
Light-Induced Charge Accumulation in PTCDI/Pentacene/Ag(111) Heterojunctions



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
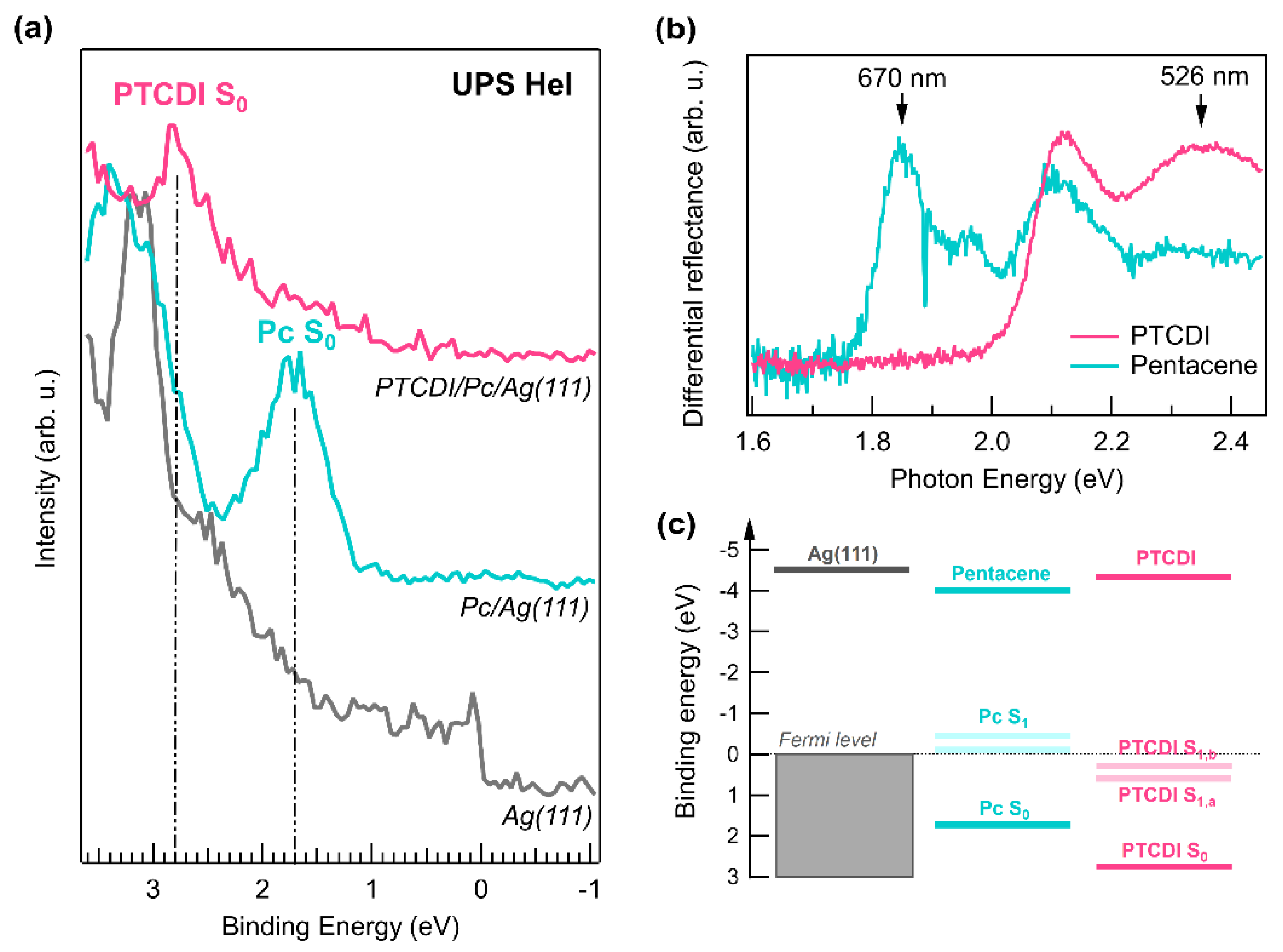
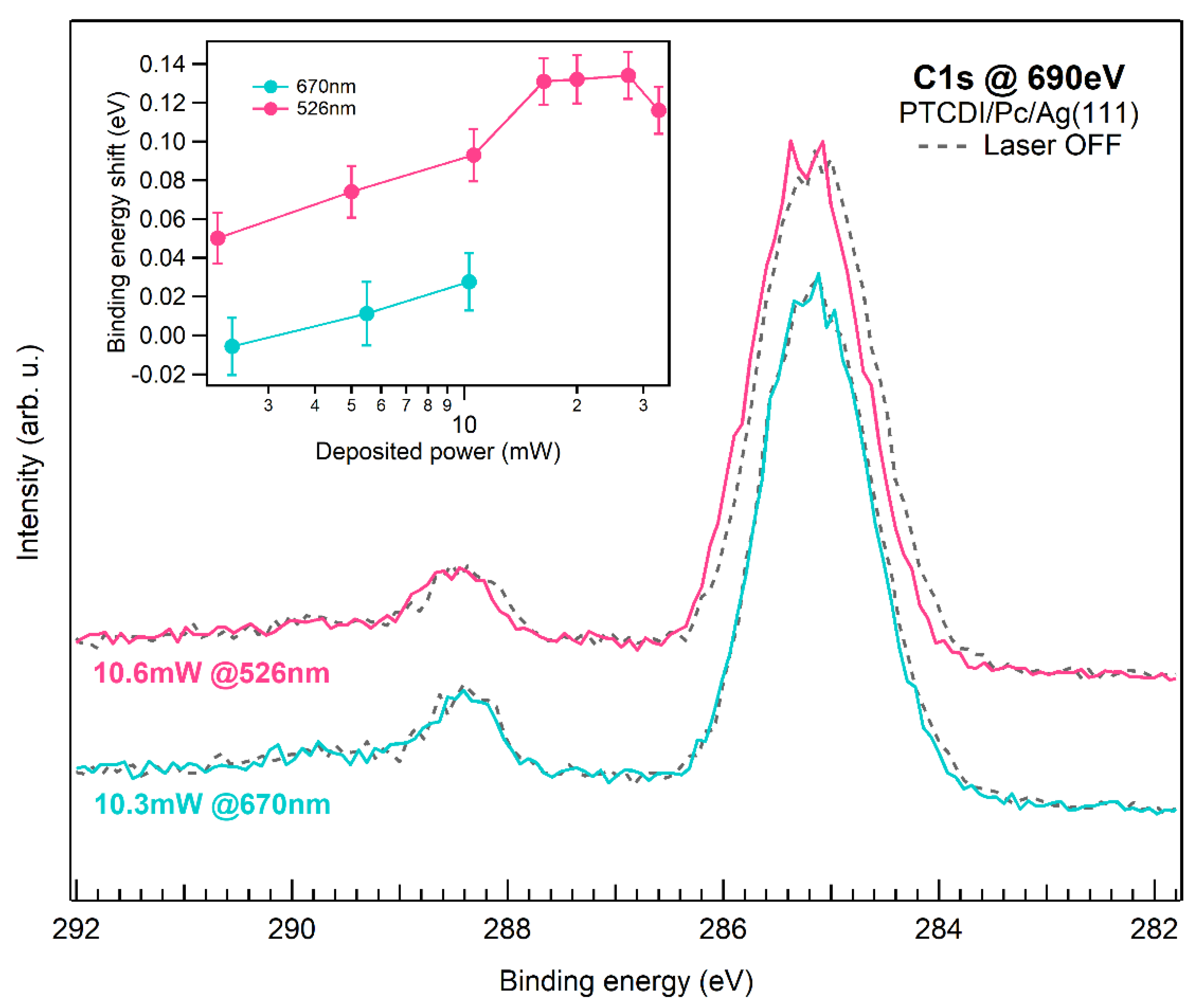
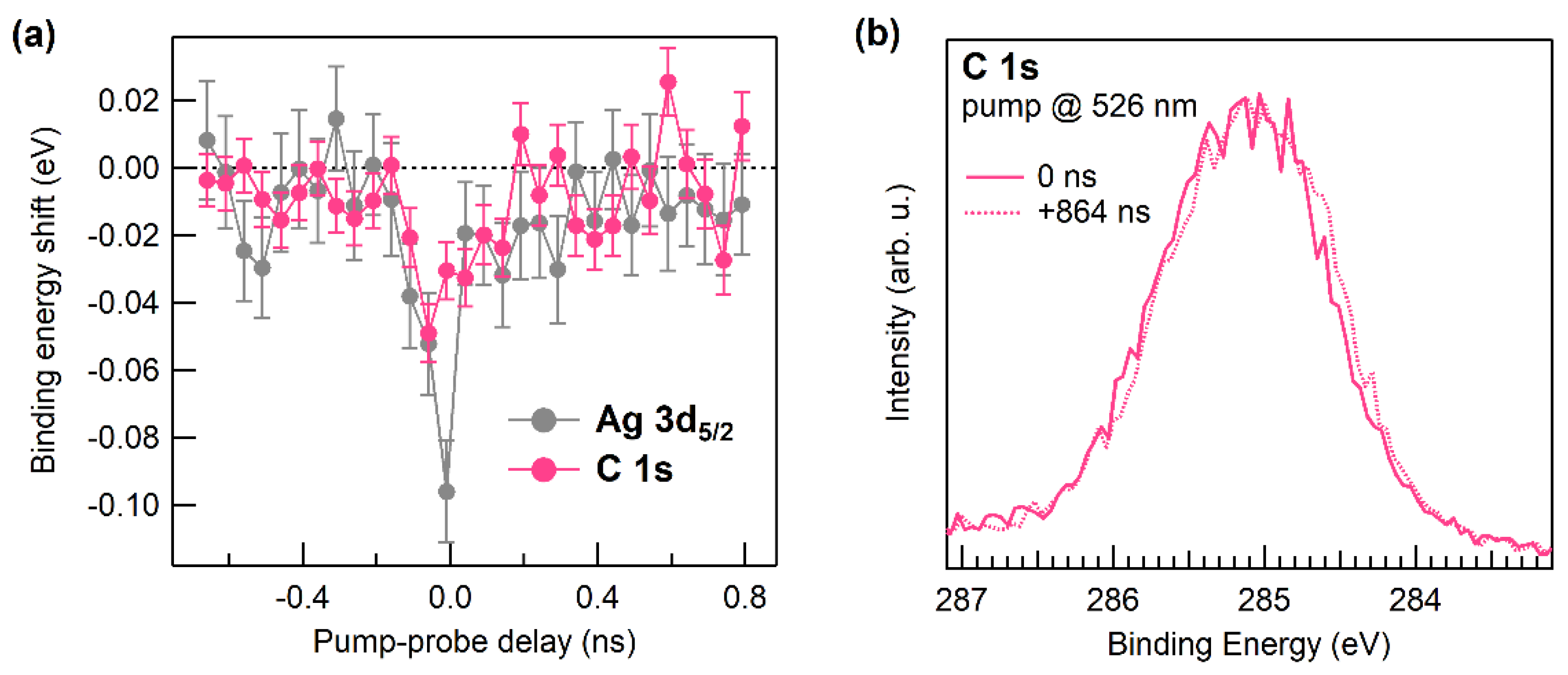
3. Results
4. Discussion and Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hanna, M.C.; Nozik, A.J. Solar conversion efficiency of photovoltaic and photoelectrolysis cells with carrier multiplication absorbers. J. Appl. Phys. 2006, 100, 074510. [Google Scholar] [CrossRef]
- Smith, M.B.; Michl, J. Recent Advances in Singlet Fission. Annu. Rev. Phys. Chem. 2013, 64, 361–386. [Google Scholar] [CrossRef]
- de la Mora, M.B.; Amelines-Sarria, O.; Monroy, B.M.; Hernández-Pérez, C.D.; Lugo, J.E. Materials for downconversion in solar cells: Perspectives and challenges. Sol. Energy Mater. Sol. Cells 2017, 165, 59–71. [Google Scholar] [CrossRef]
- Felter, K.M.; Grozema, F.C. Singlet Fission in Crystalline Organic Materials: Recent Insights and Future Directions. J. Phys. Chem. Lett. 2019, 10, 7208–7214. [Google Scholar] [CrossRef]
- Jadhav, P.J.; Mohanty, A.; Sussman, J.; Lee, J.; Baldo, M.A. Singlet Exciton Fission in Nanostructured Organic Solar Cells. Nano Lett. 2011, 11, 1495–1498. [Google Scholar] [CrossRef] [PubMed]
- Busby, E.; Xia, J.; Wu, Q.; Low, J.Z.; Song, R.; Miller, J.R.; Zhu, X.; Campos, L.M.; Sfeir, M.Y. A design strategy for intramolecular singlet fission mediated by charge-transfer states in donor–acceptor organic materials. Nat. Mater. 2015, 14, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Congreve, D.N.; Lee, J.; Thompson, N.J.; Hontz, E.; Yost, S.R.; Reusswig, P.D.; Bahlke, M.E.; Reineke, S.; Van Voorhis, T.; Baldo, M.A. External Quantum Efficiency Above 100% in a Singlet-Exciton-Fission-Based Organic Photovoltaic Cell. Science 2013, 340, 334–337. [Google Scholar] [CrossRef]
- Costantini, R.; Faber, R.; Cossaro, A.; Floreano, L.; Verdini, A.; Hättig, C.; Morgante, A.; Coriani, S.; Dell’Angela, M. Picosecond timescale tracking of pentacene triplet excitons with chemical sensitivity. Commun. Phys. 2019, 2, 56. [Google Scholar] [CrossRef]
- Paci, I.; Johnson, J.C.; Chen, X.; Rana, G.; Popović, D.; David, D.E.; Nozik, A.J.; Ratner, M.A.; Michl, J. Singlet fission for dye-sensitized solar cells: Can a suitable sensitizer be found? J. Am. Chem. Soc. 2006, 128, 16546–16553. [Google Scholar] [CrossRef]
- Zojer, E.; Taucher, T.C.; Hofmann, O.T. The Impact of Dipolar Layers on the Electronic Properties of Organic/Inorganic Hybrid Interfaces. Adv. Mater. Interfaces 2019, 6, 1900581. [Google Scholar] [CrossRef]
- Tang, J.X.; Lee, C.S.; Lee, S.T. Electronic structures of organic/organic heterojunctions: From vacuum level alignment to Fermi level pinning. J. Appl. Phys. 2007, 101, 064504. [Google Scholar] [CrossRef]
- Shu, A.L.; McClain, W.E.; Schwartz, J.; Kahn, A. Interface dipole engineering at buried organic–organic semiconductor heterojunctions. Org. Electron. 2014, 15, 2360–2366. [Google Scholar] [CrossRef]
- Hill, I.G.; Rajagopal, A.; Kahn, A.; Hu, Y. Molecular level alignment at organic semiconductor-metal interfaces. Appl. Phys. Lett. 1998, 73, 662–664. [Google Scholar] [CrossRef]
- Li, Y.; Li, P.; Lu, Z.H. Mapping Energy Levels for Organic Heterojunctions. Adv. Mater. 2017, 29, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Salomon, E.; Zhang, Q.; Barlow, S.; Marder, S.R.; Kahn, A. Substrate-dependent electronic structure of an organic heterojunction. Phys. Rev. B Condens. Matter Mater. Phys. 2008, 77, 1–6. [Google Scholar] [CrossRef]
- Himpsel, F.J. Inverse photoemission from semiconductors. Surf. Sci. Rep. 1990, 12, 3–48. [Google Scholar] [CrossRef]
- Komolov, A.S.; Lazneva, E.F.; Akhremtchik, S.N.; Chepilko, N.S.; Gavrikov, A.A. Unoccupied Electronic States at the Interface of Oligo(phenylene-vinylene) Films with Oxidized Silicon. J. Phys. Chem. C 2013, 117, 12633–12638. [Google Scholar] [CrossRef]
- Komolov, A.S.; Lazneva, E.F.; Gerasimova, N.B.; Panina, Y.A.; Baramygin, A.V.; Zashikhin, G.D.; Pshenichnyuk, S.A. Structure of vacant electronic states of an oxidized germanium surface upon deposition of perylene tetracarboxylic dianhydride films. Phys. Solid State 2016, 58, 377–381. [Google Scholar] [CrossRef]
- Komolov, A.S.; Lazneva, E.F.; Gerasimova, N.B.; Panina, Y.A.; Sobolev, V.S.; Koroleva, A.V.; Pshenichnyuk, S.A.; Asfandiarov, N.L.; Modelli, A.; Handke, B.; et al. Conduction band electronic states of ultrathin layers of thiophene/phenylene co-oligomers on an oxidized silicon surface. J. Electron. Spectros. Relat. Phenom. 2019, 235, 40–45. [Google Scholar] [CrossRef]
- Asakura, D.; Quilty, J.W.; Takubo, K.; Hirata, S.; Mizokawa, T.; Muraoka, Y.; Hiroi, Z. Photoemission Study of YBa2Cu3Oy Thin Films under Light Illumination. Phys. Rev. Lett. 2004, 93, 247006. [Google Scholar] [CrossRef]
- Hiroshiba, N.; Hayakawa, R.; Chikyow, T.; Yamashita, Y.; Yoshikawa, H.; Kobayashi, K.; Morimoto, K.; Matsuishi, K.; Wakayama, Y. Energy-level alignments and photo-induced carrier processes at the heteromolecular interface of quaterrylene and N,N′-dioctyl-3,4,9,10- perylenedicarboximide. Phys. Chem. Chem. Phys. 2011, 13, 6280–6285. [Google Scholar] [CrossRef]
- Kronik, L. Surface photovoltage phenomena: Theory, experiment, and applications. Surf. Sci. Rep. 1999, 37, 1–206. [Google Scholar] [CrossRef]
- Yang, S.-L.; Sobota, J.A.; Kirchmann, P.S.; Shen, Z.-X. Electron propagation from a photo-excited surface: Implications for time-resolved photoemission. Appl. Phys. A 2014, 116, 85–90. [Google Scholar] [CrossRef]
- Stadtmüller, B.; Emmerich, S.; Jungkenn, D.; Haag, N.; Rollinger, M.; Eich, S.; Maniraj, M.; Aeschlimann, M.; Cinchetti, M.; Mathias, S. Strong modification of the transport level alignment in organic materials after optical excitation. Nat. Commun. 2019, 10, 1470. [Google Scholar] [CrossRef]
- Costantini, R.; Grazioli, C.; Cossaro, A.; Floreano, L.; Morgante, A.; Dell’Angela, M.; Dell’Angela, M. Pump–Probe X-ray Photoemission Reveals Light-Induced Carrier Accumulation in Organic Heterojunctions. J. Phys. Chem. C 2020, 124, 26603–26612. [Google Scholar] [CrossRef]
- Roth, F.; Neppl, S.; Shavorskiy, A.; Arion, T.; Mahl, J.; Seo, H.O.; Bluhm, H.; Hussain, Z.; Gessner, O.; Eberhardt, W. Efficient charge generation from triplet excitons in metal-organic heterojunctions. Phys. Rev. B 2019, 99, 1–6. [Google Scholar] [CrossRef]
- Biber, M.; Aydoğan, Ş.; Çaldıran, Z.; Çakmak, B.; Karacalı, T.; Türüt, A. The influence of annealing temperature and time on the efficiency of pentacene: PTCDI organic solar cells. Results Phys. 2017, 7, 3444–3448. [Google Scholar] [CrossRef]
- Karak, S.; Reddy, V.S.; Ray, S.K.; Dhar, A. Organic photovoltaic devices based on pentacene/N,N′-dioctyl-3,4,9,10-perylenedicarboximide heterojunctions. Org. Electron. 2009, 10, 1006–1010. [Google Scholar] [CrossRef]
- Chou, W.Y.; Chang, J.; Yen, C.T.; Lin, Y.S.; Tang, F.C.; Liu, S.J.; Cheng, H.L.; Lien-Chung Hsu, S.; Chen, J.S. The importance of p-n junction interfaces for efficient small molecule-based organic solar cells. Phys. Chem. Chem. Phys. 2012, 14, 5284–5288. [Google Scholar] [CrossRef] [PubMed]
- Costantini, R.; Stredansky, M.; Cvetko, D.; Kladnik, G.; Verdini, A.; Sigalotti, P.; Cilento, F.; Salvador, F.; De Luisa, A.; Benedetti, D.; et al. ANCHOR-SUNDYN: A novel endstation for time resolved spectroscopy at the ALOISA beamline. J. Electron. Spectros. Relat. Phenom. 2018, 229, 7–12. [Google Scholar] [CrossRef]
- Forker, R.; Fritz, T. Optical differential reflectance spectroscopy of ultrathin epitaxial organic films. Phys. Chem. Chem. Phys. 2009, 11, 2142. [Google Scholar] [CrossRef]
- Jaeckel, B.; Sambur, J.B.; Parkinson, B.A. The influence of metal work function on the barrier heights of metal/pentacene junctions. J. Appl. Phys. 2008, 103, 063719. [Google Scholar] [CrossRef]
- Pedio, M.; Doyle, B.; Mahne, N.; Giglia, A.; Borgatti, F.; Nannarone, S.; Henze, S.K.M.; Temirov, R.; Tautz, F.S.; Casalis, L.; et al. Growth of pentacene on Ag (111) surface: A NEXAFS study. Appl. Surf. Sci. 2007, 254, 103–107. [Google Scholar] [CrossRef]
- Mete, E.; Demiroğlu, İ.; Danışman, M.F.; Ellialtıoğlu, Ş. Pentacene Multilayers on Ag (111) Surface. J. Phys. Chem. C 2010, 114, 2724–2729. [Google Scholar] [CrossRef][Green Version]
- Cumpson, P.J.; Seah, M.P. Elastic Scattering Corrections in AES and XPS. II. Estimating Attenuation Lengths and Conditions Required for their Valid Use in Overlayer/Substrate Experiments. Surf. Interface Anal. 1997, 25, 430–446. [Google Scholar] [CrossRef]
- Han, S.H.; Yoo, S.; Kippelen, B.; Levi, D. Precise determination of optical properties of pentacene thin films grown on various substrates: Gauss-Lorentz model with effective medium approach. Appl. Phys. B Lasers Opt. 2011, 104, 139–144. [Google Scholar] [CrossRef]
- Faltermeier, D.; Gompf, B.; Dressel, M.; Tripathi, A.K.; Pflaum, J. Optical properties of pentacene thin films and single crystals. Phys. Rev. B Condens. Matter Mater. Phys. 2006, 74, 1–6. [Google Scholar] [CrossRef]
- Fujiki, A.; Miyake, Y.; Oshikane, Y.; Akai-Kasaya, M.; Saito, A.; Kuwahara, Y. STM-induced light emission from thin films of perylene derivatives on the HOPG and Au substrates. Nanoscale Res. Lett. 2011, 6, 1–8. [Google Scholar] [CrossRef]
- Schouwink, P.; Schäfer, A.; Seidel, C.; Fuchs, H. The influence of molecular aggregation on the device properties of organic light emitting diodes. Thin Solid Films 2000, 372, 163–168. [Google Scholar] [CrossRef]
- Oltean, M.; Calborean, A.; Mile, G.; Vidrighin, M.; Iosin, M.; Leopold, L.; Maniu, D.; Leopold, N.; Chis, V. Absorption spectra of PTCDI: A combined UV–Vis and TD-DFT study. Spectrochim. Acta Part A Mol. Biomol. Spectrosopy 2012, 97, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Emanuelsson, C.; Johansson, L.S.O.; Zhang, H.M. Photoelectron spectroscopy studies of PTCDI on Sn/Si(111)-23 × 23. Chem. Phys. 2020, 539, 110973. [Google Scholar] [CrossRef]
- Dell’Angela, M.; Anniyev, T.; Beye, M.; Coffee, R.; Föhlisch, A.; Gladh, J.; Kaya, S.; Katayama, T.; Krupin, O.; Nilsson, A.; et al. Vacuum space charge effects in sub-picosecond soft X-ray photoemission on a molecular adsorbate layer. Struct. Dyn. 2015, 2, 025101. [Google Scholar] [CrossRef] [PubMed]
- Deibel, C.; Strobel, T.; Dyakonov, V. Role of the Charge Transfer State in Organic Donor-Acceptor Solar Cells. Adv. Mater. 2010, 22, 4097–4111. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.-L.; Ligges, M.; Jailaubekov, A.; Kaake, L.; Miaja-Avila, L.; Zhu, X.-Y. Observing the Multiexciton State in Singlet Fission and Ensuing Ultrafast Multielectron Transfer. Science 2011, 334, 1541–1545. [Google Scholar] [CrossRef]
- Conrad-Burton, F.S.; Liu, T.; Geyer, F.; Costantini, R.; Schlaus, A.P.; Spencer, M.S.; Wang, J.; Sánchez, R.H.; Zhang, B.; Xu, Q.; et al. Controlling Singlet Fission by Molecular Contortion. J. Am. Chem. Soc. 2019, 141, 13143–13147. [Google Scholar] [CrossRef]
- Eaton, S.W.; Shoer, L.E.; Karlen, S.D.; Dyar, S.M.; Margulies, E.A.; Veldkamp, B.S.; Ramanan, C.; Hartzler, D.A.; Savikhin, S.; Marks, T.J.; et al. Singlet Exciton Fission in Polycrystalline Thin Films of a Slip-Stacked Perylenediimide. J. Am. Chem. Soc. 2013, 135, 14701–14712. [Google Scholar] [CrossRef]
- Le, A.K.; Bender, J.A.; Arias, D.H.; Cotton, D.E.; Johnson, J.C.; Roberts, S.T. Singlet Fission Involves an Interplay between Energetic Driving Force and Electronic Coupling in Perylenediimide Films. J. Am. Chem. Soc. 2018, 140, 814–826. [Google Scholar] [CrossRef]
- Zhu, X.; Wang, K.; He, J.; Zhang, L.; Yu, H.; He, D.; Hu, B. Exploring Deep and Shallow Trap States in a Non-Fullerene Acceptor ITIC-Based Organic Bulk Heterojunction Photovoltaic System. J. Phys. Chem. C 2019, 123, 20691–20697. [Google Scholar] [CrossRef]
- Haneef, H.F.; Zeidell, A.M.; Jurchescu, O.D. Charge carrier traps in organic semiconductors: A review on the underlying physics and impact on electronic devices. J. Mater. Chem. C 2020, 8, 759–787. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costantini, R.; Cossaro, A.; Morgante, A.; Dell’Angela, M. Light-Induced Charge Accumulation in PTCDI/Pentacene/Ag(111) Heterojunctions. Chemistry 2021, 3, 744-752. https://doi.org/10.3390/chemistry3030053
Costantini R, Cossaro A, Morgante A, Dell’Angela M. Light-Induced Charge Accumulation in PTCDI/Pentacene/Ag(111) Heterojunctions. Chemistry. 2021; 3(3):744-752. https://doi.org/10.3390/chemistry3030053
Chicago/Turabian StyleCostantini, Roberto, Albano Cossaro, Alberto Morgante, and Martina Dell’Angela. 2021. "Light-Induced Charge Accumulation in PTCDI/Pentacene/Ag(111) Heterojunctions" Chemistry 3, no. 3: 744-752. https://doi.org/10.3390/chemistry3030053
APA StyleCostantini, R., Cossaro, A., Morgante, A., & Dell’Angela, M. (2021). Light-Induced Charge Accumulation in PTCDI/Pentacene/Ag(111) Heterojunctions. Chemistry, 3(3), 744-752. https://doi.org/10.3390/chemistry3030053