
The Non-Anhydrous, Minimally Basic Synthesis of the Dopamine D2 Agonist [18F]MCL-524

 ,
,
Abstract
:
1. Introduction
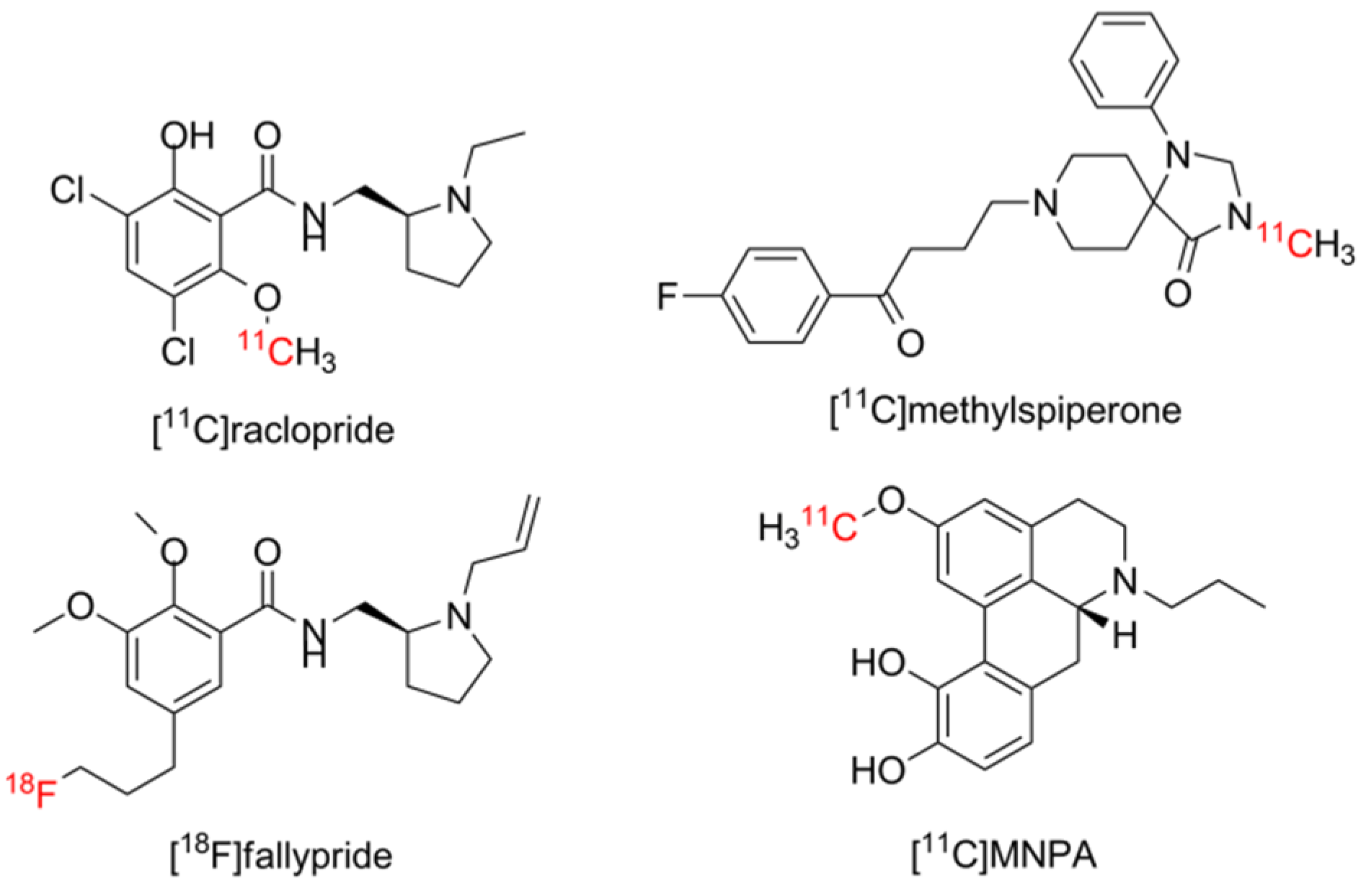
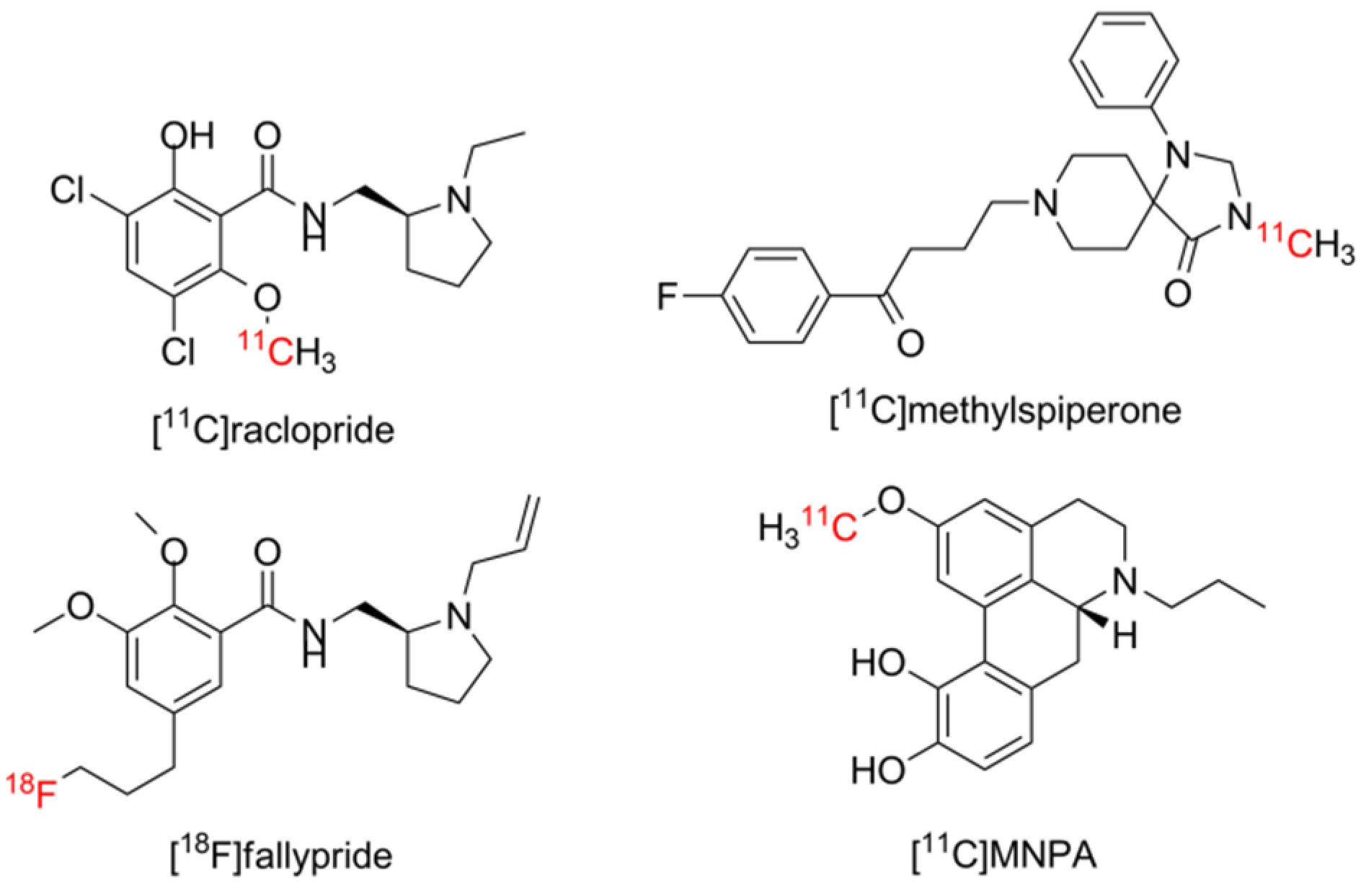
1.1. Aporphines
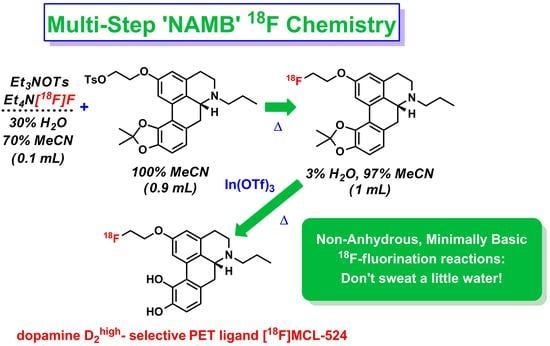
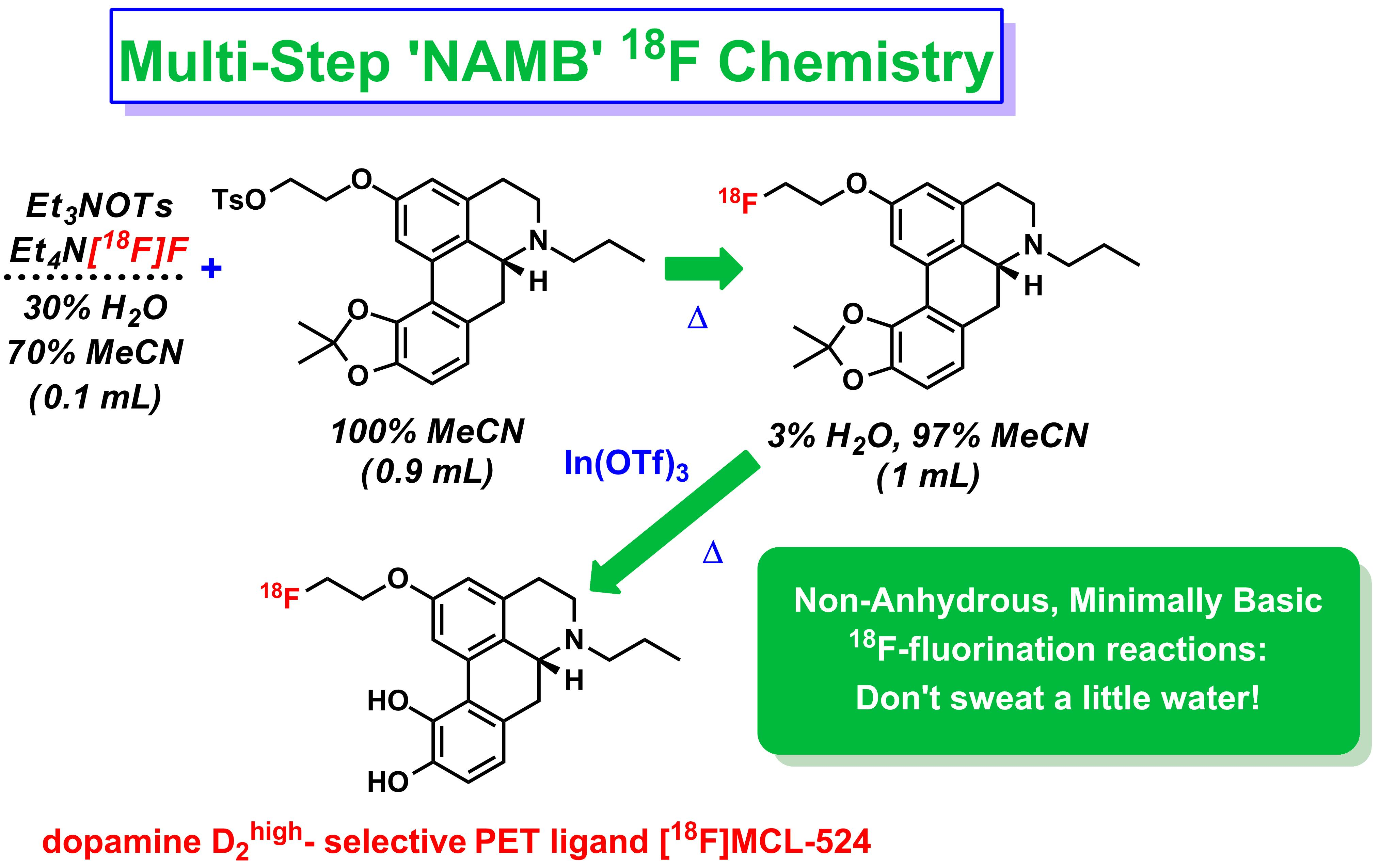
1.2. NAMB 18F Chemistry
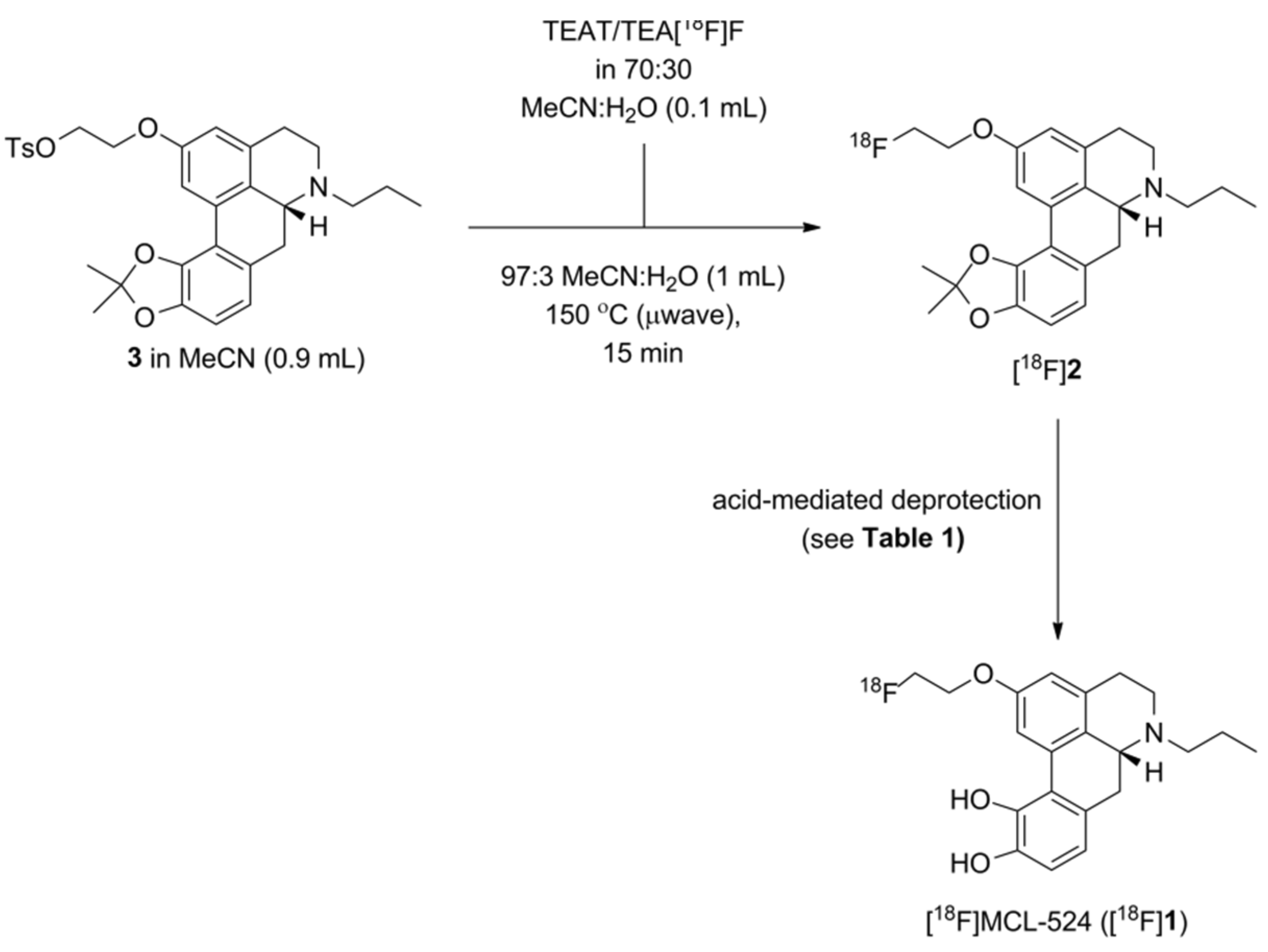
1.3. Preperation of [18F]MCL-524
2. Materials and Methods
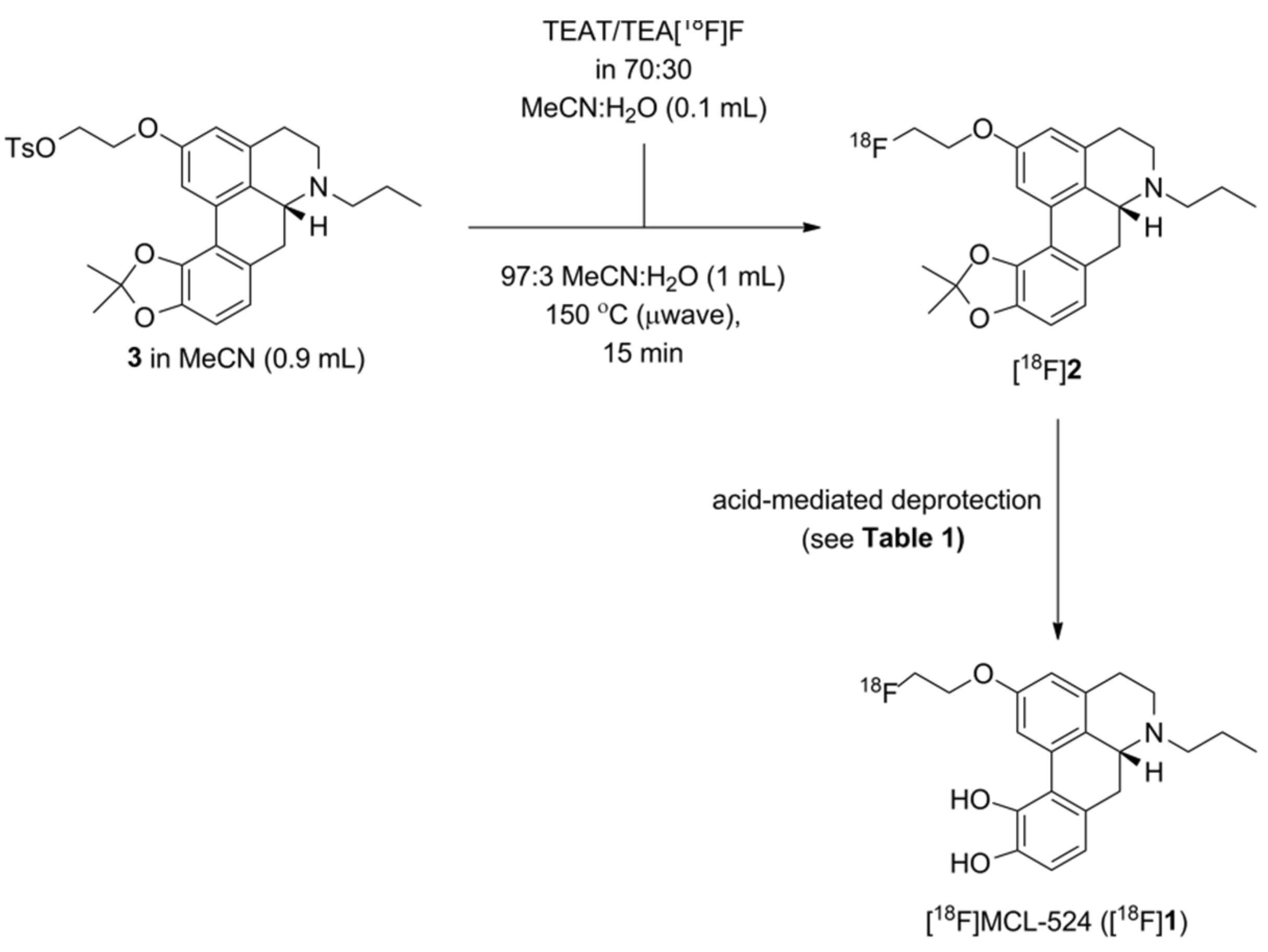
Radiosynthesis of [18F]MCL-524 ([18F]1) via In(OTf)3-Mediated Deprotection
3. Results
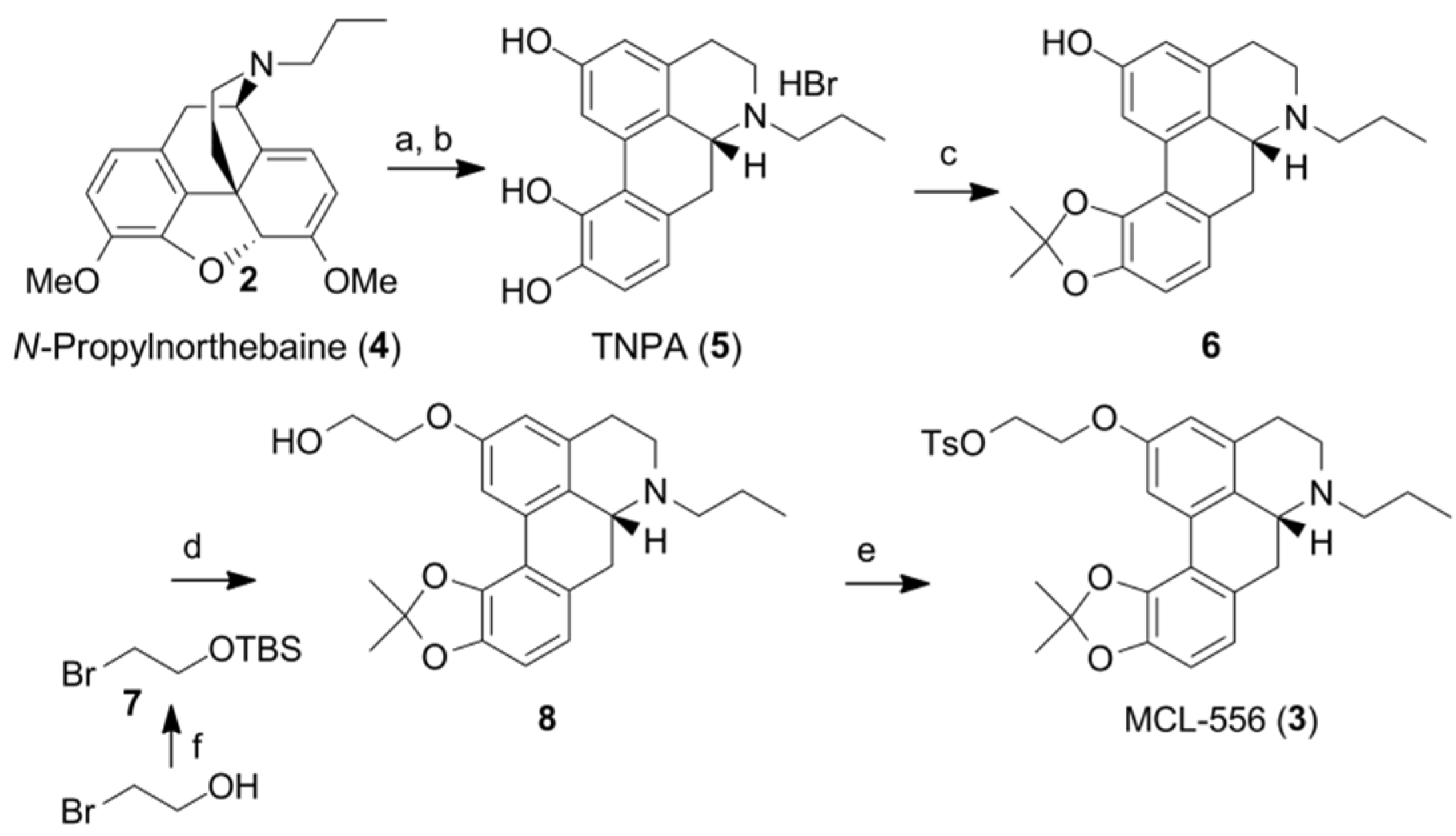
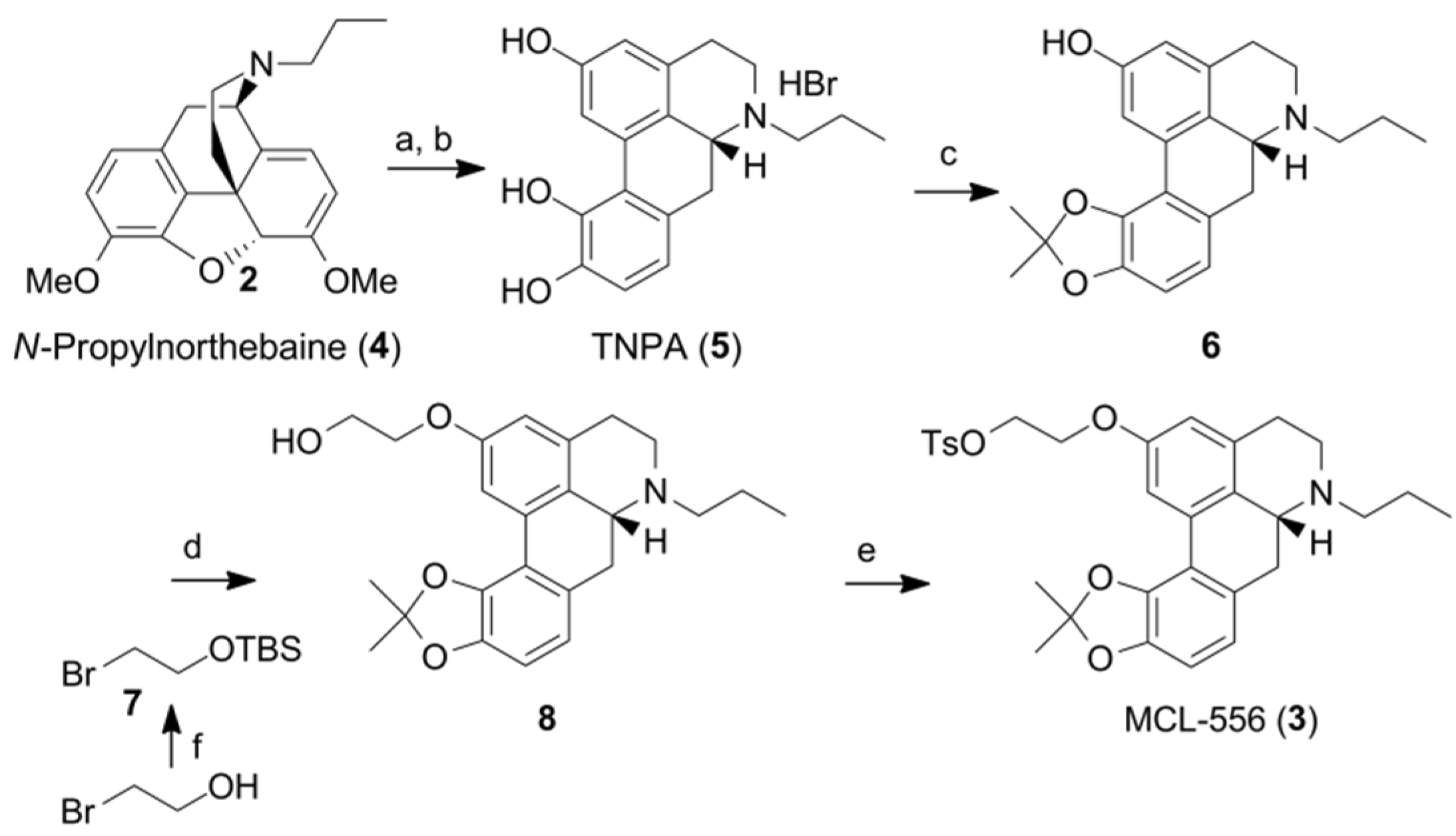
3.1. Summary of Non-Radioactive Synthesis
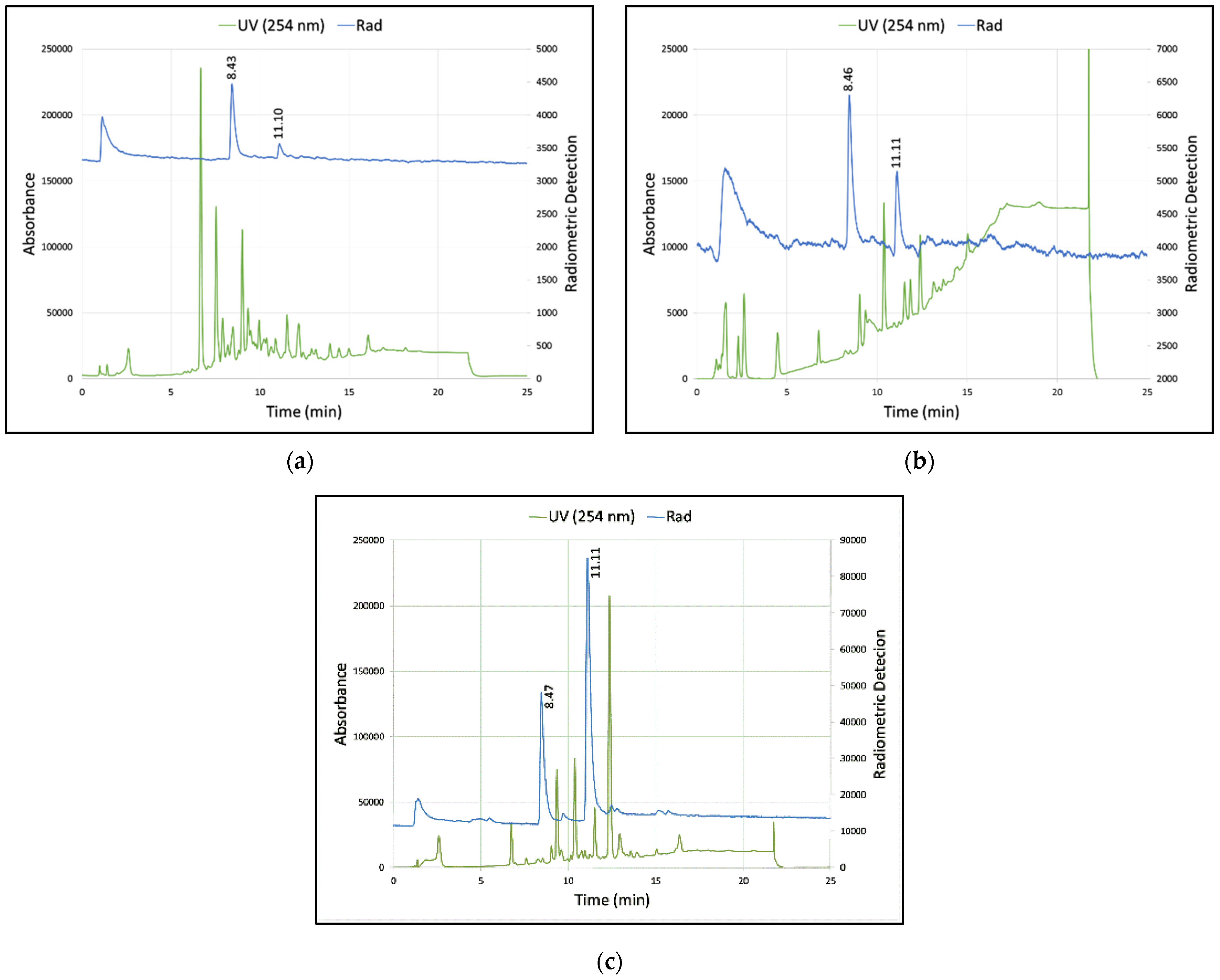
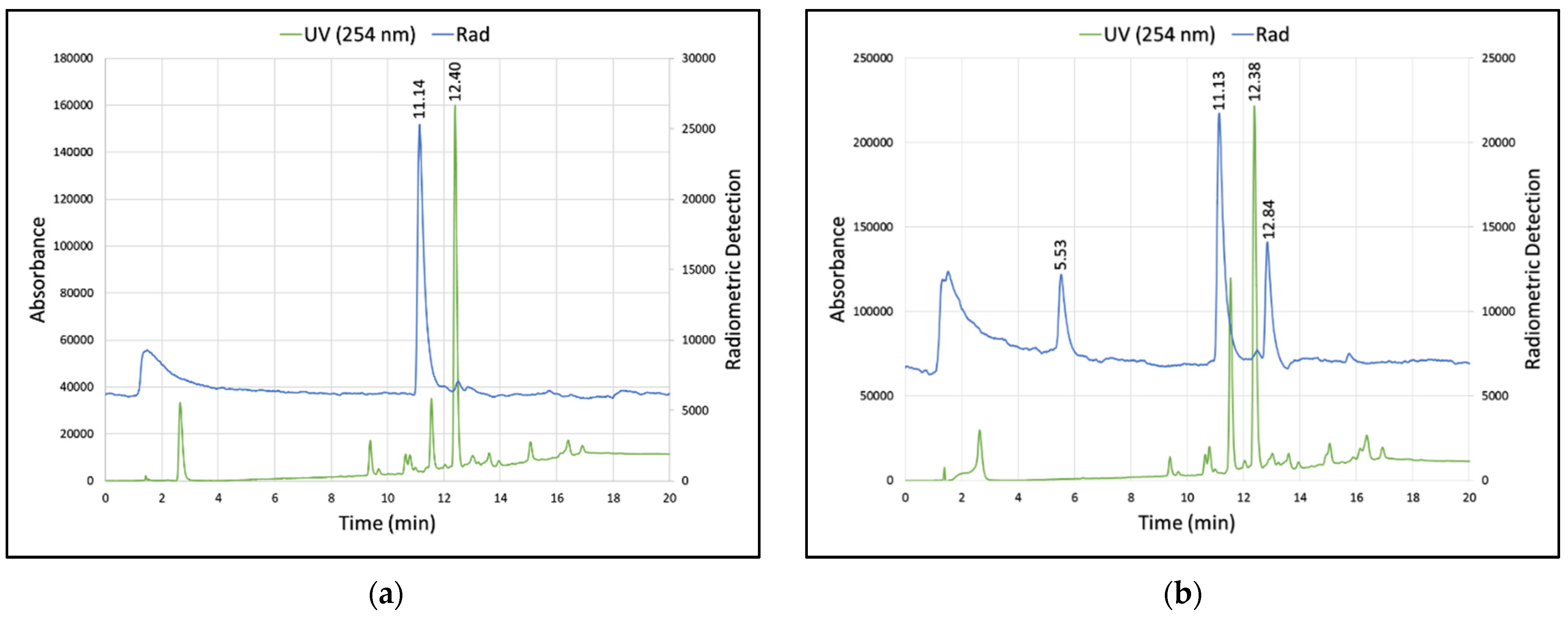
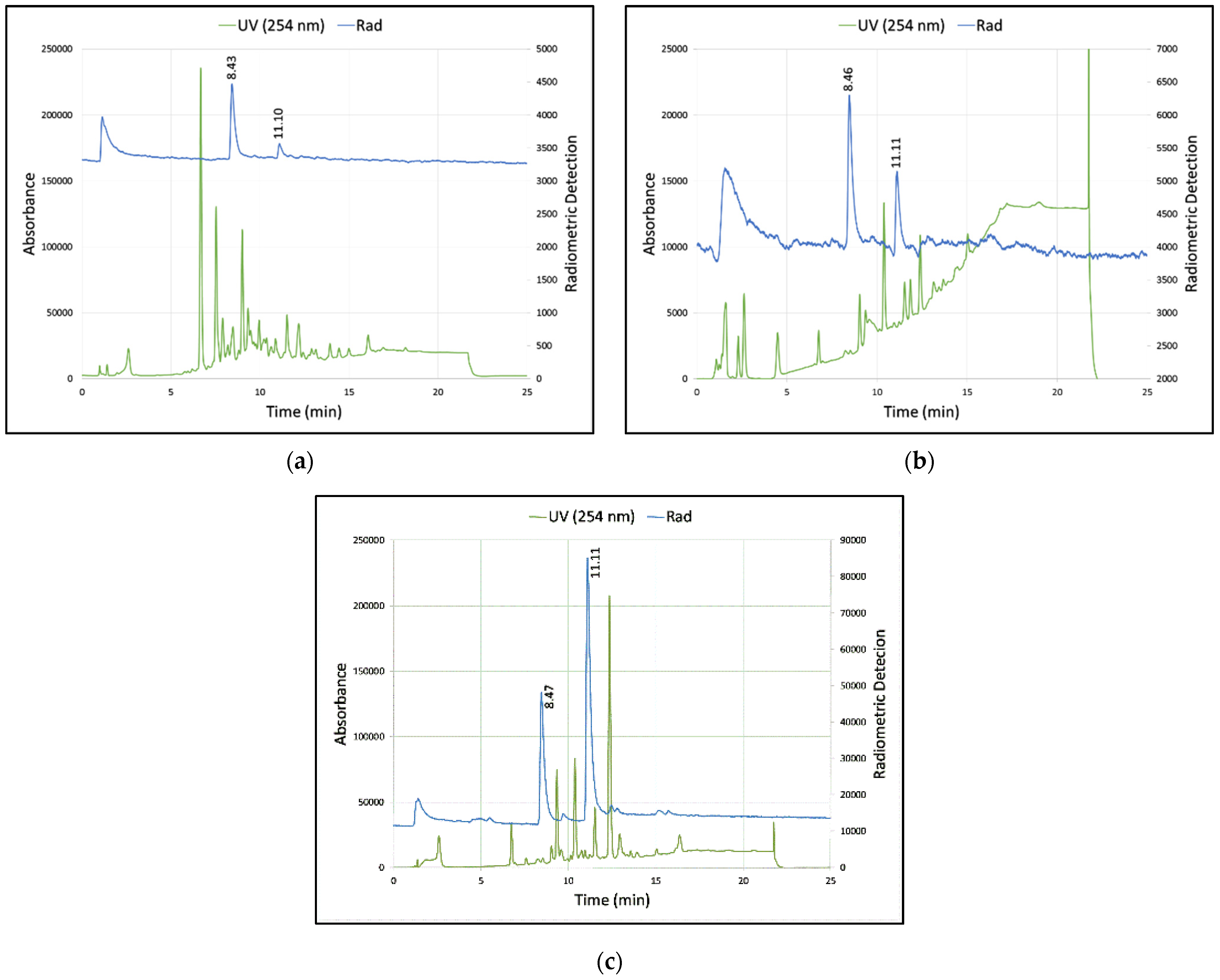
3.2. Summary of Radioactive Synthesis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Beaulieu, J.M.; Gainetdinov, R.R. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 2011, 63, 182–217. [Google Scholar] [CrossRef] [Green Version]
- Blum, K.; Sheridan, P.J.; Wood, R.C.; Braverman, E.R.; Chen, T.J.; Cull, J.G.; Comings, D.E. The D2 dopamine receptor gene as a determinant of reward deficiency syndrome. J. R. Soc. Med. 1996, 89, 396–400. [Google Scholar] [CrossRef] [Green Version]
- Noble, E.P. The DRD2 gene in psychiatric and neurological disorders and its phenotypes. Pharmacogenomics 2000, 1, 309–333. [Google Scholar] [CrossRef]
- Volkow, N.D.; Morales, M. The Brain on Drugs: From Reward to Addiction. Cell 2015, 162, 712–725. [Google Scholar] [CrossRef] [Green Version]
- Volkow, N.D.; Wang, G.J.; Fowler, J.S.; Tomasi, D.; Telang, F. Addiction: Beyond dopamine reward circuitry. Proc. Natl. Acad. Sci. USA 2011, 108, 15037–15042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volkow, N.D.; Fowler, J.S.; Wang, G.J.; Baler, R.; Telang, F. Imaging dopamine’s role in drug abuse and addiction. Neuropharmacology 2009, 56 (Suppl. 1), 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeman, M.V.; Seeman, P. Is schizophrenia a dopamine supersensitivity psychotic reaction? Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 48, 155–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeman, P. Are dopamine D2 receptors out of control in psychosis? Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 46, 146–152. [Google Scholar] [CrossRef]
- Seeman, P. Schizophrenia and dopamine receptors. Eur. Neuropsychopharmacol. 2013, 23, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Seeman, P. All roads to schizophrenia lead to dopamine supersensitivity and elevated dopamine D2(high) receptors. CNS Neurosci. Ther. 2011, 17, 118–132. [Google Scholar] [CrossRef] [PubMed]
- Seeman, P. Antiparkinson therapeutic potencies correlate with their affinities at dopamine D2(High) receptors. Synapse 2007, 61, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Seeman, P.; Schwarz, J.; Chen, J.F.; Szechtman, H.; Perreault, M.; McKnight, G.S.; Roder, J.C.; Quirion, R.; Boksa, P.; Srivastava, L.K.; et al. Psychosis pathways converge via D2high dopamine receptors. Synapse 2006, 60, 319–346. [Google Scholar] [CrossRef] [PubMed]
- Seeman, P. Targeting the dopamine D2 receptor in schizophrenia. Expert Opin. Ther. Targets 2006, 10, 515–531. [Google Scholar] [CrossRef] [PubMed]
- Nakata, Y.; Kanahara, N.; Iyo, M. Dopamine supersensitivity psychosis in schizophrenia: Concepts and implications in clinical practice. J. Psychopharmacol. 2017, 31, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Sibley, D.R.; De Lean, A.; Creese, I. Anterior pituitary dopamine receptors. Demonstration of interconvertible high and low affinity states of the D-2 dopamine receptor. J. Biol. Chem. 1982, 257, 6351–6361. [Google Scholar] [CrossRef]
- Sibley, D.R.; Creese, I. Regulation of ligand binding to pituitary D-2 dopaminergic receptors. Effects of divalent cations and functional group modification. J. Biol. Chem. 1983, 258, 4957–4965. [Google Scholar] [CrossRef]
- Seeman, P.; Weinshenker, D.; Quirion, R.; Srivastava, L.K.; Bhardwaj, S.K.; Grandy, D.K.; Premont, R.T.; Sotnikova, T.D.; Boksa, P.; El-Ghundi, M.; et al. Dopamine supersensitivity correlates with D2High states, implying many paths to psychosis. Proc. Natl. Acad. Sci. USA 2005, 102, 3513–3518. [Google Scholar] [CrossRef] [Green Version]
- Seeman, P.; Tallerico, T.; Ko, F. Dopamine displaces [3H]domperidone from high-affinity sites of the dopamine D2 receptor, but not [3H]raclopride or [3H]spiperone in isotonic medium: Implications for human positron emission tomography. Synapse 2003, 49, 209–215. [Google Scholar] [CrossRef]
- Skinbjerg, M.; Sibley, D.R.; Javitch, J.A.; Abi-Dargham, A. Imaging the high-affinity state of the dopamine D2 receptor in vivo: Fact or fiction? Biochem. Pharmacol. 2012, 83, 193–198. [Google Scholar] [CrossRef] [Green Version]
- Shalgunov, V.; van Waarde, A.; Booij, J.; Michel, M.C.; Dierckx, R.; Elsinga, P.H. Hunting for the high-affinity state of G-protein-coupled receptors with agonist tracers: Theoretical and practical considerations for positron emission tomography imaging. Med. Res. Rev. 2019, 39, 1014–1052. [Google Scholar] [CrossRef]
- van Wieringen, J.P.; Booij, J.; Shalgunov, V.; Elsinga, P.; Michel, M.C. Agonist high- and low-affinity states of dopamine D(2) receptors: Methods of detection and clinical implications. Naunyn Schmiedebergs Arch. Pharmacol. 2013, 386, 135–154. [Google Scholar] [CrossRef]
- Zhang, L.; Villalobos, A. Strategies to facilitate the discovery of novel CNS PET ligands. EJNMMI Radiopharm. Chem. 2017, 1, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokoloff, P.; Giros, B.; Martres, M.P.; Bouthenet, M.L.; Schwartz, J.C. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 1990, 347, 146–151. [Google Scholar] [CrossRef]
- Breier, A.; Su, T.P.; Saunders, R.; Carson, R.E.; Kolachana, B.S.; De Bartolomeis, A.; Weinberger, D.R.; Weisenfeld, N.; Malhotra, A.K.; Eckelman, W.C.; et al. Schizophrenia is Associated with Elevated Amphetamine-Induced Synaptic Dopamine Concentrations: Evidence from a Novel Positron Emission Tomography Method. Proc. Natl. Acad. Sci. USA 1997, 94, 2569–2574. [Google Scholar] [CrossRef] [Green Version]
- Narendran, R.; Hwang, D.R.; Slifstein, M.; Hwang, Y.; Huang, Y.; Ekelund, J.; Guillin, O.; Scher, E.; Martinez, D.; Laruelle, M. Measurement of the proportion of D2 receptors configured in state of high affinity for agonists in vivo: A positron emission tomography study using [11C]N-propyl-norapomorphine and [11C]raclopride in baboons. J. Pharmacol. Exp. Ther. 2005, 315, 80–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volkow, N.D.; Wang, G.J.; Fowler, J.S.; Logan, J.; Schlyer, D.; Hitzemann, R.; Lieberman, J.; Angrist, B.; Pappas, N.; MacGregor, R.; et al. Imaging endogenous dopamine competition with [11C]raclopride in the human brain. Synapse 1994, 16, 255–262. [Google Scholar] [CrossRef]
- Dewey, S.L.; Smith, G.S.; Logan, J.; Brodie, J.D.; Fowler, J.S.; Wolf, A.P. Striatal binding of the PET ligand 11C-raclopride is altered by drugs that modify synaptic dopamine levels. Synapse 1993, 13, 350–356. [Google Scholar] [CrossRef]
- Mukherjee, J.; Yang, Z.Y.; Brown, T.; Lew, R.; Wernick, M.; Ouyang, X.; Yasillo, N.; Chen, C.T.; Mintzer, R.; Cooper, M. Preliminary assessment of extrastriatal dopamine D-2 receptor binding in the rodent and nonhuman primate brains using the high affinity radioligand, 18F-fallypride. Nucl. Med. Biol. 1999, 26, 519–527. [Google Scholar] [CrossRef]
- Hartvig, P.; Eckernas, S.A.; Ekblom, B.; Lindstrom, L.; Lundqvist, H.; Axelsson, S.; Fasth, K.J.; Gullberg, P.; Langstrom, B. Receptor binding and selectivity of three 11C-labelled dopamine receptor antagonists in the brain of rhesus monkeys studied with positron emission tomography. Acta Neurol. Scand. 1988, 77, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Finnema, S.J.; Seneca, N.; Farde, L.; Shchukin, E.; Sovago, J.; Gulyas, B.; Wikstrom, H.V.; Innis, R.B.; Neumeyer, J.L.; Halldin, C. A preliminary PET evaluation of the new dopamine D-2 receptor agonist C-11 MNPA in cynomolgus monkey. Nucl. Med. Biol. 2005, 32, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Subburaju, S.; Sromek, A.W.; Seeman, P.; Neumeyer, J.L. New Dopamine D2 Receptor Agonist, [3H]MCL-536, for Detecting Dopamine D2high Receptors in Vivo. ACS Chem. Neurosci. 2018, 9, 1283–1289. [Google Scholar] [CrossRef]
- Sromek, A.W.; Si, Y.G.; Zhang, T.; George, S.R.; Seeman, P.; Neumeyer, J.L. Synthesis and Evaluation of Fluorinated Aporphines: Potential Positron Emission Tomography Ligands for D2 Receptors. ACS Med. Chem. Lett. 2011, 2, 189–194. [Google Scholar] [CrossRef]
- Finnema, S.J.; Stepanov, V.; Nakao, R.; Sromek, A.W.; Zhang, T.; Neumeyer, J.L.; George, S.R.; Seeman, P.; Stabin, M.G.; Jonsson, C.; et al. 18F-MCL-524, an 18F-Labeled Dopamine D2 and D3 Receptor Agonist Sensitive to Dopamine: A Preliminary PET Study. J. Nucl. Med. 2014, 55, 1164–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.J.; Oh, S.J.; Chi, D.Y.; Moon, D.H.; Ryu, J.S. High Yielding F-18 Fluorination Method by Fine Control of the Base. B Kor. Chem. Soc. 2012, 33, 2177–2180. [Google Scholar] [CrossRef] [Green Version]
- Sergeev, M.E.; Morgia, F.; Lazari, M.; Wang, C., Jr.; van Dam, R.M. Titania-catalyzed radiofluorination of tosylated precursors in highly aqueous medium. J. Am. Chem. Soc. 2015, 137, 5686–5694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richarz, R.; Krapf, P.; Zarrad, F.; Urusova, E.A.; Neumaier, B.; Zlatopolskiy, B.D. Neither azeotropic drying, nor base nor other additives: A minimalist approach to 18F-labeling. Org. Biomol. Chem. 2014, 12, 8094–8099. [Google Scholar] [CrossRef] [PubMed]
- Basuli, F.; Zhang, X.; Woodroofe, C.C.; Jagoda, E.M.; Choyke, P.L.; Swenson, R.E. Fast indirect fluorine-18 labeling of protein/peptide using the useful 6-fluoronicotinic acid-2,3,5,6-tetrafluorophenyl prosthetic group: A method comparable to direct fluorination. J. Labelled Comp. Radiopharm. 2017, 60, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Kniess, T.; Laube, M.; Steinbach, J. “Hydrous 18F-fluoroethylation”—Leaving off the azeotropic drying. Appl. Radiat. Isot. 2017, 127, 260–268. [Google Scholar] [CrossRef]
- Kwon, Y.D.; Son, J.; Yun, M.J.; Chun, J.H. Azeotropic drying-free aliphatic radiofluorination to produce PET radiotracers in a mixed organic solvent system. Tetrahedron Lett. 2018, 59, 2848–2852. [Google Scholar] [CrossRef]
- Inkster, J.A.H.; Akurathi, V.; Sromek, A.W.; Chen, Y.; Neumeyer, J.L.; Packard, A.B. A new radiosynthesisof the D2 agonist [18F]MCL-524 using tetraethylammonium tosylateas a phase transfer catalyst and In(OTf)3 for catechol deprotection. In Proceedings of the 22nd International Symposium on Radiopharmaceutical Sciences, Dresden, Germany, 14–19 May 2017. [Google Scholar]
- Inkster, J.A.H.; Akurathi, V.; Sromek, A.W.; Neumeyer, J.L.; Packard, A.B. Tetraethylammoniumperchlorate and tosylate as non-basic anion exchange reagents and 18F-fluorinationphase-transfer catalysts: Application to the synthesis of [18F]fallypride. In Proceedings of the 22nd International Symposium on Radiopharmaceutical Sciences, Dresden, Germany, 14–19 May 2017. [Google Scholar]
- Inkster, J.; Akurathi, V.; Chen, Y.; Sromek, A.; Neumeyer, J.; Packard, A. 18F chemistry without azeotropic distillations: Tetraethylammonium salts as combined anion exchange reagents and phase transfer catalysts. J. Nucl. Med. 2016, 57, 328. [Google Scholar]
- Orlovskaya, V.; Antuganov, D.; Fedorova, O.; Timofeev, V.; Krasikova, R. Tetrabutylammonium tosylate as inert phase-transfer catalyst: The key to high efficiency S(N)2 radiofluorinations. Appl. Radiat. Isot. 2020, 163, 109195. [Google Scholar] [CrossRef] [PubMed]
- Orlovskaya, V.; Fedorova, O.; Nadporojskii, M.; Krasikova, R. A fully automated azeotropic drying free synthesis of O-(2-[18F]fluoroethyl)-l-tyrosine ([18F]FET) using tetrabutylammonium tosylate. Appl. Radiat. Isot. 2019, 152, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, O.; Orlovskaya, V.; Nadporojskii, M.; Krasikova, R. Automated synthesis of the 16 alpha- F-18 fluoroestradiol (F-18 FES): Minimization of precursor amount and resulting benefits. Radiochim. Acta 2020, 108, 979–988. [Google Scholar] [CrossRef]
- Inkster, J.A.H.; Akurathi, V.; Sromek, A.W.; Chen, Y.; Neumeyer, J.L.; Packard, A.B. A non-anhydrous, minimally basic protocol for the simplification of nucleophilic 18F-fluorination chemistry. Sci. Rep. 2020, 10, 6818. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Kim, K. Synthesis of the new thebaine derivatives by the Diels-Alder reaction with northebaine. Taehan Hwahakhoe Chi 1988, 32, 371–376. [Google Scholar]
- Gao, Y.G.; Baldessarini, R.J.; Kula, N.S.; Neumeyer, J.L. Synthesis and dopamine receptor affinities of enantiomers of 2-substituted apomorphines and their N-n-propyl analogues. J. Med. Chem. 1990, 33, 1800–1805. [Google Scholar] [CrossRef] [PubMed]
- Steiger, C.; Finnema, S.J.; Raus, L.; Schou, M.; Nakao, R.; Suzuki, K.; Pike, V.W.; Wikstroem, H.V.; Halldin, C. A two-step one-pot radiosynthesis of the potent dopamine D2/D3 agonist PET radioligand [11C]MNPA. J. Labelled Compd. Radiopharm. 2009, 52, 158–165. [Google Scholar] [CrossRef]
- Si, Y.G.; Gardner, M.P.; Tarazi, F.I.; Baldessarini, R.J.; Neumeyer, J.L. Synthesis and binding studies of 2-O- and 11-O-substituted N-alkylnoraporphines. Bioorg. Med. Chem. Lett. 2008, 18, 3971–3973. [Google Scholar] [CrossRef]
- Golden, K.C.; Gregg, B.T.; Quinn, J.F. Mild, versatile, and chemoselective indium(III) triflate-catalyzed deprotection of acetonides under microwave heating conditions. Tetrahedron Lett. 2010, 51, 4010–4013. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Reagent Added (Volume) | Time (min) | Temp (°C) | Conversion (%) a | DC-RCY (%) | Molar Activity b (GBq/µmol) |
|---|---|---|---|---|---|---|
| 1 | 1:1 6 N H2SO4:MeOH (0.2 mL) | 5 | 100 | 5 | - c | - |
| 2 | 1:1 6 N H2SO4:MeOH (0.2 mL) | 10 | 150 | 86 | - c | - |
| 3 | In(OTf)3 d in H2O (0.1 mL) | 20 | 150 | 33, 35, 38 e | 6, 5, 9 e | 0.9, 0.7, 0.3 e |
| 4 | 90% TFA (1 mL) | 5 | 100 | 67 | 8 | 1.6 |
| 5 | 90% TFA (1 mL) | 10 | 100 | 83 | 14 | 2.0 |
| 6 | Yb(OTf)3 d·xH2O in H2O (0.1 mL) | 20 | 150 | 0 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inkster, J.A.H.; Sromek, A.W.; Akurathi, V.; Neumeyer, J.L.; Packard, A.B. The Non-Anhydrous, Minimally Basic Synthesis of the Dopamine D2 Agonist [18F]MCL-524. Chemistry 2021, 3, 1047-1056. https://doi.org/10.3390/chemistry3030075
Inkster JAH, Sromek AW, Akurathi V, Neumeyer JL, Packard AB. The Non-Anhydrous, Minimally Basic Synthesis of the Dopamine D2 Agonist [18F]MCL-524. Chemistry. 2021; 3(3):1047-1056. https://doi.org/10.3390/chemistry3030075
Chicago/Turabian StyleInkster, James A. H., Anna W. Sromek, Vamsidhar Akurathi, John L. Neumeyer, and Alan B. Packard. 2021. "The Non-Anhydrous, Minimally Basic Synthesis of the Dopamine D2 Agonist [18F]MCL-524" Chemistry 3, no. 3: 1047-1056. https://doi.org/10.3390/chemistry3030075
APA StyleInkster, J. A. H., Sromek, A. W., Akurathi, V., Neumeyer, J. L., & Packard, A. B. (2021). The Non-Anhydrous, Minimally Basic Synthesis of the Dopamine D2 Agonist [18F]MCL-524. Chemistry, 3(3), 1047-1056. https://doi.org/10.3390/chemistry3030075