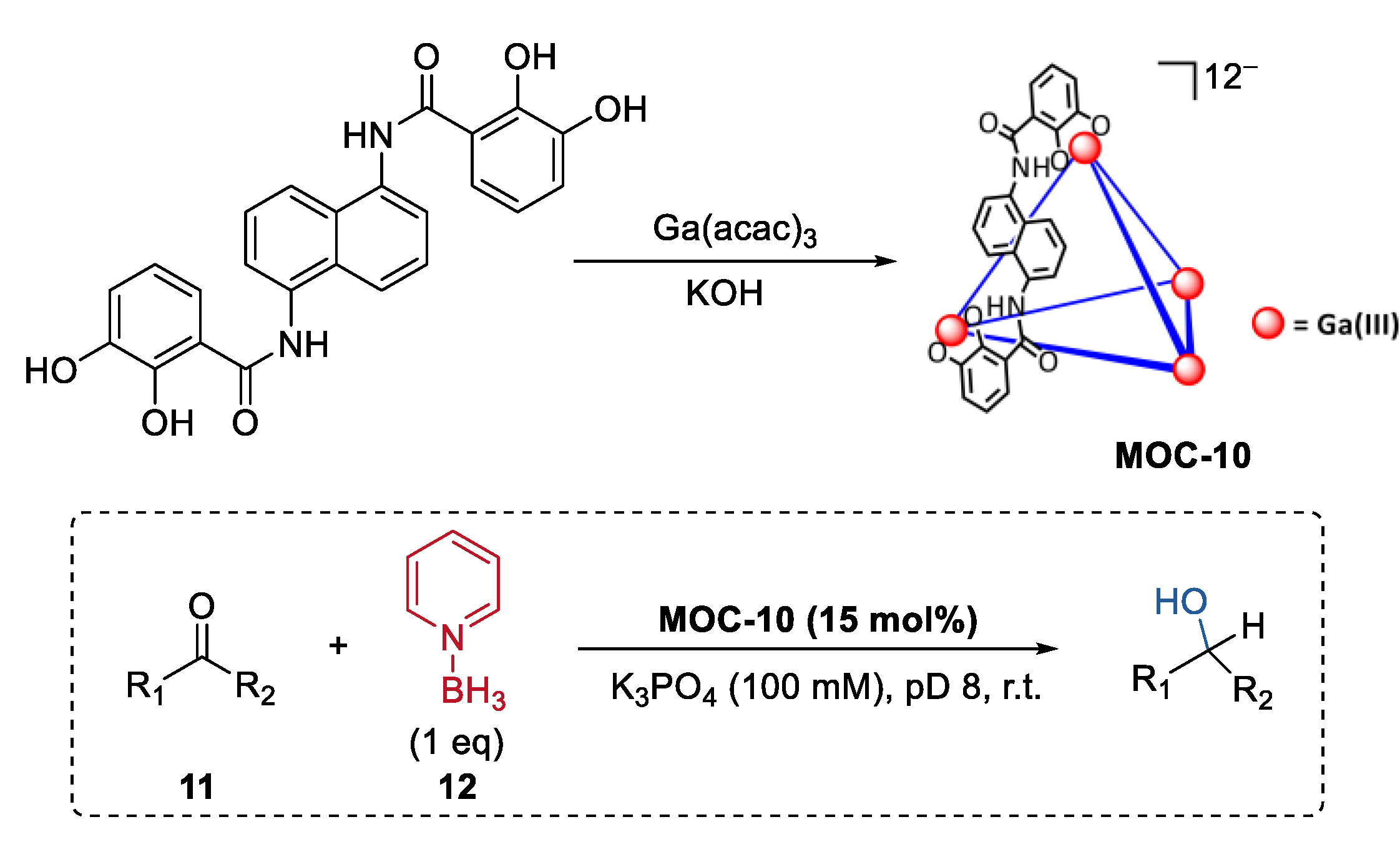
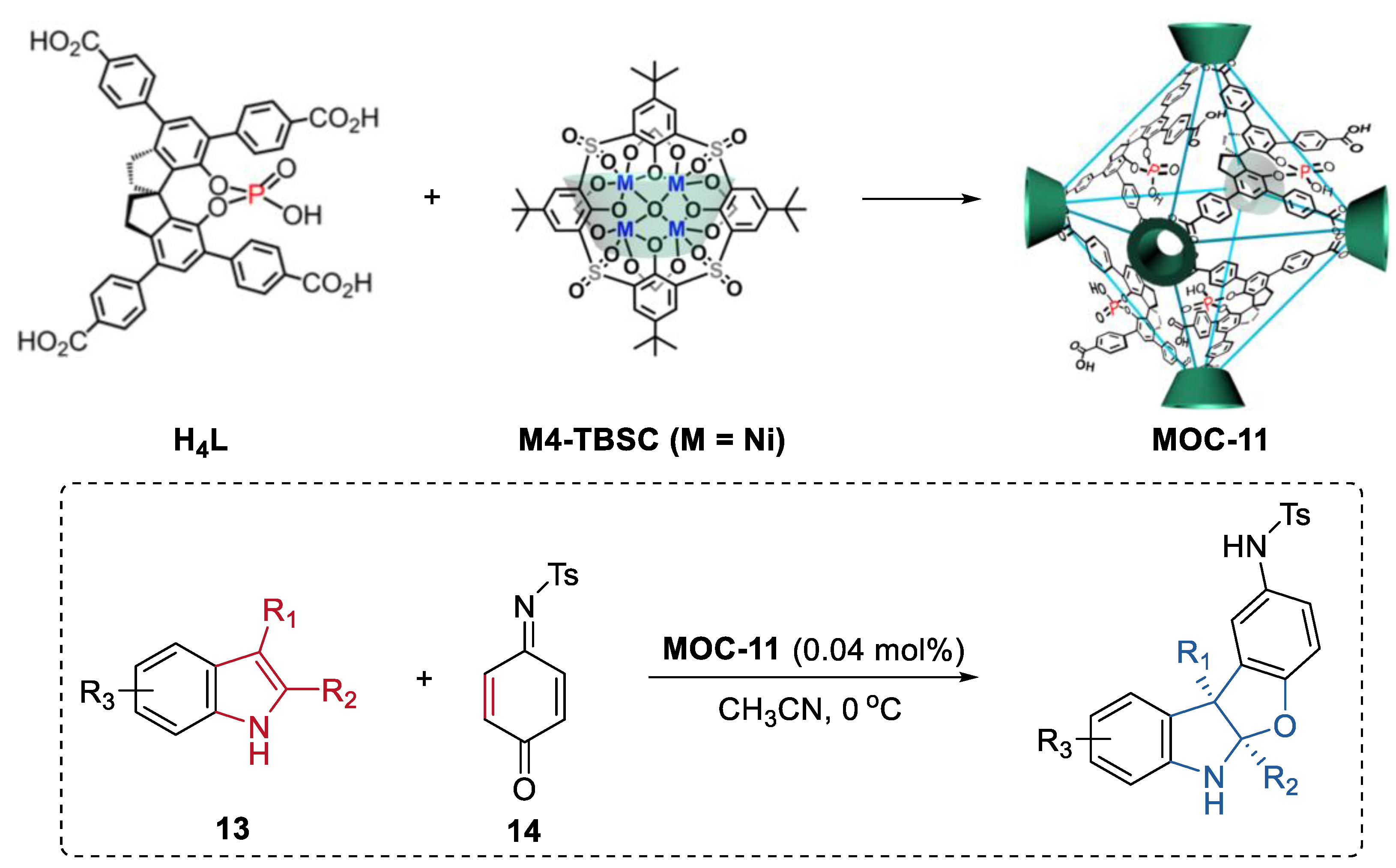
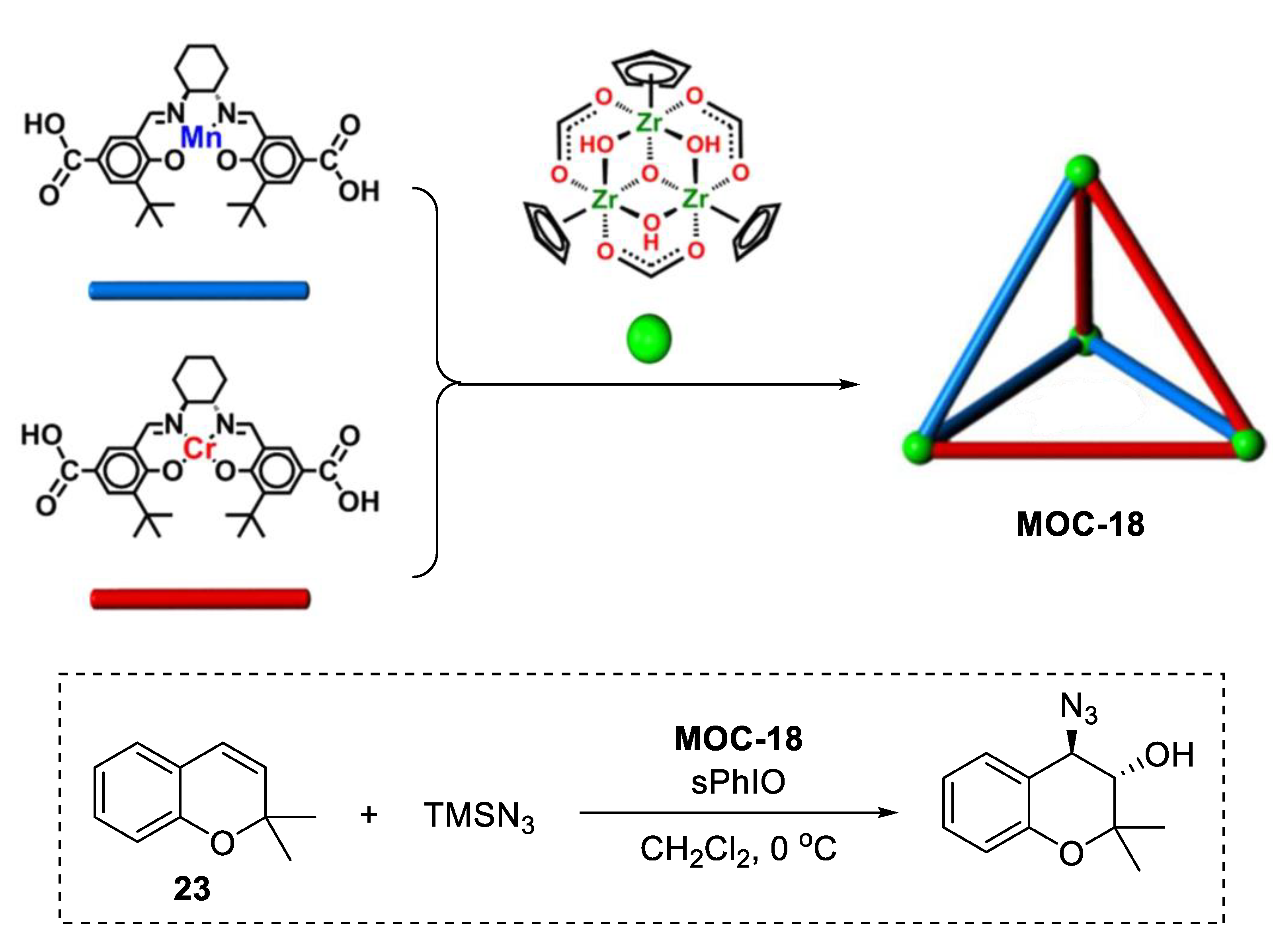
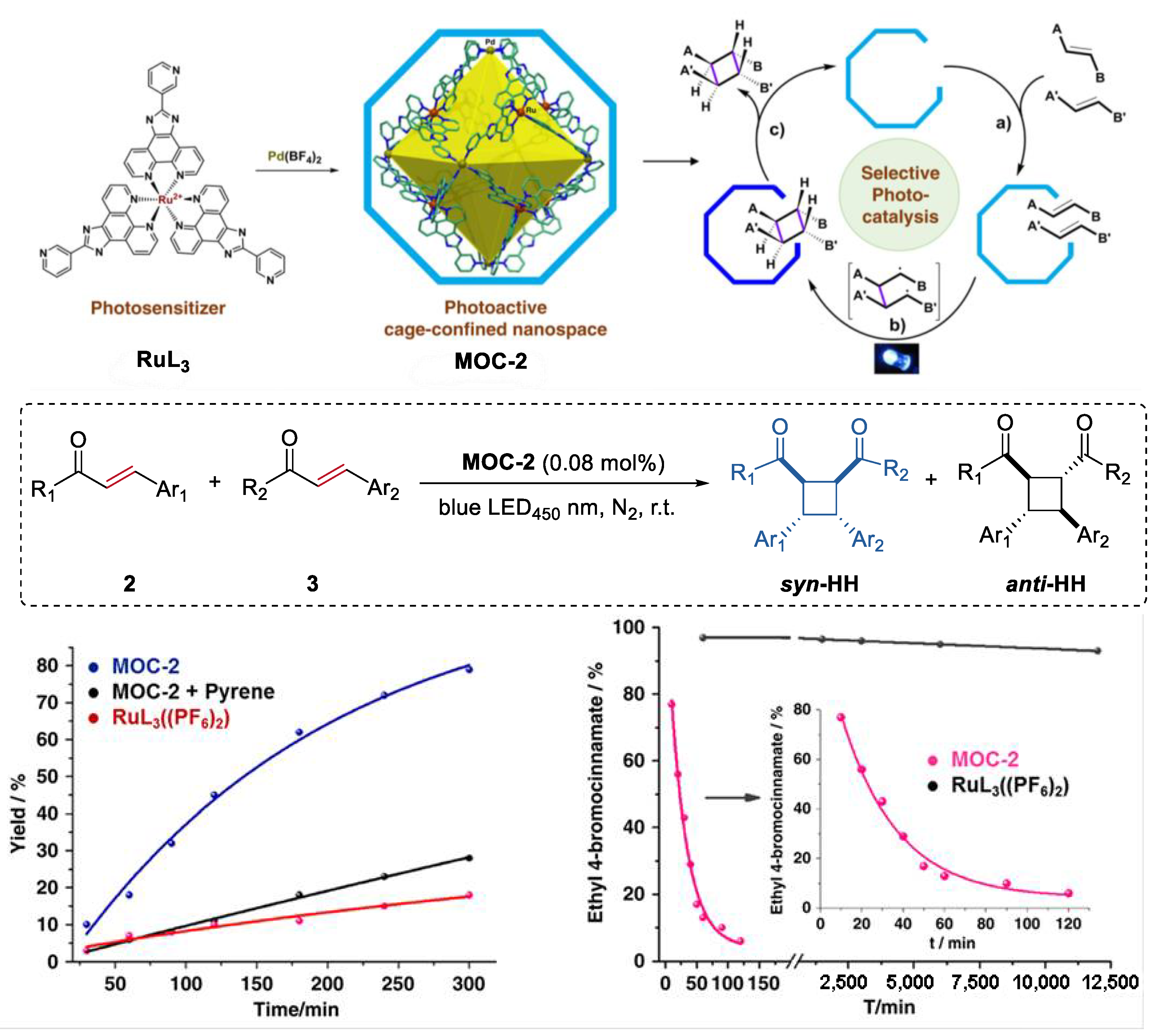
3. Improvement of Selectivity by Metal–Organic Cages
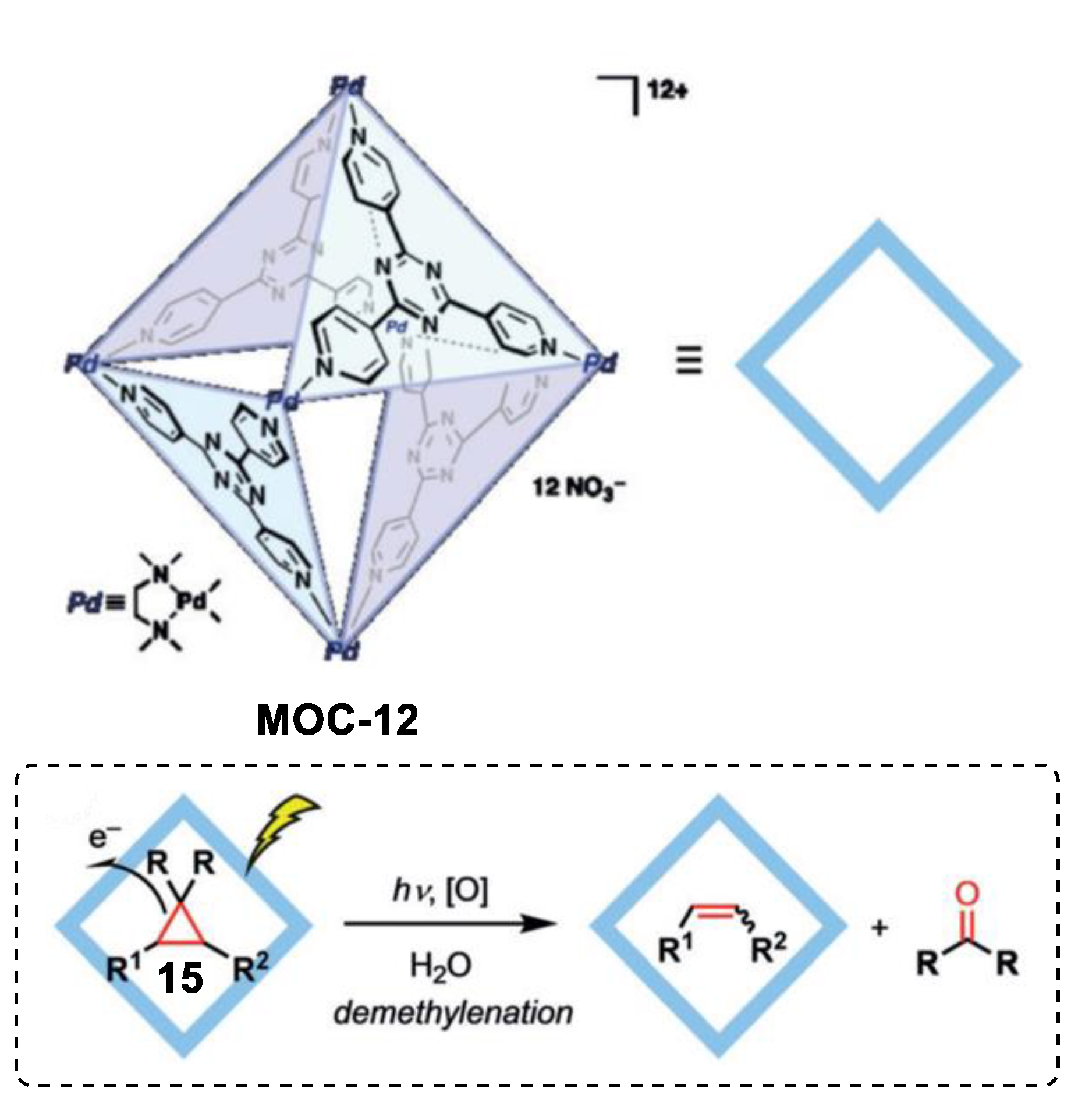
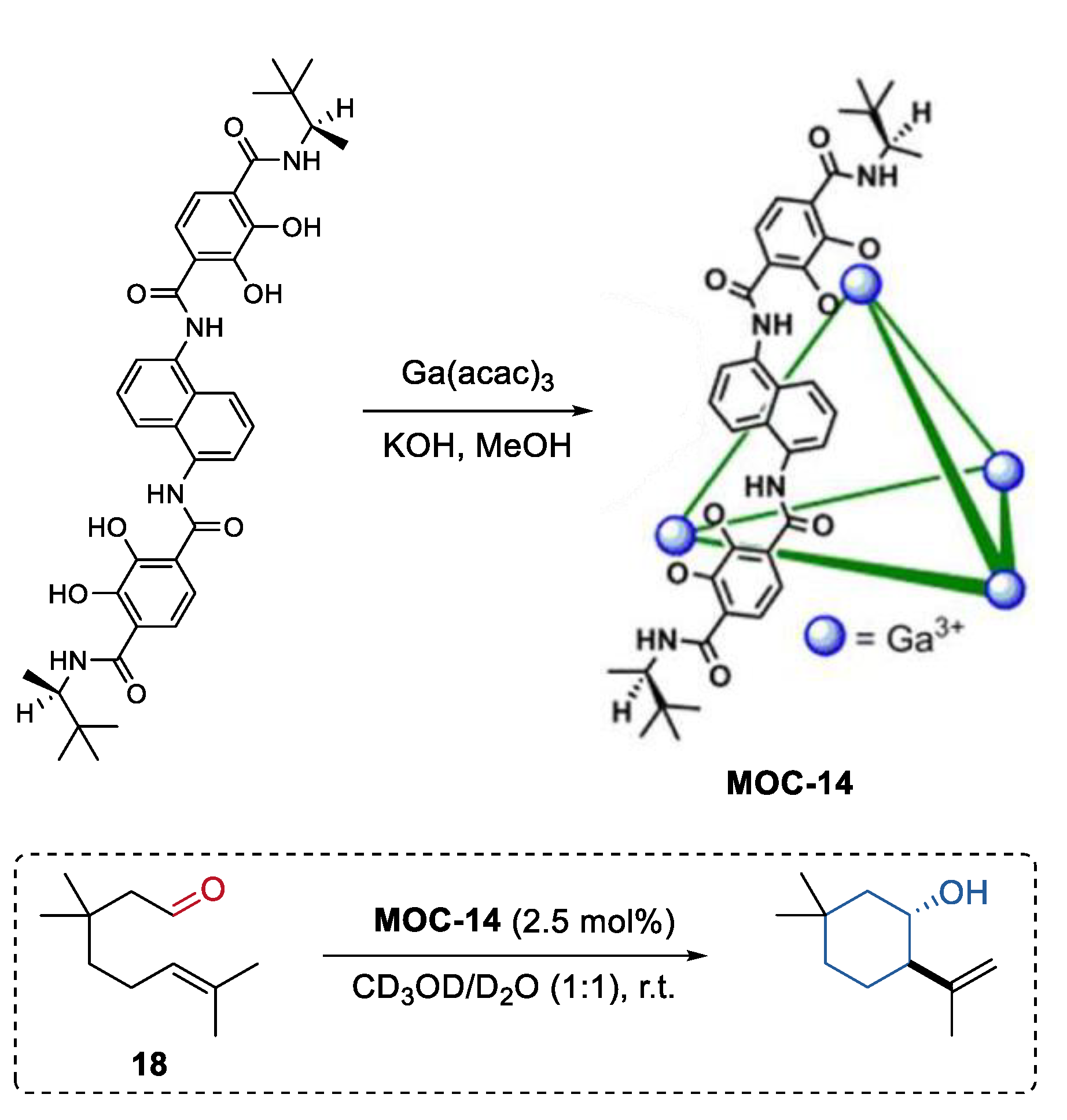
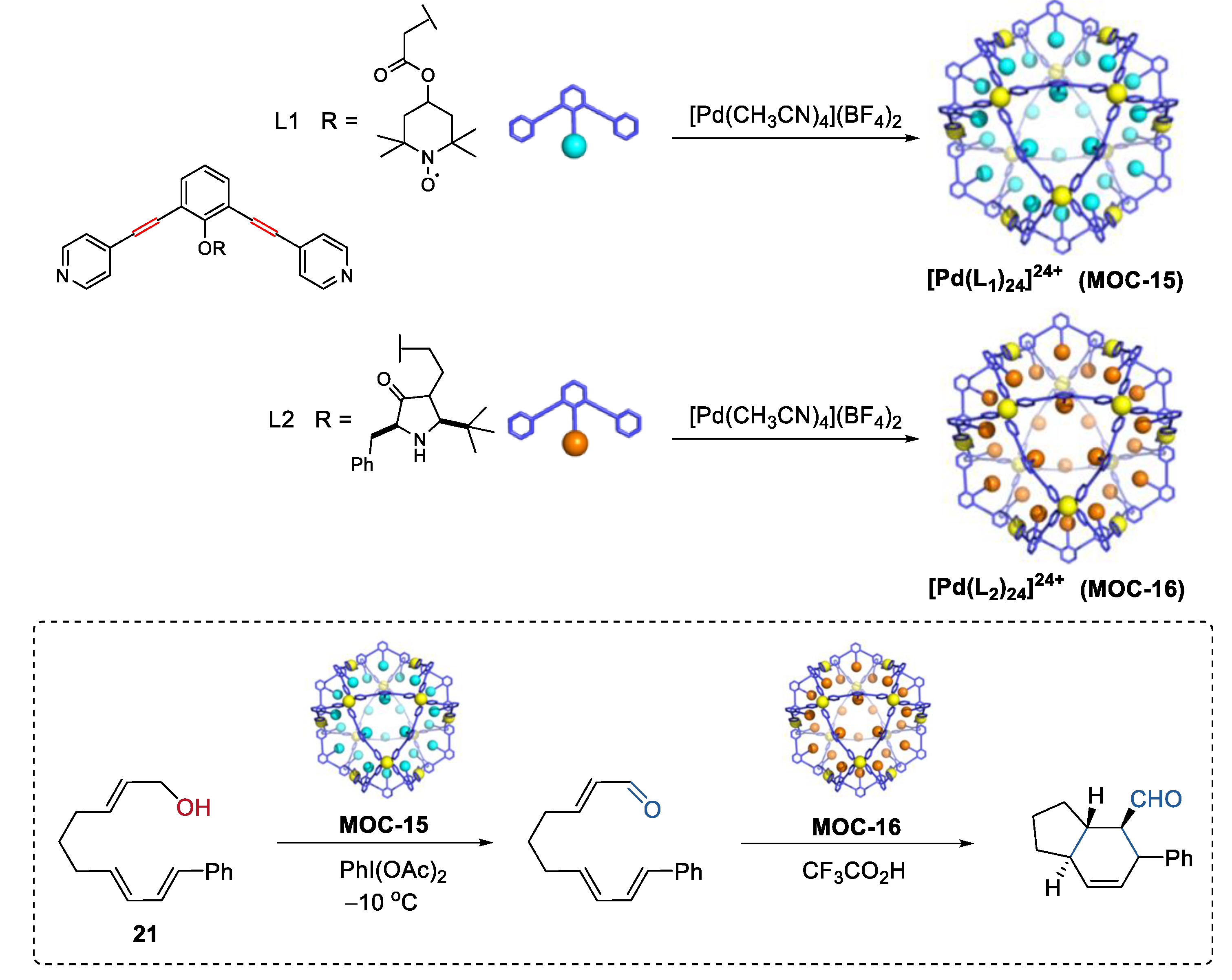
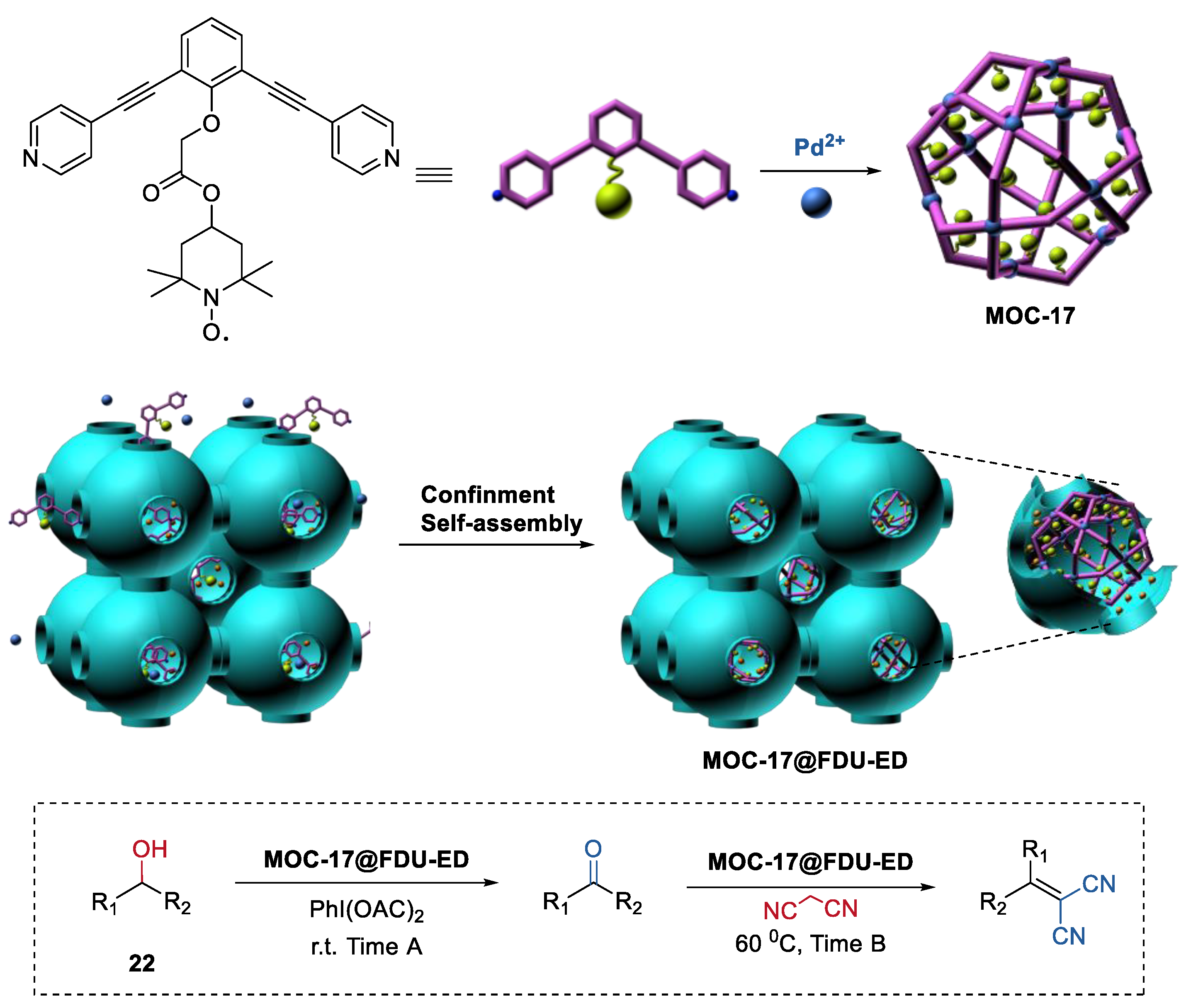
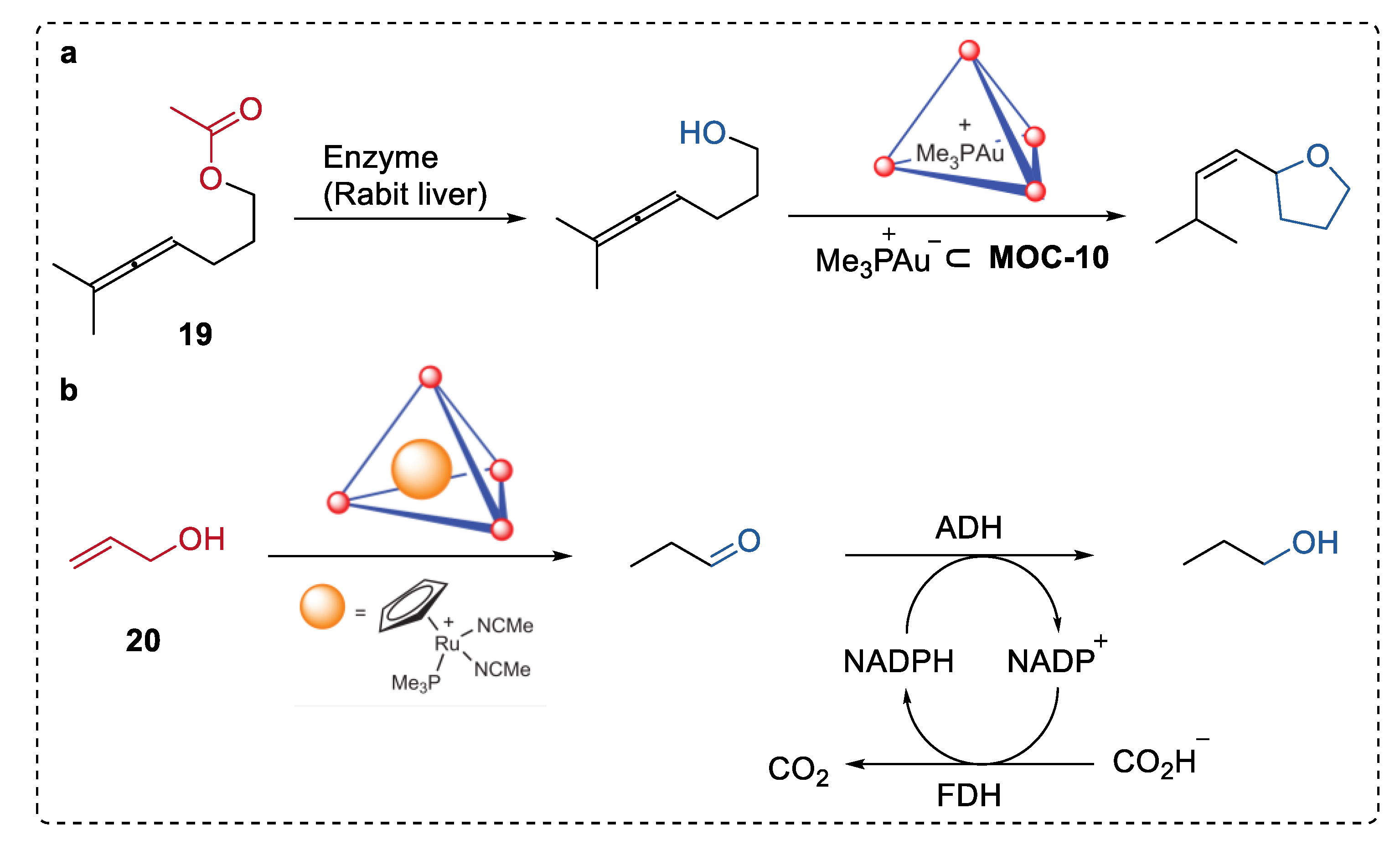
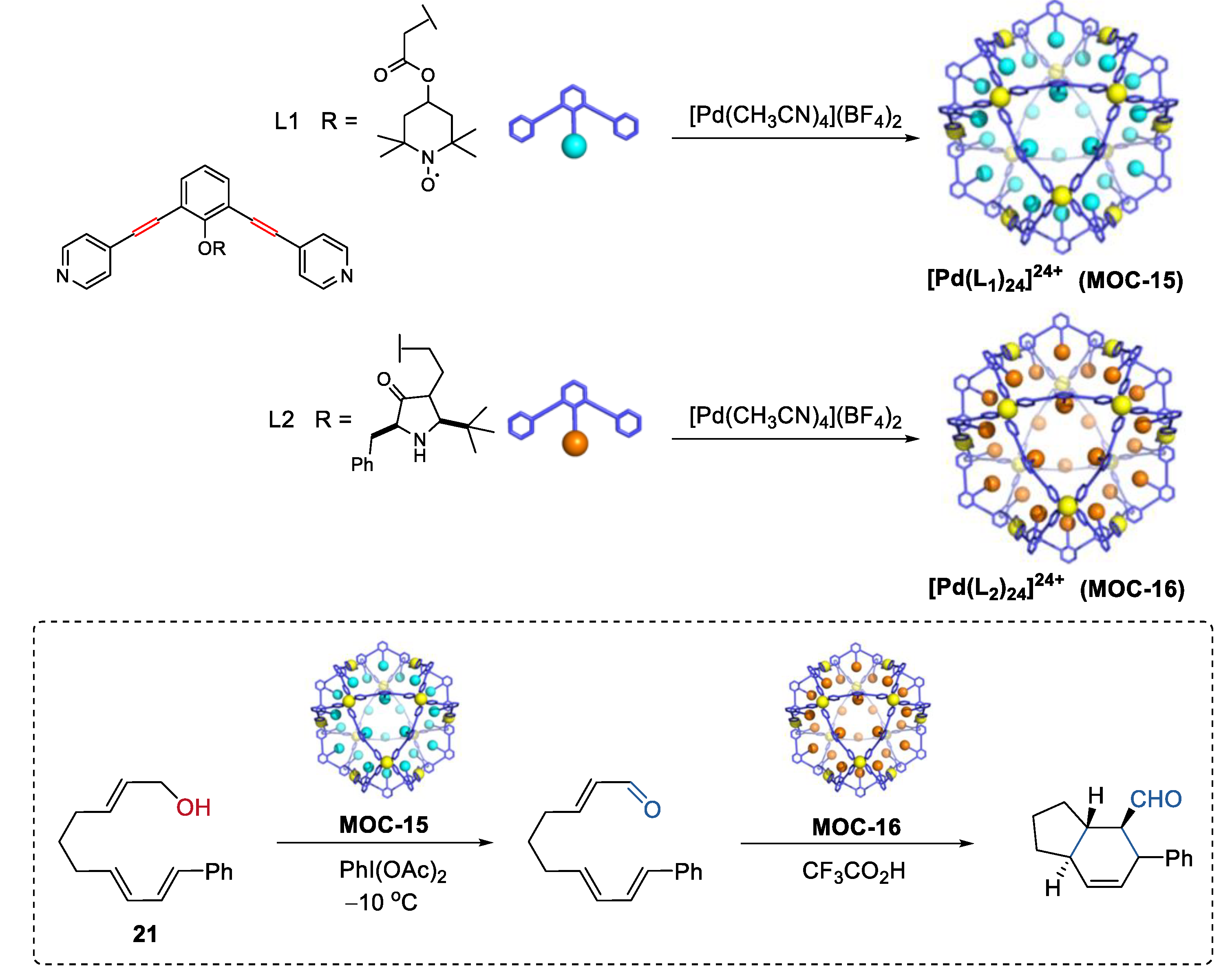
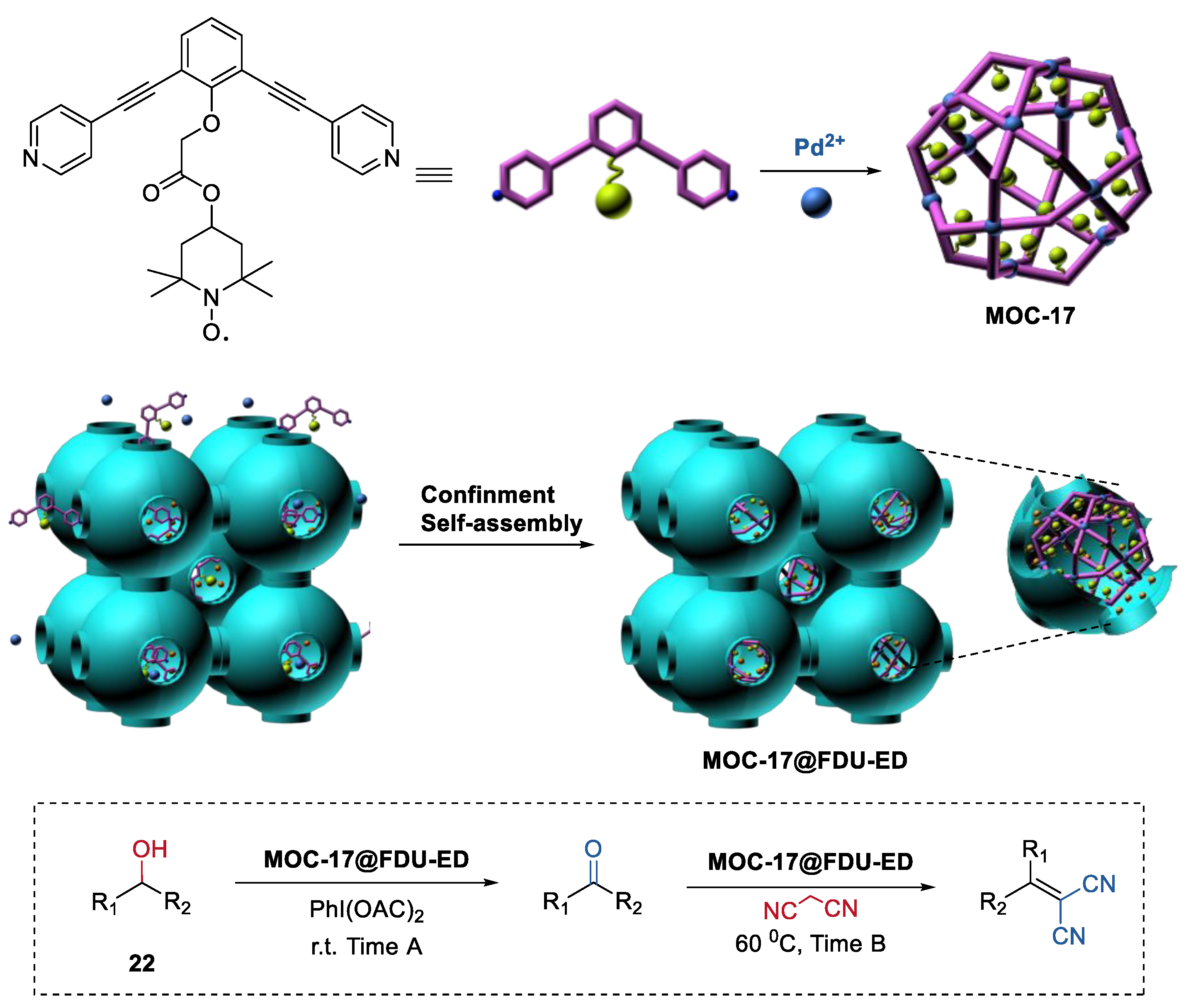
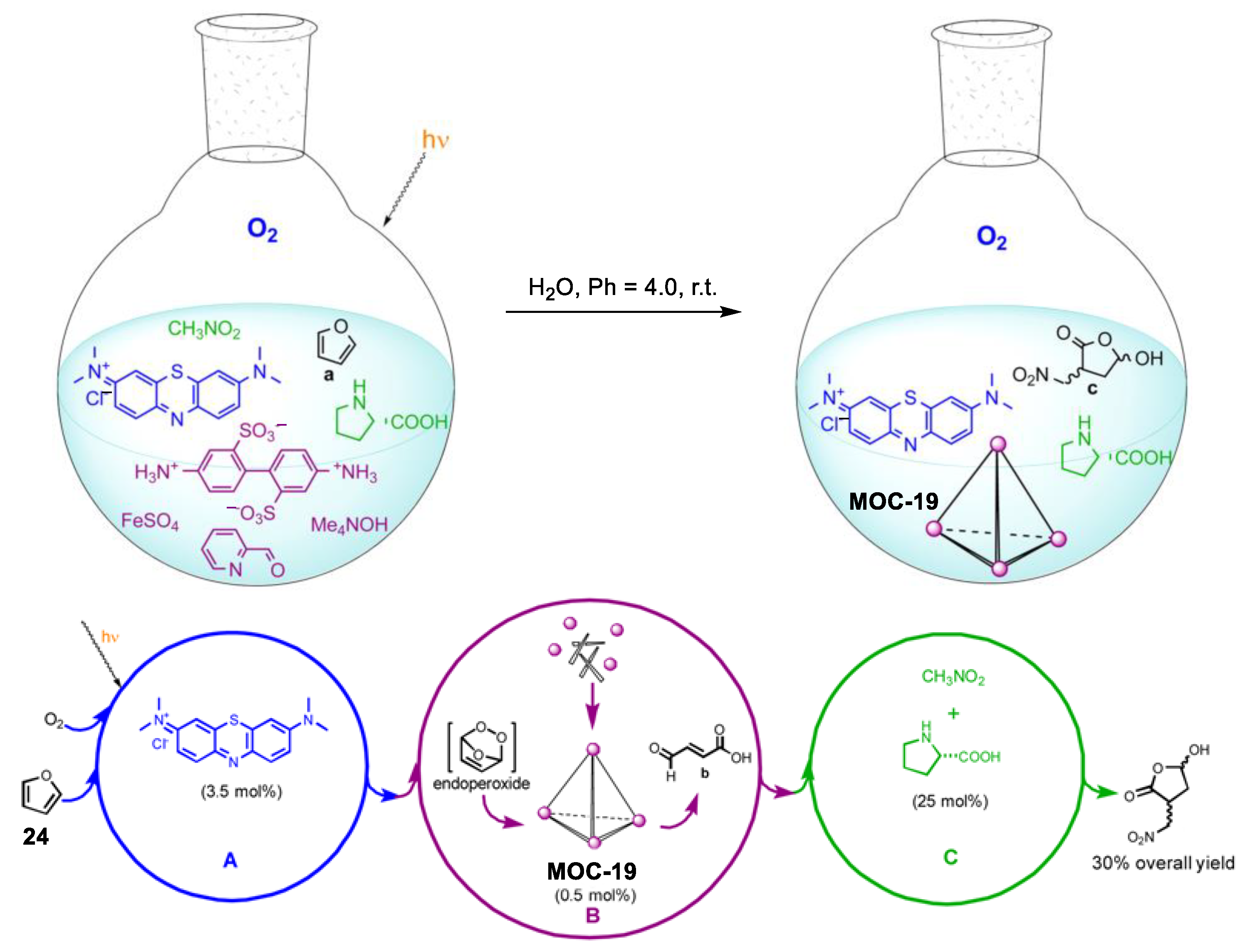
4. Tandem or Cascade Reactions Catalyzed by Metal–Organic Cages
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Anastas, P.; Eghbali, N. Green chemistry: Principles and practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A. The e factor 25 years on: The rise of green chemistry and sustainability. Green Chem. 2017, 19, 18–43. [Google Scholar] [CrossRef]
- Song, J.; Han, B. Green chemistry: A tool for the sustainable development of the chemical industry. Natl. Sci. Rev. 2014, 2, 255–256. [Google Scholar] [CrossRef] [Green Version]
- Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P.W. Supramolecular catalysis. Part 1: Non-covalent interactions as a tool for building and modifying homogeneous catalysts. Chem. Soc. Rev. 2014, 43, 1660–1733. [Google Scholar] [CrossRef] [PubMed]
- Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P.W.N.M. Supramolecular catalysis. Part 2: Artificial enzyme mimics. Chem. Soc. Rev. 2014, 43, 1734–1787. [Google Scholar] [CrossRef]
- Morimoto, M.; Bierschenk, S.M.; Xia, K.T.; Bergman, R.G.; Raymond, K.N.; Toste, F.D. Advances in supramolecular host-mediated reactivity. Nat. Catal. 2020, 3, 969–984. [Google Scholar] [CrossRef]
- Cullen, W.; Misuraca, M.C.; Hunter, C.A.; Williams, N.H.; Ward, M.D. Highly efficient catalysis of the kemp elimination in the cavity of a cubic coordination cage. Nat. Chem. 2016, 8, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Grommet, A.B.; Feller, M.; Klajn, R. Chemical reactivity under nanoconfinement. Nat. Nanotechnol. 2020, 15, 256–271. [Google Scholar] [CrossRef]
- Meeuwissen, J.; Reek, J.N. Supramolecular catalysis beyond enzyme mimics. Nat. Chem. 2010, 2, 615–621. [Google Scholar] [CrossRef]
- Zhang, G.; Mastalerz, M. Organic cage compounds—From shape-persistency to function. Chem. Soc. Rev. 2014, 43, 1934–1947. [Google Scholar] [CrossRef]
- Jordan, J.H.; Gibb, B.C. Molecular containers assembled through the hydrophobic effect. Chem. Soc. Rev. 2015, 44, 547–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajami, D.; Liu, L.; Rebek, J., Jr. Soft templates in encapsulation complexes. Chem. Soc. Rev. 2015, 44, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Fujita, D.; Ueda, Y.; Sato, S.; Mizuno, N.; Kumasaka, T.; Fujita, M. Self-assembly of tetravalent goldberg polyhedra from 144 small components. Nature 2016, 540, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, A.J.; Rowland, C.A.; Bloch, E.D. Permanently microporous metal–Organic polyhedra. Chem. Rev. 2020, 120, 8987–9014. [Google Scholar] [CrossRef]
- Brown, C.J.; Toste, F.D.; Bergman, R.G.; Raymond, K.N. Supramolecular catalysis in metal–Ligand cluster hosts. Chem. Rev. 2015, 115, 3012–3035. [Google Scholar] [CrossRef]
- Lee, S.; Jeong, H.; Nam, D.; Lah, M.S.; Choe, W. The rise of metal–Organic polyhedra. Chem. Soc. Rev. 2021, 50, 528–555. [Google Scholar] [CrossRef]
- Chen, L.-J.; Yang, H.-B. Construction of stimuli-responsive functional materials via hierarchical self-assembly involving coordination interactions. Acc. Chem. Res. 2018, 51, 2699–2710. [Google Scholar] [CrossRef]
- Pullen, S.; Tessarolo, J.; Clever, G.H. Increasing structural and functional complexity in self-assembled coordination cages. Chem. Sci. 2021, 12, 7269–7293. [Google Scholar] [CrossRef]
- Chen, L.; Chen, Q.H.; Wu, M.Y.; Jiang, F.L.; Hong, M.C. Controllable coordination-driven self-assembly: From discrete metallocages to infinite cage-based frameworks. Acc. Chem. Res. 2015, 48, 201–210. [Google Scholar] [CrossRef]
- Xu, L.; Wang, Y.-X.; Chen, L.-J.; Yang, H.-B. Construction of multiferrocenyl metallacycles and metallacages via coordination-driven self-assembly: From structure to functions. Chem. Soc. Rev. 2015, 44, 2148–2167. [Google Scholar] [CrossRef]
- Mouarrawis, V.; Plessius, R.; van der Vlugt, J.I.; Reek, J.N.H. Confinement effects in catalysis using well-defined materials and cages. Front. Chem. 2018, 6, 623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vardhan, H.; Verpoort, F. Metal-organic polyhedra: Catalysis and reactive intermediates. Adv. Synth. Catal. 2015, 357, 1351–1368. [Google Scholar] [CrossRef]
- Xue, Y.; Hang, X.; Ding, J.; Li, B.; Zhu, R.; Pang, H.; Xu, Q. Catalysis within coordination cages. Coord. Chem. Rev. 2021, 430, 213656. [Google Scholar] [CrossRef]
- Jin, Y.; Zhang, Q.; Zhang, Y.; Duan, C. Electron transfer in the confined environments of metal-organic coordination supramolecular systems. Chem. Soc. Rev. 2020, 49, 5561–5600. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.M.; Bergman, R.G.; Raymond, K.N.; Toste, F.D. Self-assembled tetrahedral hosts as supramolecular catalysts. Acc. Chem. Res. 2018, 51, 2447–2455. [Google Scholar] [CrossRef]
- Zhao, L.; Jing, X.; Li, X.; Guo, X.; Zeng, L.; He, C.; Duan, C. Catalytic properties of chemical transformation within the confined pockets of werner-type capsules. Coord. Chem. Rev. 2019, 378, 151–187. [Google Scholar] [CrossRef]
- Vazquez-Gonzalez, M.; Wang, C.; Willner, I. Biocatalytic cascades operating on macromolecular scaffolds and in confined environments. Nat. Catal. 2020, 3, 256–273. [Google Scholar] [CrossRef]
- Lohr, T.L.; Marks, T.J. Orthogonal tandem catalysis. Nat. Chem. 2015, 7, 477–482. [Google Scholar] [CrossRef]
- Catti, L.; Zhang, Q.; Tiefenbacher, K. Advantages of catalysis in self-assembled molecular capsules. Chem. Eur. J. 2016, 22, 9060–9066. [Google Scholar] [CrossRef]
- Gaeta, C.; La Manna, P.; De Rosa, M.; Soriente, A.; Talotta, C.; Neri, P. Supramolecular catalysis with self-assembled capsules and cages: What happens in confined spaces. ChemCatChem 2021, 13, 1638–1658. [Google Scholar] [CrossRef]
- Fang, Y.; Powell, J.A.; Li, E.; Wang, Q.; Perry, Z.; Kirchon, A.; Yang, X.; Xiao, Z.; Zhu, C.; Zhang, L.; et al. Catalytic reactions within the cavity of coordination cages. Chem. Soc. Rev. 2019, 48, 4707–4730. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Jordan, J.H.; Hu, X.Y.; Wang, L. Supramolecular strategies for controlling reactivity within confined nanospaces. Angew. Chem. Int. Ed. 2020, 59, 13712–13721. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.Q.; Liu, Y.; Cui, Y. Supramolecular chirality in metal-organic complexes. Acc. Chem. Res. 2021, 54, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Koblenz, T.S.; Wassenaar, J.; Reek, J.N.H. Reactivity within a confined self-assembled nanospace. Chem. Soc. Rev. 2008, 37, 247–262. [Google Scholar] [CrossRef] [PubMed]
- Cook, T.R.; Stang, P.J. Recent developments in the preparation and chemistry of metallacycles and metallacages via coordination. Chem. Rev. 2015, 115, 7001–7045. [Google Scholar] [CrossRef]
- Percastegui, E.G.; Ronson, T.K.; Nitschke, J.R. Design and applications of water-soluble coordination cages. Chem. Rev. 2020, 120, 13480–13544. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Gao, W.X.; Lin, L.; Jin, G.X. Recent advances in the construction and applications of heterometallic macrocycles and cages. Coord. Chem. Rev. 2017, 344, 323–344. [Google Scholar] [CrossRef]
- McConnell, A.J. Metallosupramolecular cages: From design principles and characterisation techniques to applications. Chem. Soc. Rev. 2022, 51, 2957–2971. [Google Scholar] [CrossRef]
- Takezawa, H.; Shitozawa, K.; Fujita, M. Enhanced reactivity of twisted amides inside a molecular cage. Nat. Chem. 2020, 12, 574–578. [Google Scholar] [CrossRef]
- Ibukuro, F.; Kusukawa, T.; Fujita, M. A thermally switchable molecular lock. Guest-templated synthesis of a kinetically stable nanosized cage. J. Am. Chem. Soc. 1998, 120, 8561–8562. [Google Scholar] [CrossRef]
- Takezawa, H.; Akiba, S.; Murase, T.; Fujita, M. Cavity-directed chromism of phthalein dyes. J. Am. Chem. Soc. 2015, 137, 7043–7046. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Oguro, D.; Miyazawa, M.; Oka, H.; Yamaguchi, K.; Ogura, K. Self-assembly of ten molecules into nanometre-sized organic host frameworks. Nature 1995, 378, 469–471. [Google Scholar] [CrossRef]
- Takezawa, H.; Murase, T.; Fujita, M. Temporary and permanent trapping of the metastable twisted conformer of an overcrowded chromic alkene via encapsulation. J. Am. Chem. Soc. 2012, 134, 17420–17423. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.S.; Wu, K.; Yin, C.; Li, K.; Huang, Y.; Ruan, J.; Feng, X.; Hu, P.; Su, C.Y. Cage-confined photocatalysis for wide-scope unusually selective [2 + 2] cycloaddition through visible-light triplet sensitization. Nat. Commun. 2020, 11, 4675. [Google Scholar] [CrossRef]
- Li, K.; Zhang, L.-Y.; Yan, C.; Wei, S.-C.; Pan, M.; Zhang, L.; Su, C.-Y. Stepwise assembly of pd6(rul3)8 nanoscale rhombododecahedral metal–organic cages via metalloligand strategy for guest trapping and protection. J. Am. Chem. Soc. 2014, 136, 4456–4459. [Google Scholar] [CrossRef]
- Chen, S.; Li, K.; Zhao, F.; Zhang, L.; Pan, M.; Fan, Y.-Z.; Guo, J.; Shi, J.; Su, C.-Y. A metal-organic cage incorporating multiple light harvesting and catalytic centres for photochemical hydrogen production. Nat. Commun. 2016, 7, 13169. [Google Scholar] [CrossRef] [Green Version]
- Yoshizawa, M.; Tamura, M.; Fujita, M. Diels-alder in aqueous molecular hosts: Unusual regioselectivity and efficient catalysis. Science 2006, 312, 251–254. [Google Scholar] [CrossRef]
- Gonell, S.; Reek, J.N.H. Gold-catalyzed cycloisomerization reactions within guanidinium M12L24 nanospheres: The effect of local concentrations. ChemCatChem 2019, 11, 1458–1464. [Google Scholar] [CrossRef] [Green Version]
- Gramage-Doria, R.; Hessels, J.; Leenders, S.H.; Troppner, O.; Durr, M.; Ivanovic-Burmazovic, I.; Reek, J.N. Gold(I) catalysis at extreme concentrations inside self-assembled nanospheres. Angew. Chem. Int. Ed. 2014, 53, 13380–13384. [Google Scholar] [CrossRef]
- Cauteruccio, S.; Loos, A.; Bossi, A.; Blanco Jaimes, M.C.; Dova, D.; Rominger, F.; Prager, S.; Dreuw, A.; Licandro, E.; Hashmi, A.S.K. Gold(I) complexes of tetrathiaheterohelicene phosphanes. Inorg. Chem. 2013, 52, 7995–8004. [Google Scholar] [CrossRef]
- Wang, Q.Q.; Gonell, S.; Leenders, S.H.; Durr, M.; Ivanovic-Burmazovic, I.; Reek, J.N. Self-assembled nanospheres with multiple endohedral binding sites pre-organize catalysts and substrates for highly efficient reactions. Nat. Chem. 2016, 8, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Schug, K.A.; Lindner, W. Noncovalent binding between guanidinium and anionic groups: Focus on biological- and synthetic-based arginine/guanidinium interactions with phosph[on]ate and sulf[on]ate residues. Chem. Rev. 2005, 105, 67–114. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Poole, D., 3rd; Mathew, S.; Yan, N.; Hessels, J.; Orth, N.; Ivanovic-Burmazovic, I.; Reek, J.N.H. Control over electrochemical water oxidation catalysis by preorganization of molecular ruthenium catalysts in self-assembled nanospheres. Angew. Chem. Int. Ed. 2018, 57, 11247–11251. [Google Scholar] [CrossRef] [PubMed]
- Daubignard, J.; Lutz, M.; Detz, R.J.; de Bruin, B.; Reek, J.N.H. Origin of the selectivity and activity in the Rhodium-catalyzed asymmetric hydrogenation using supramolecular ligands. ACS Catal. 2019, 9, 7535–7547. [Google Scholar] [CrossRef] [Green Version]
- Whitehead, M.; Turega, S.; Stephenson, A.; Hunter, C.A.; Ward, M.D. Quantification of solvent effects on molecular recognition in polyhedral coordination cage hosts. Chem. Sci. 2013, 4, 2744–2751. [Google Scholar] [CrossRef] [Green Version]
- Turega, S.; Cullen, W.; Whitehead, M.; Hunter, C.A.; Ward, M.D. Mapping the internal recognition surface of an octanuclear coordination cage using guest libraries. J. Am. Chem. Soc. 2014, 136, 8475–8483. [Google Scholar] [CrossRef]
- Cullen, W.; Metherell, A.J.; Wragg, A.B.; Taylor, C.G.P.; Williams, N.H.; Ward, M.D. Catalysis in a cationic coordination cage using a cavity-bound guest and surface-bound anions: Inhibition, activation, and autocatalysis. J. Am. Chem. Soc. 2018, 140, 2821–2828. [Google Scholar] [CrossRef] [Green Version]
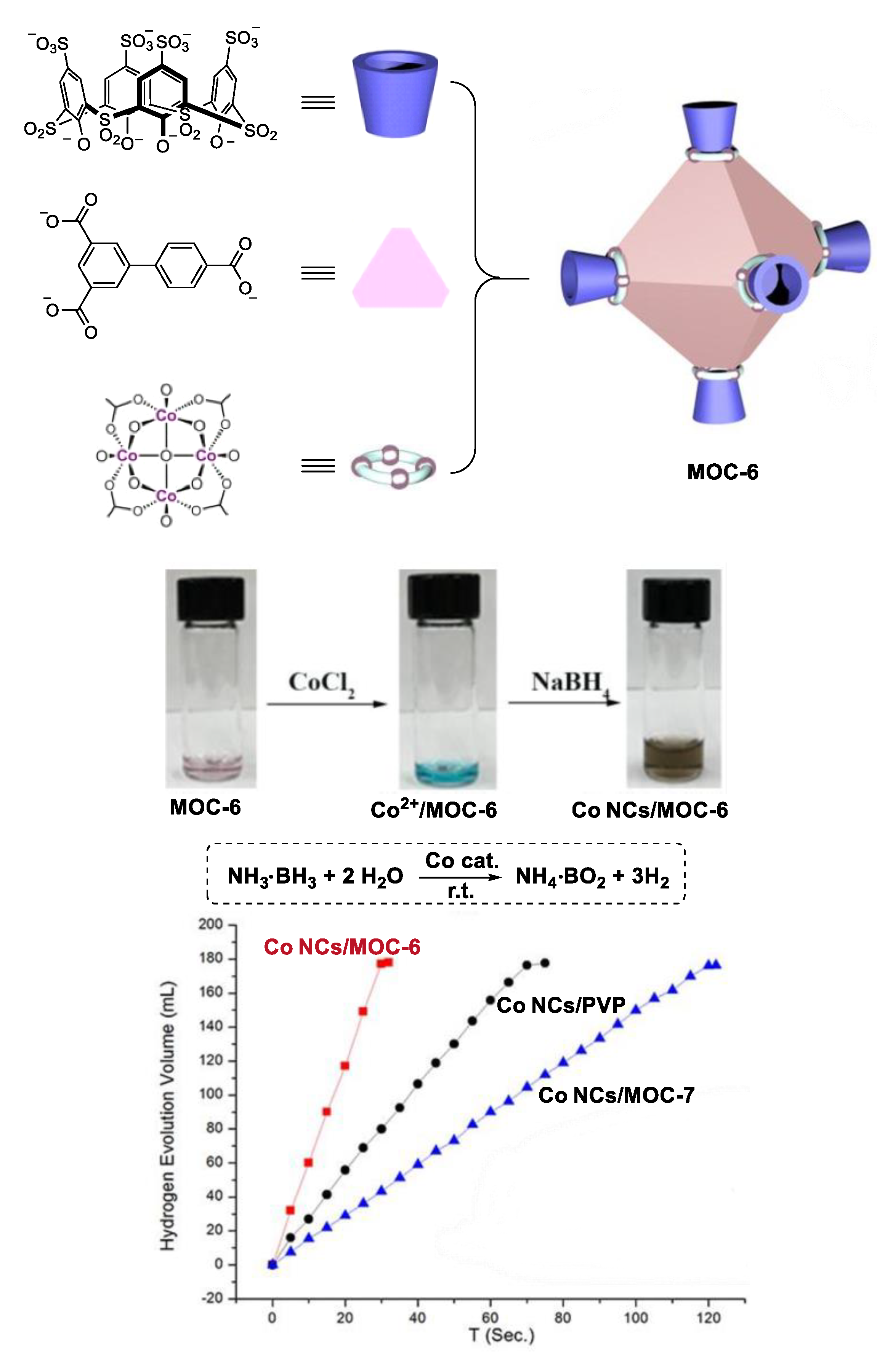
- Fang, Y.; Xiao, Z.; Li, J.; Lollar, C.; Liu, L.; Lian, X.; Yuan, S.; Banerjee, S.; Zhang, P.; Zhou, H.C. Formation of a highly reactive cobalt nanocluster crystal within a highly negatively charged porous coordination cage. Angew. Chem. Int. Ed. 2018, 57, 5283–5287. [Google Scholar] [CrossRef]
- Fang, Y.; Li, J.; Togo, T.; Jin, F.; Xiao, Z.; Liu, L.; Drake, H.; Lian, X.; Zhou, H.-C. Ultra-small face-centered-cubic ru nanoparticles confined within a porous coordination cage for dehydrogenation. Chem 2018, 4, 555–563. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Lavendomme, R.; Burghaus, O.; Nitschke, J.R. A Zn4L6 capsule with enhanced catalytic C-C bond formation activity upon c60 binding. Angew. Chem. Int. Ed. 2019, 58, 9073–9077. [Google Scholar] [CrossRef]
- Lu, Z.; Ronson, T.K.; Nitschke, J.R. Reversible reduction drives anion ejection and C60 binding within an FeII4L6 cage. Chem. Sci. 2020, 11, 1097–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartwig, J.F. Catalyst-controlled site-selective bond activation. Acc. Chem. Res. 2017, 50, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Bender, T.A.; Bergman, R.G.; Raymond, K.N.; Toste, F.D. A supramolecular strategy for selective catalytic hydrogenation independent of remote chain length. J. Am. Chem. Soc. 2019, 141, 11806–11810. [Google Scholar] [CrossRef] [PubMed]
- Hart-Cooper, W.M.; Zhao, C.; Triano, R.M.; Yaghoubi, P.; Ozores, H.L.; Burford, K.N.; Toste, F.D.; Bergman, R.G.; Raymond, K.N. The effect of host structure on the selectivity and mechanism of supramolecular catalysis of prins cyclizations. Chem. Sci. 2015, 6, 1383–1393. [Google Scholar] [CrossRef] [Green Version]
- Hughes, D.L. Biocatalysis in drug development—Highlights of the recent patent literature. Org. Process Res. Dev. 2018, 22, 1063–1080. [Google Scholar] [CrossRef]
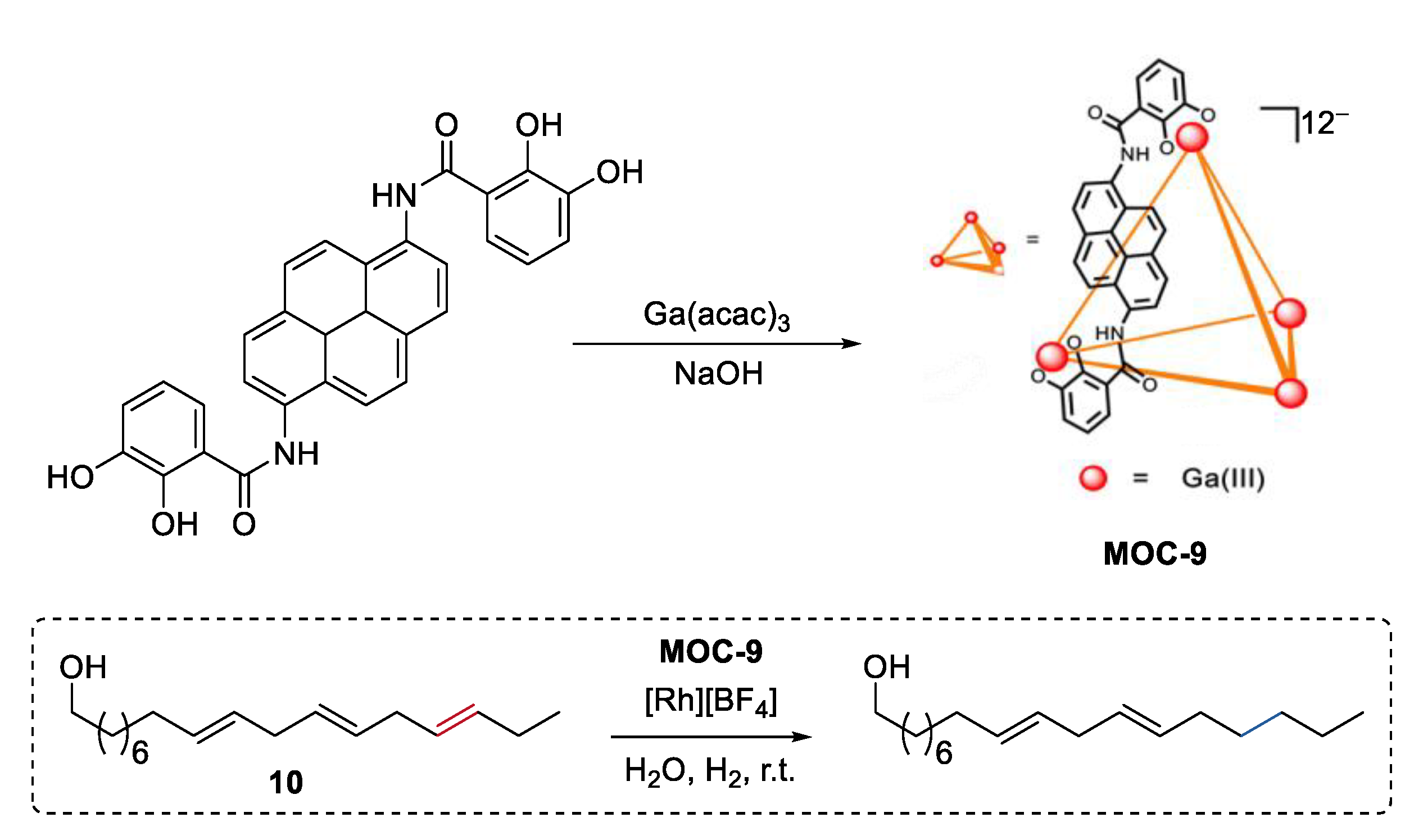
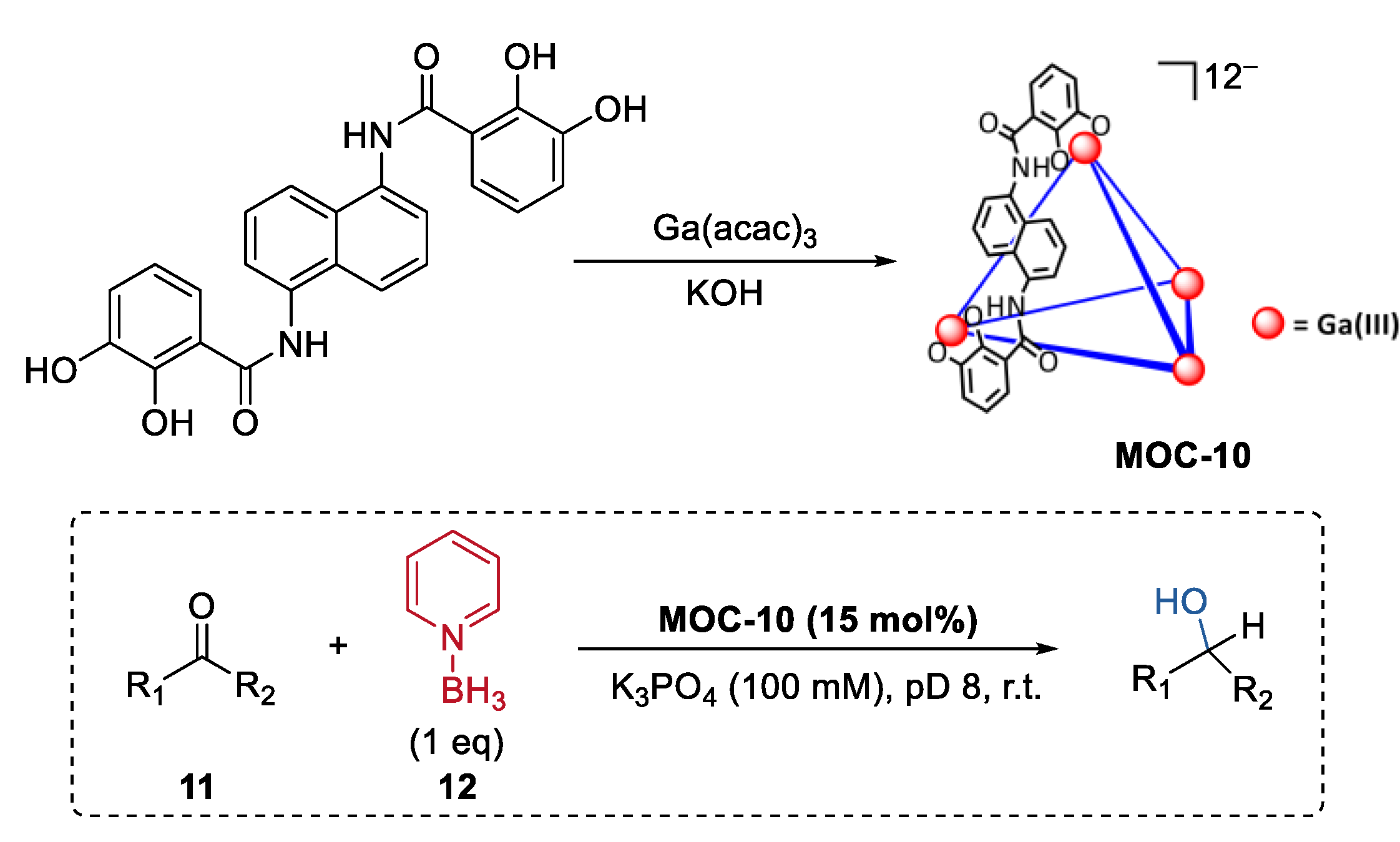
- Morimoto, M.; Cao, W.; Bergman, R.G.; Raymond, K.N.; Toste, F.D. Chemoselective and site-selective reductions catalyzed by a supramolecular host and a pyridine-borane cofactor. J. Am. Chem. Soc. 2021, 143, 2108–2114. [Google Scholar] [CrossRef]
- Bierschenk, S.M.; Bergman, R.G.; Raymond, K.N.; Toste, F.D. A nanovessel-catalyzed three-component aza-darzens reaction. J. Am. Chem. Soc. 2020, 142, 733–737. [Google Scholar] [CrossRef]
- Xu, G.; Leloux, S.; Zhang, P.; Meijide Suárez, J.; Zhang, Y.; Derat, E.; Ménand, M.; Bistri-Aslanoff, O.; Roland, S.; Leyssens, T.; et al. Capturing the monomeric (L)CuH in nhc-capped cyclodextrin: Cavity-controlled chemoselective hydrosilylation of α,β-unsaturated ketones. Angew. Chem. Int. Ed. 2020, 59, 7591–7597. [Google Scholar] [CrossRef] [PubMed]
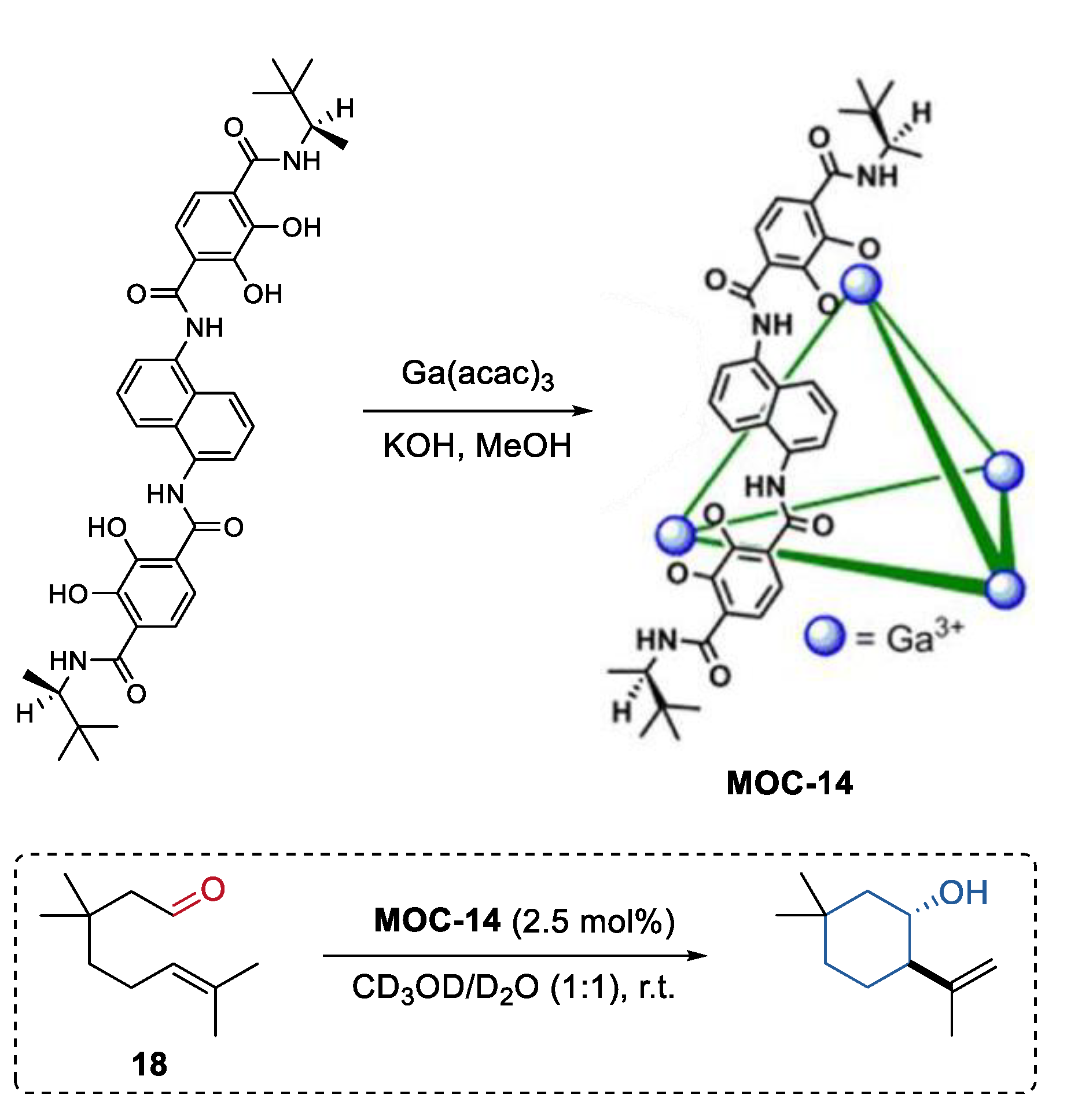
- Chu, D.; Gong, W.; Jiang, H.; Tang, X.; Cui, Y.; Liu, Y. Boosting enantioselectivity of chiral molecular catalysts with supramolecular metal–organic cages. CCS Chem. 2021, 4, 1692–1700. [Google Scholar] [CrossRef]
- Gong, W.; Chu, D.; Jiang, H.; Chen, X.; Cui, Y.; Liu, Y. Permanent porous hydrogen-bonded frameworks with two types of bronsted acid sites for heterogeneous asymmetric catalysis. Nat. Commun. 2019, 10, 600. [Google Scholar] [CrossRef]
- Hu, F.; Liu, C.; Wu, M.; Pang, J.; Jiang, F.; Yuan, D.; Hong, M. An ultrastable and easily regenerated hydrogen-bonded organic molecular framework with permanent porosity. Angew. Chem. Int. Ed. 2017, 56, 2101–2104. [Google Scholar] [CrossRef] [PubMed]
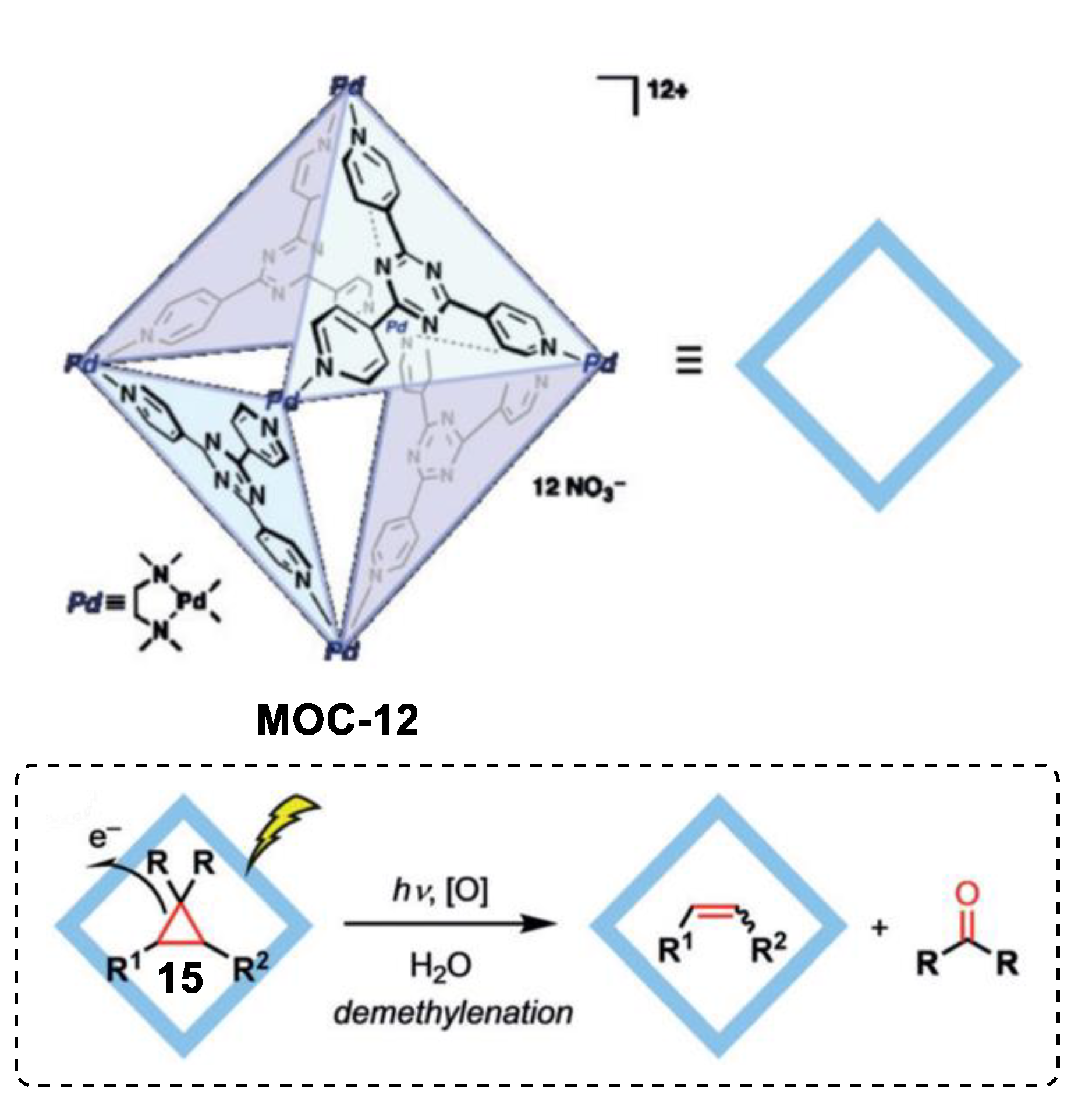
- Cullen, W.; Takezawa, H.; Fujita, M. Demethylenation of cyclopropanes via photoinduced guest-to-host electron transfer in an m6 l4 cage. Angew. Chem. Int. Ed. 2019, 58, 9171–9173. [Google Scholar] [CrossRef] [PubMed]
- Furutani, Y.; Kandori, H.; Kawano, M.; Nakabayashi, K.; Yoshizawa, M.; Fujita, M. In situ spectroscopic, electrochemical, and theoretical studies of the photoinduced host−guest electron transfer that precedes unusual host-mediated alkane photooxidation. J. Am. Chem. Soc. 2009, 131, 4764–4768. [Google Scholar] [CrossRef] [PubMed]
- Murase, T.; Nishijima, Y.; Fujita, M. Unusual photoreaction of triquinacene within self-assembled hosts. Chem. Asian J. 2012, 7, 826–829. [Google Scholar] [CrossRef]
- Takezawa, H.; Kanda, T.; Nanjo, H.; Fujita, M. Site-selective functionalization of linear diterpenoids through u-shaped folding in a confined artificial cavity. J. Am. Chem. Soc. 2019, 141, 5112–5115. [Google Scholar] [CrossRef]
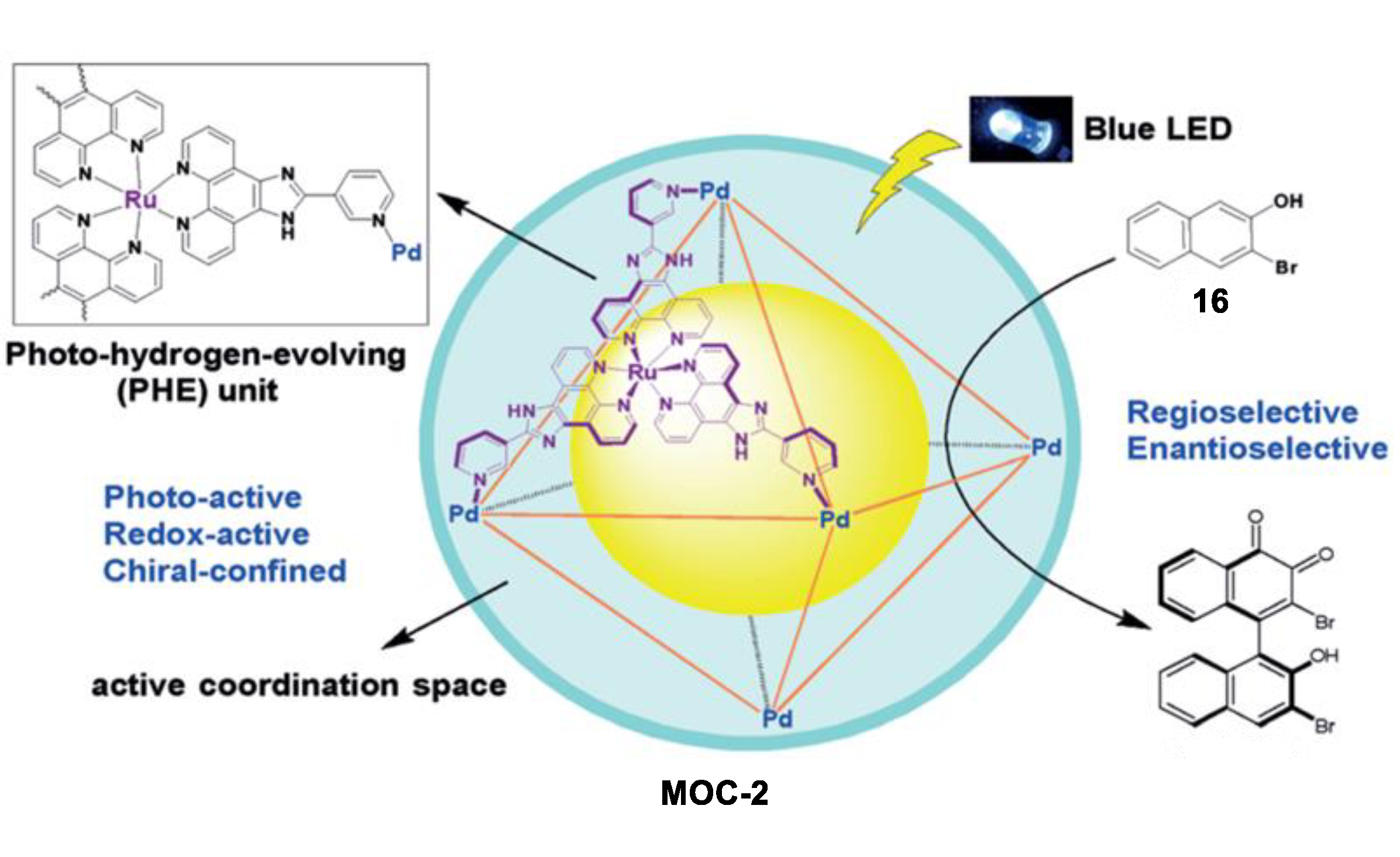
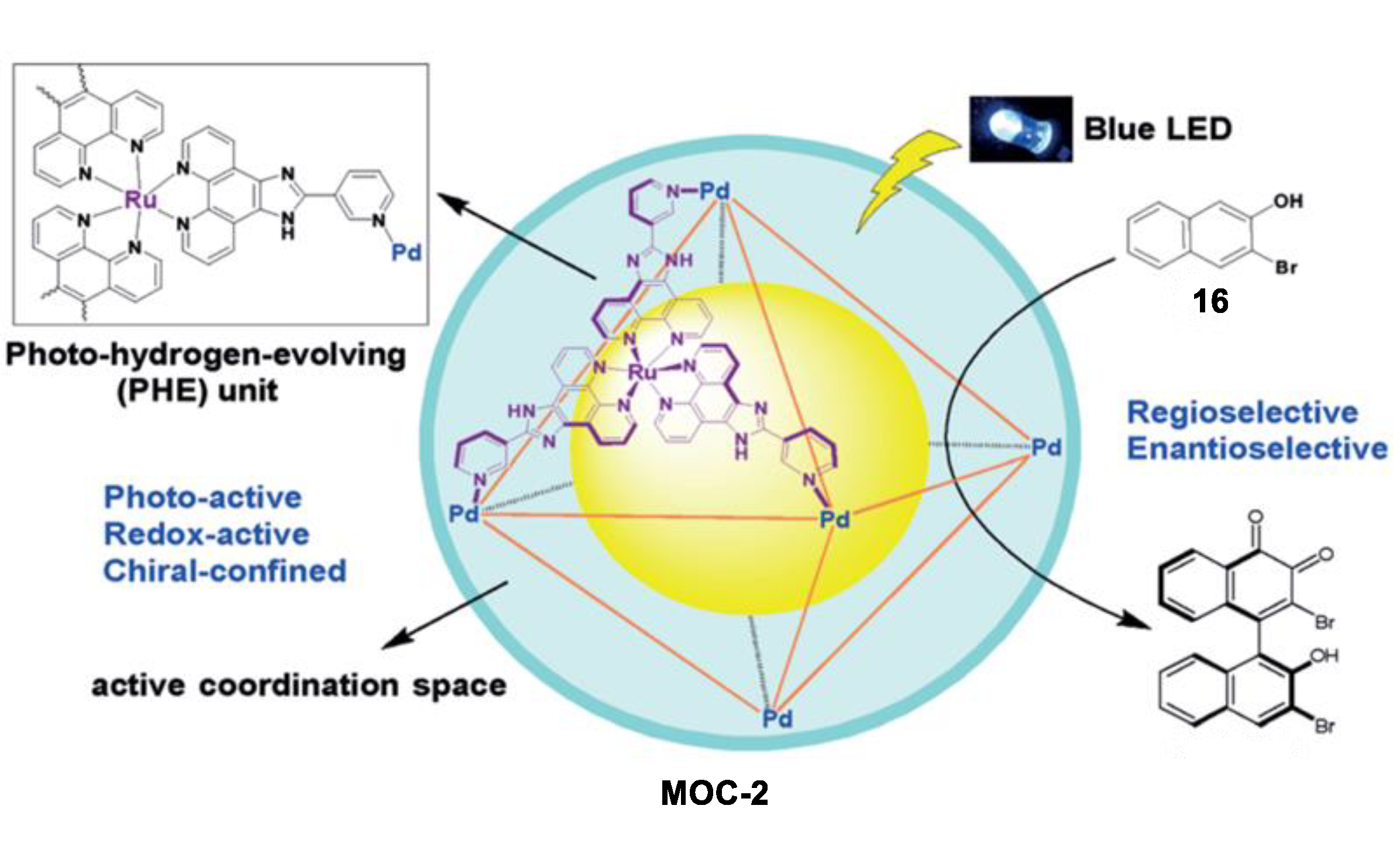
- Guo, J.; Xu, Y.W.; Li, K.; Xiao, L.M.; Chen, S.; Wu, K.; Chen, X.D.; Fan, Y.Z.; Liu, J.M.; Su, C.Y. Regio- and enantioselective photodimerization within the confined space of a homochiral ruthenium/palladium heterometallic coordination cage. Angew. Chem. Int. Ed. 2017, 56, 3852–3856. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Li, K.; Hou, Y.-J.; Pan, M.; Zhang, L.-Y.; Chen, L.; Su, C.-Y. Homochiral d4-symmetric metal–Organic cages from stereogenic Ru(II) metalloligands for effective enantioseparation of atropisomeric molecules. Nat. Commun. 2016, 7, 10487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jongkind, L.J.; Caumes, X.; Hartendorp, A.P.T.; Reek, J.N.H. Ligand template strategies for catalyst encapsulation. Acc. Chem. Res. 2018, 51, 2115–2128. [Google Scholar] [CrossRef]
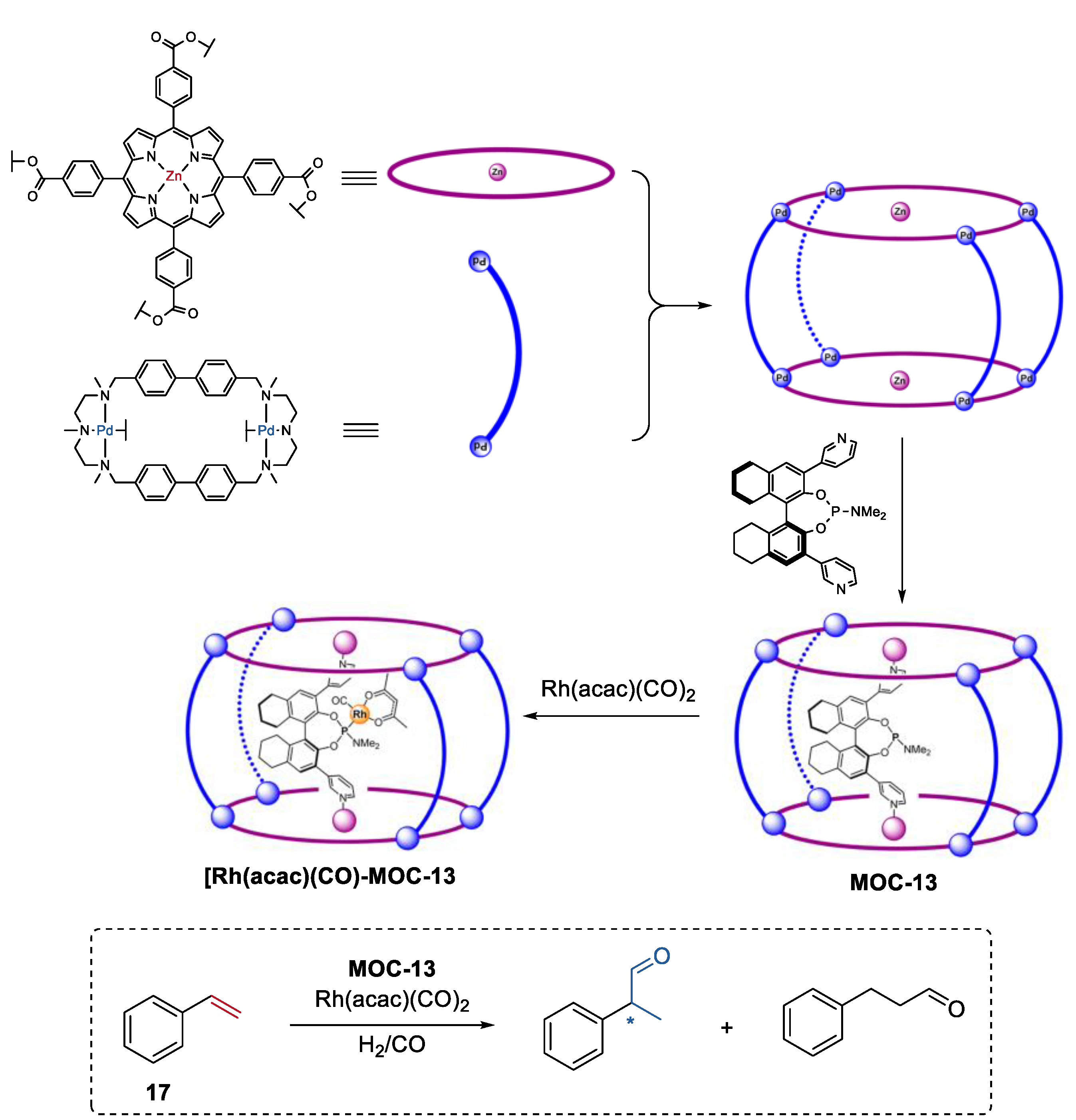
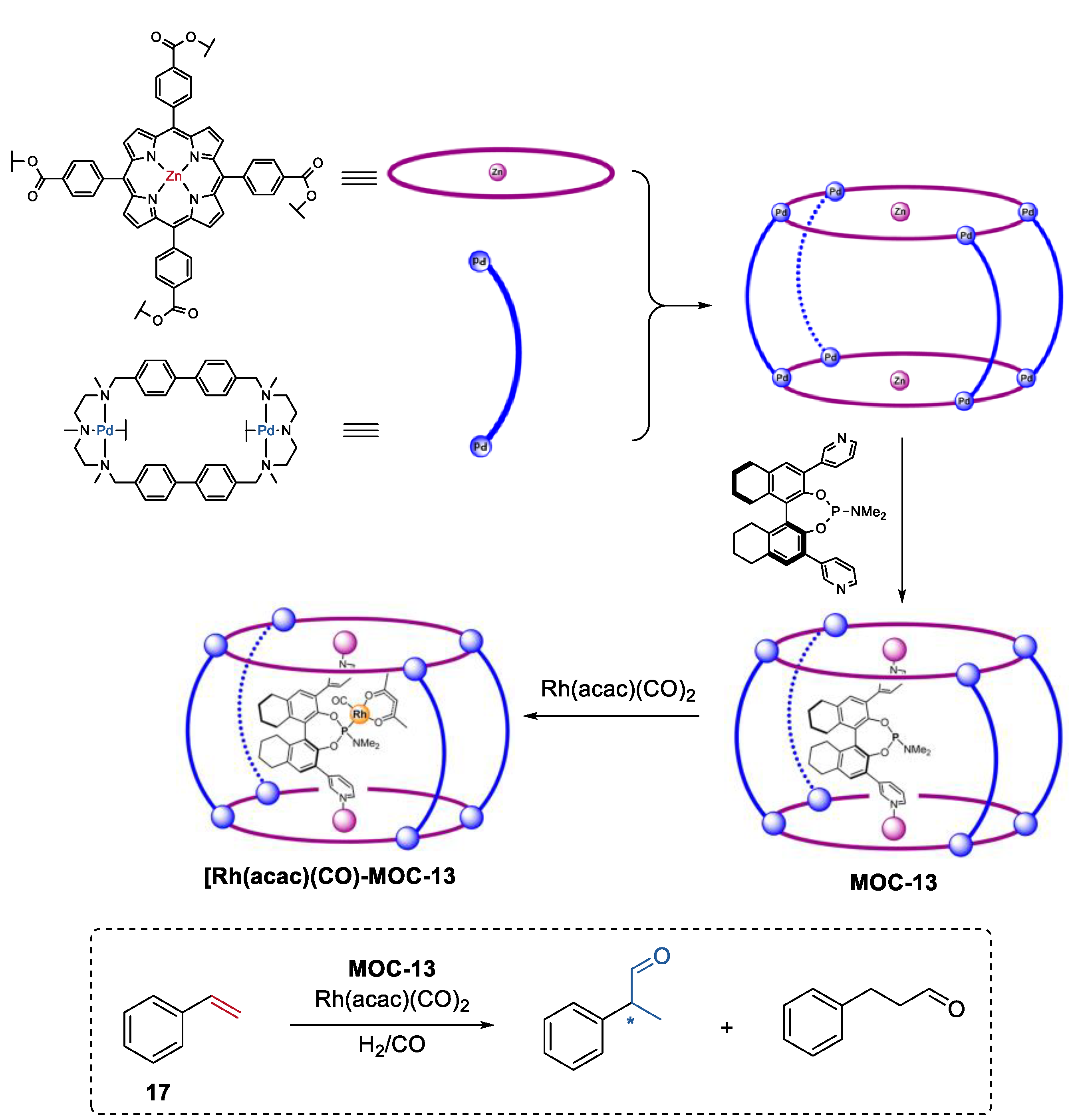
- Garcia-Simon, C.; Gramage-Doria, R.; Raoufmoghaddam, S.; Parella, T.; Costas, M.; Ribas, X.; Reek, J.N. Enantioselective hydroformylation by a rh-catalyst entrapped in a supramolecular metallocage. J. Am. Chem. Soc. 2015, 137, 2680–2687. [Google Scholar] [CrossRef]
- García-Simón, C.; Garcia-Borràs, M.; Gómez, L.; Parella, T.; Osuna, S.; Juanhuix, J.; Imaz, I.; Maspoch, D.; Costas, M.; Ribas, X. Sponge-like molecular cage for purification of fullerenes. Nat. Commun. 2014, 5, 5557. [Google Scholar] [CrossRef]
- Zhao, C.; Sun, Q.-F.; Hart-Cooper, W.M.; DiPasquale, A.G.; Toste, F.D.; Bergman, R.G.; Raymond, K.N. Chiral amide directed assembly of a diastereo- and enantiopure supramolecular host and its application to enantioselective catalysis of neutral substrates. J. Am. Chem. Soc. 2013, 135, 18802–18805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.J.; Clary, K.N.; Bergman, R.G.; Raymond, K.N.; Toste, F.D. A supramolecular approach to combining enzymatic and transition metal catalysis. Nat. Chem. 2013, 5, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Ito, H.; Fujita, D.; Fujita, M. Permeable self-assembled molecular containers for catalyst isolation enabling two-step cascade reactions. J. Am. Chem. Soc. 2017, 139, 6090–6093. [Google Scholar] [CrossRef] [PubMed]
- Vogler, T.; Studer, A. Applications of tempo in synthesis. Synthesis 2008, 2008, 1979–1993. [Google Scholar]
- Jones, S.B.; Simmons, B.; Mastracchio, A.; MacMillan, D.W.C. Collective synthesis of natural products by means of organocascade catalysis. Nature 2011, 475, 183–188. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Iida, J.; Suzuki, K.; Kawano, M.; Ozeki, T.; Fujita, M. Fluorous nanodroplets structurally confined in an organopalladium sphere. Science 2006, 313, 1273–1276. [Google Scholar] [CrossRef] [Green Version]
- Zhu, F.-F.; Chen, L.-J.; Chen, S.; Wu, G.-Y.; Jiang, W.-L.; Shen, J.-C.; Qin, Y.; Xu, L.; Yang, H.-B. Confinement self-assembly of metal-organic cages within mesoporous carbon for one-pot sequential reactions. Chem 2020, 6, 2395–2406. [Google Scholar] [CrossRef]
- Chen, L.-J.; Chen, S.; Qin, Y.; Xu, L.; Yin, G.-Q.; Zhu, J.-L.; Zhu, F.-F.; Zheng, W.; Li, X.; Yang, H.-B. Construction of porphyrin-containing metallacycle with improved stability and activity within mesoporous carbon. J. Am. Chem. Soc. 2018, 140, 5049–5052. [Google Scholar] [CrossRef]
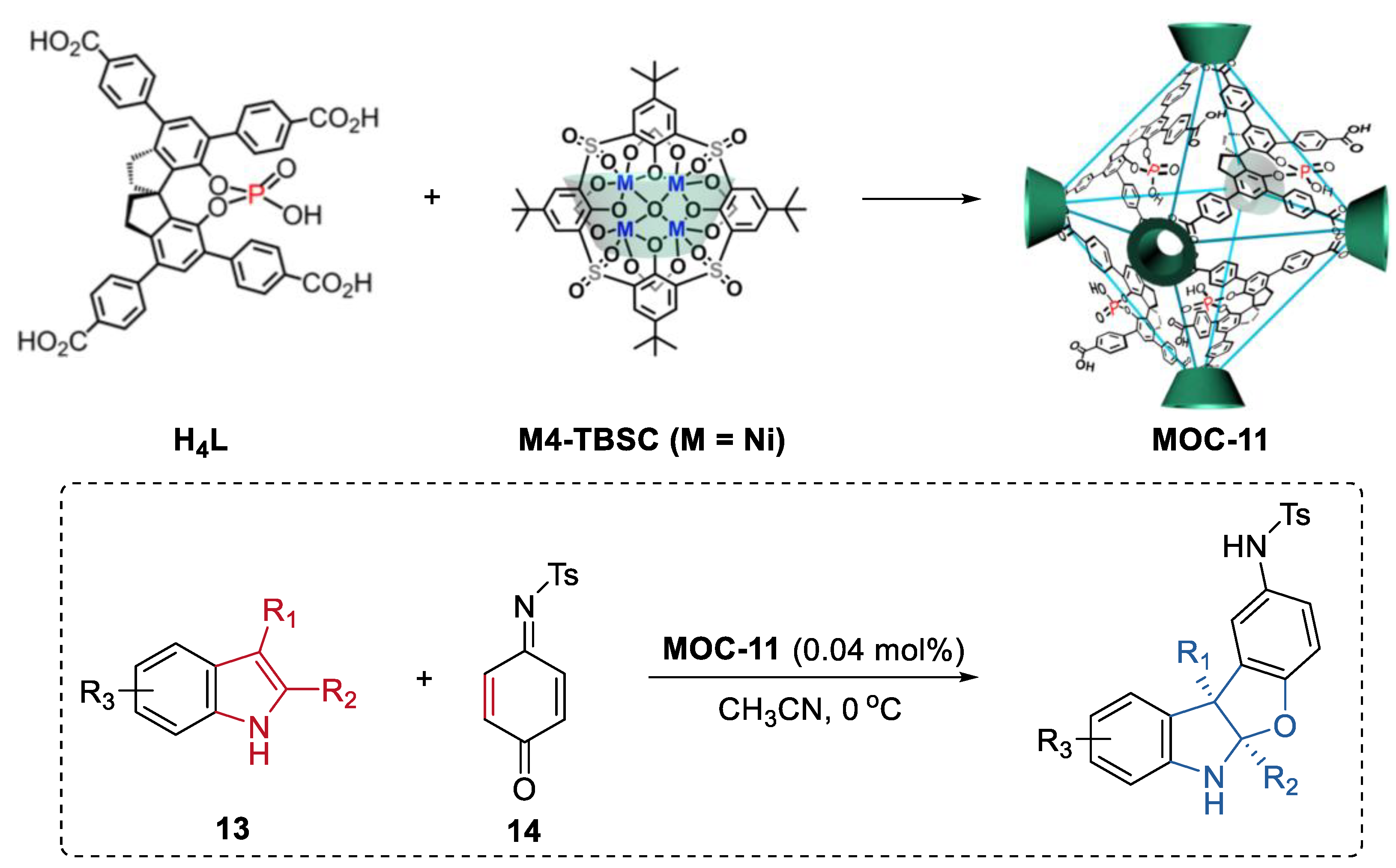
- Jiao, J.; Tan, C.; Li, Z.; Liu, Y.; Han, X.; Cui, Y. Design and assembly of chiral coordination cages for asymmetric sequential reactions. J. Am. Chem. Soc. 2018, 140, 2251–2259. [Google Scholar] [CrossRef]
- Srinivasan, K.; Michaud, P.; Kochi, J.K. Epoxidation of olefins with cationic (salen)manganese(III) complexes. The modulation of catalytic activity by substituents. J. Am. Chem. Soc. 1986, 108, 2309–2320. [Google Scholar] [CrossRef]
- Wende, R.C.; Schreiner, P.R. Evolution of asymmetric organocatalysis: Multi- and retrocatalysis. Green Chem. 2012, 14, 1821–1849. [Google Scholar] [CrossRef] [Green Version]
- Salles, A.G., Jr.; Zarra, S.; Turner, R.M.; Nitschke, J.R. A self-organizing chemical assembly line. J. Am. Chem. Soc. 2013, 135, 19143–19146. [Google Scholar] [CrossRef] [PubMed]
- Holloway, L.R.; Bogie, P.M.; Lyon, Y.; Ngai, C.; Miller, T.F.; Julian, R.R.; Hooley, R.J. Tandem reactivity of a self-assembled cage catalyst with endohedral acid groups. J. Am. Chem. Soc. 2018, 140, 8078–8081. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.A.; Holloway, L.R.; Miller, T.F.; Lyon, Y.; Julian, R.R.; Hooley, R.J. Electronic effects on narcissistic self-sorting in multicomponent self-assembly of fe-iminopyridine meso-helicates. Inorg. Chem. 2016, 55, 9805–9815. [Google Scholar] [CrossRef]
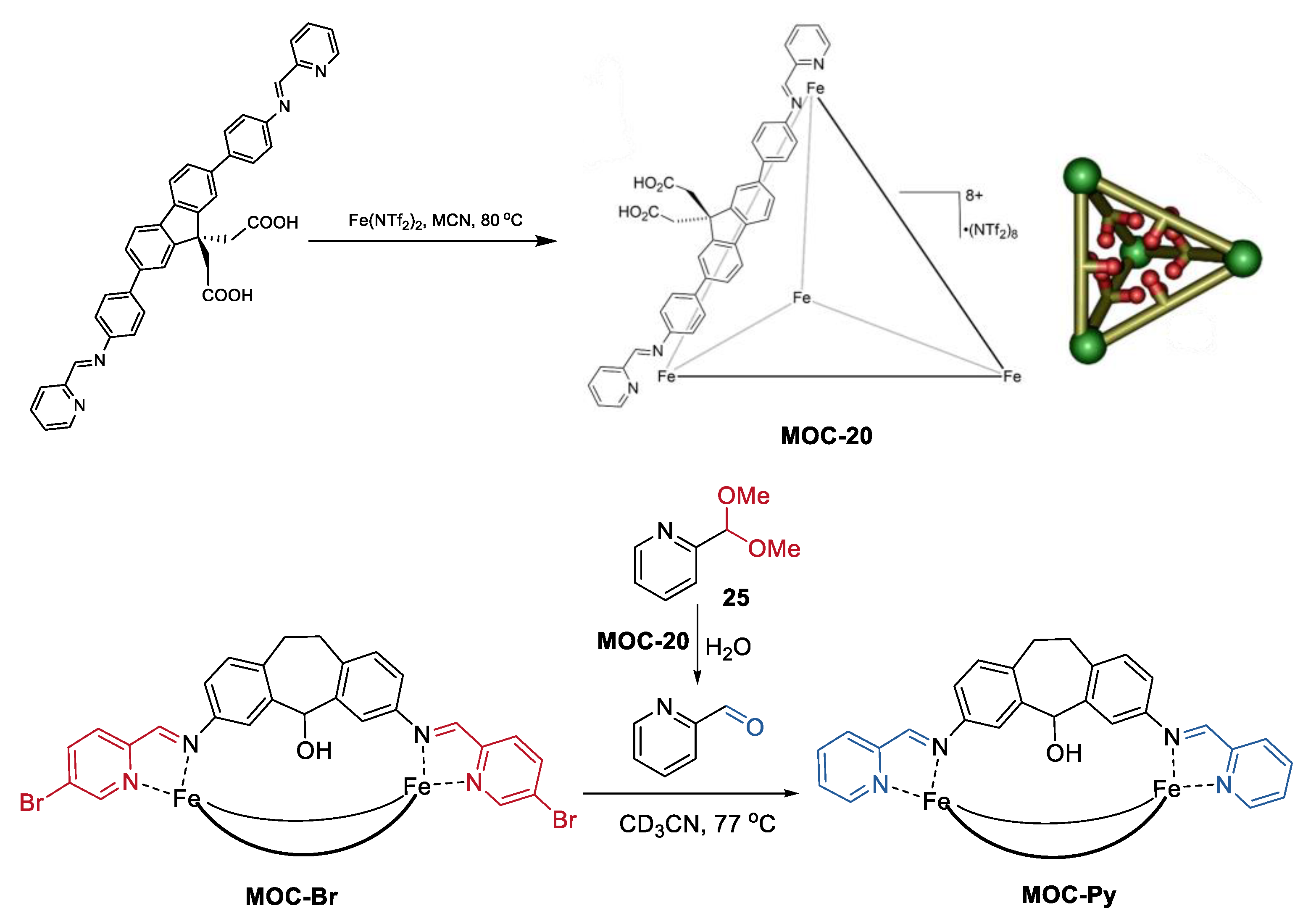
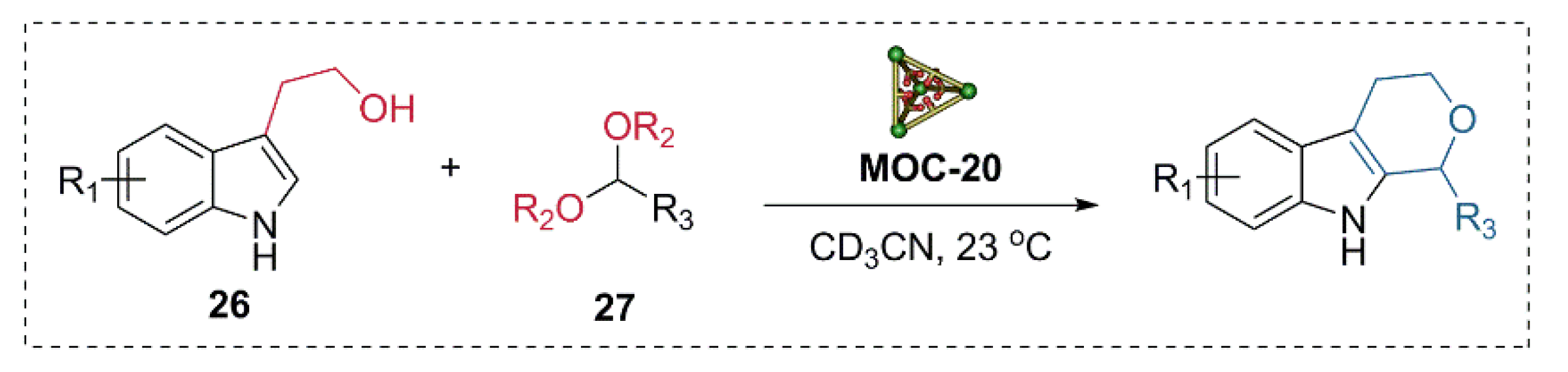
- Ngai, C.; Sanchez-Marsetti, C.M.; Harman, W.H.; Hooley, R.J. Supramolecular catalysis of the oxa-pictet-spengler reaction with an endohedrally functionalized self-assembled cage complex. Angew. Chem. Int. Ed. 2020, 59, 23505–23509. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.; Chen, L.-J. Metal–Organic Cages: Applications in Organic Reactions. Chemistry 2022, 4, 494-519. https://doi.org/10.3390/chemistry4020036
Chen S, Chen L-J. Metal–Organic Cages: Applications in Organic Reactions. Chemistry. 2022; 4(2):494-519. https://doi.org/10.3390/chemistry4020036
Chicago/Turabian StyleChen, Shangjun, and Li-Jun Chen. 2022. "Metal–Organic Cages: Applications in Organic Reactions" Chemistry 4, no. 2: 494-519. https://doi.org/10.3390/chemistry4020036
APA StyleChen, S., & Chen, L.-J. (2022). Metal–Organic Cages: Applications in Organic Reactions. Chemistry, 4(2), 494-519. https://doi.org/10.3390/chemistry4020036