Synthesis and Photostability of Cyclooctatetraene-Substituted Free Base Porphyrins

 , , , ,
, , , ,
Abstract
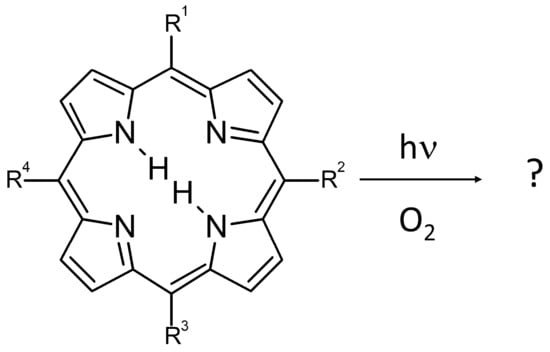
1. Introduction
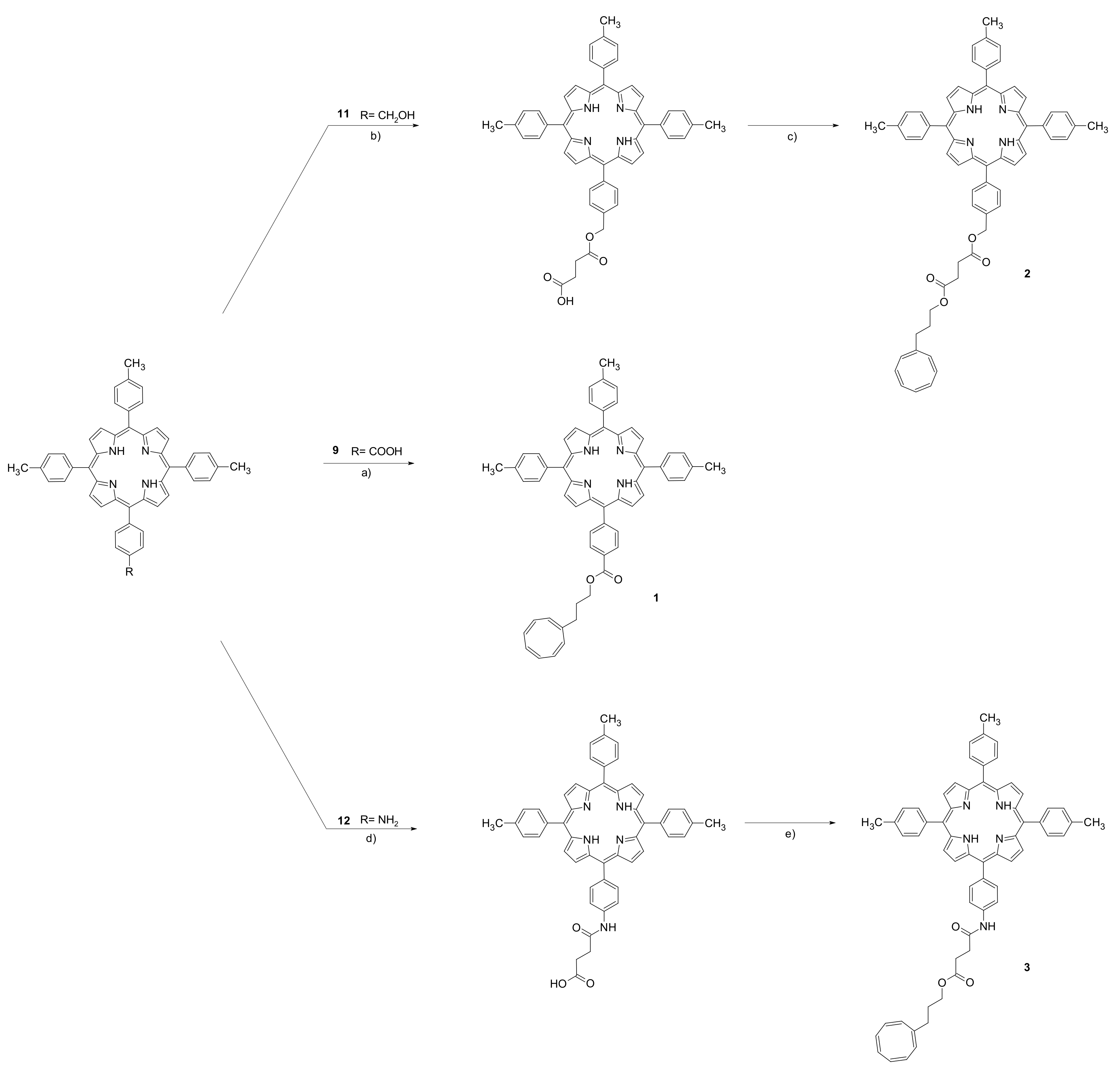
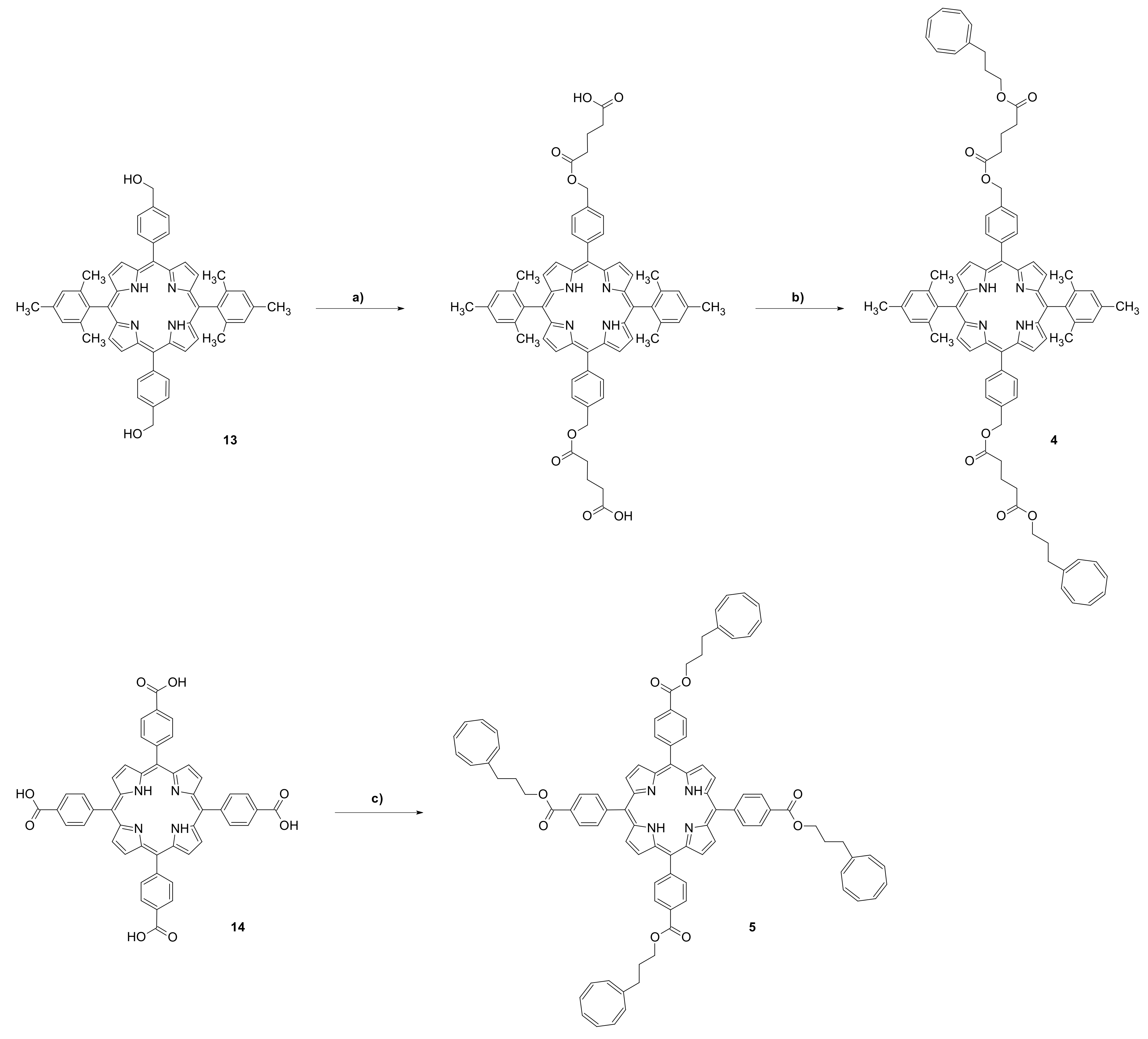
2. Materials and Methods
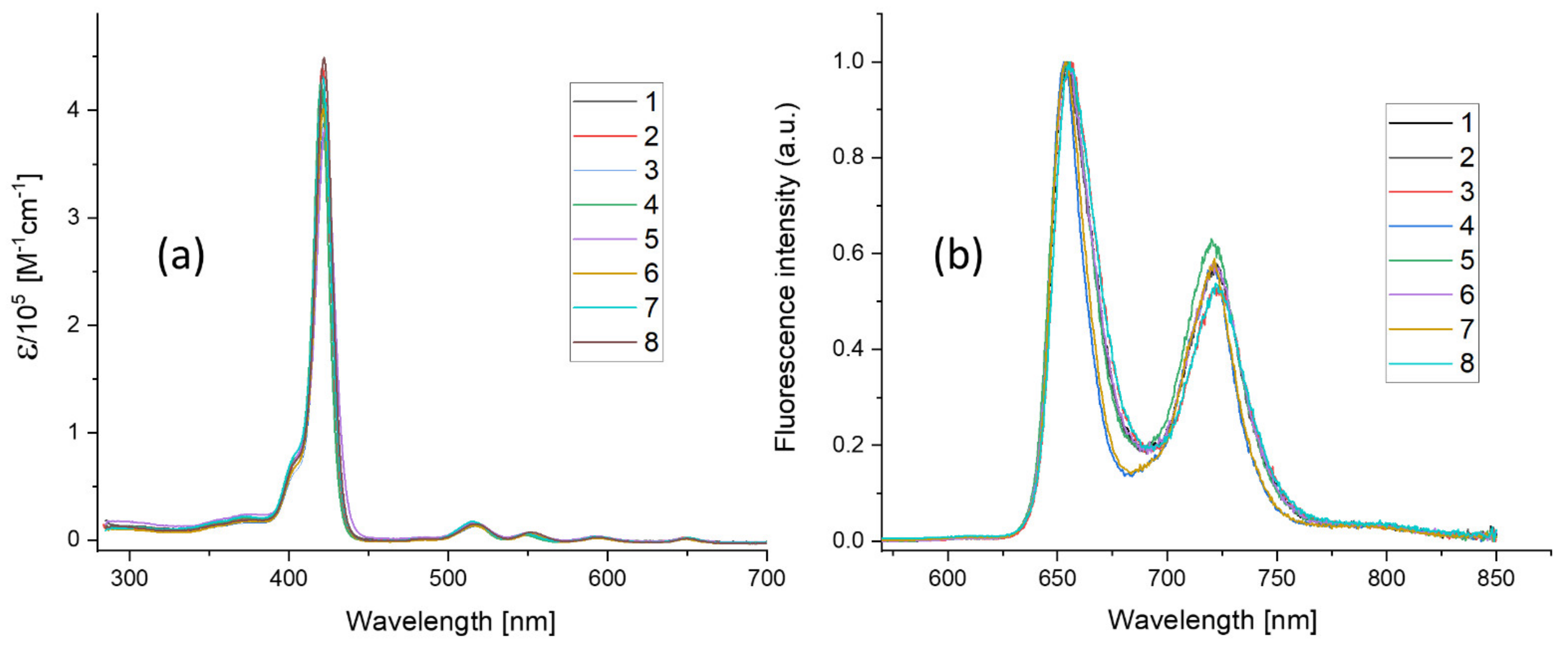
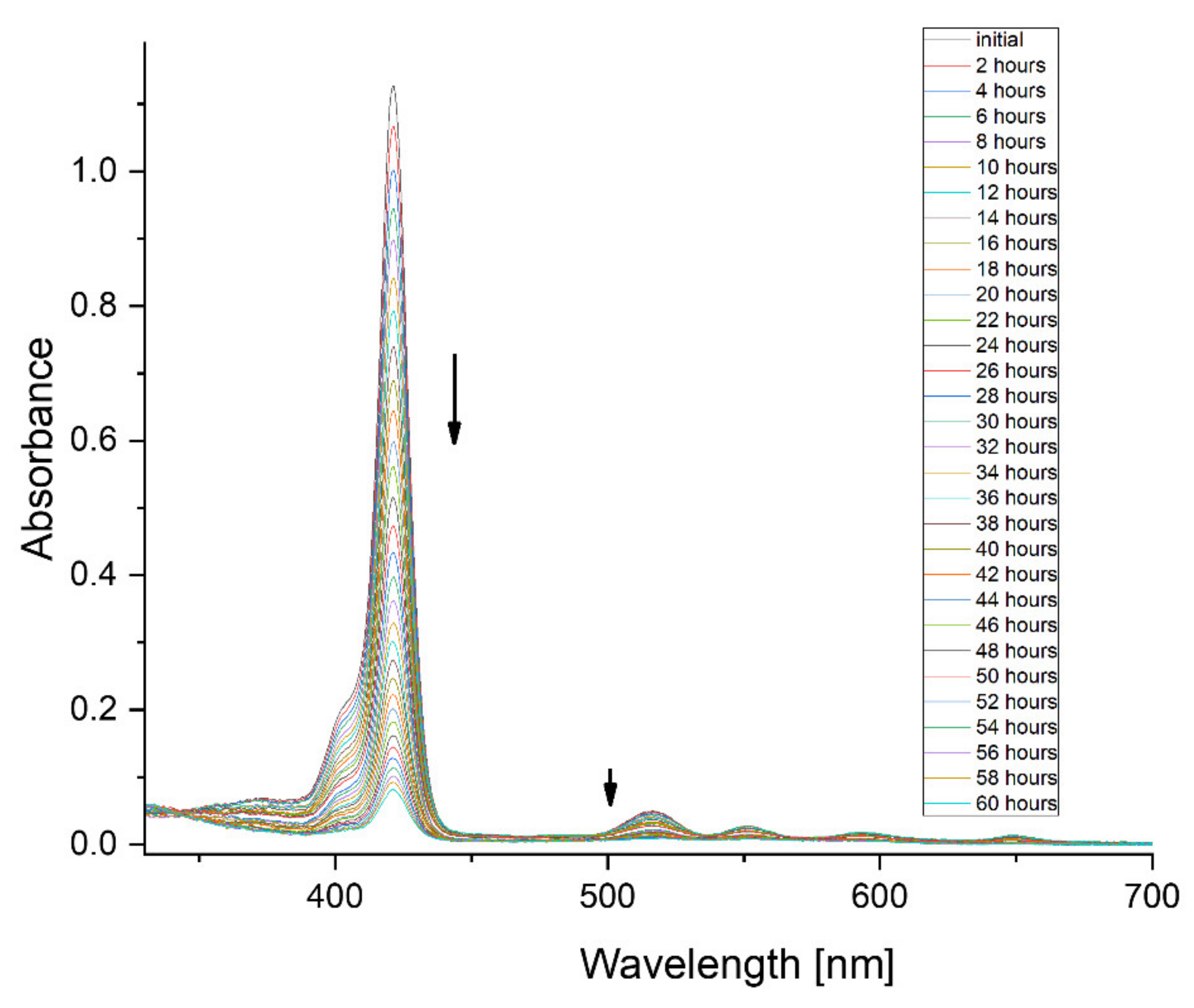
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Demchenko, A.P. Photobleaching of organic fluorophores: Quantitative characterization, mechanisms, protection. Methods Appl. Fluoresc. 2020, 8, 022001. [Google Scholar] [CrossRef] [PubMed]
- Ha, T.; Tinnefeld, P. Photophysics of Fluorescent Probes for Single-Molecule Biophysics and Super-Resolution Imaging. Ann. Rev. Phys. Chem. 2012, 63, 595–617. [Google Scholar] [CrossRef] [PubMed]
- Bregnhøj, M.; Prete, M.; Turkovic, V.; Petersen, A.U.; Nielsen, M.B.; Madsen, M.; Ogilby, P.R. Oxygen-dependent photophysics and photochemistry of prototypical compounds for organic photovoltaics: Inhibiting degradation initiated by singlet oxygen at a molecular level. Methods Appl. Fluoresc. 2019, 8, 014001. [Google Scholar] [CrossRef]
- Grabenhorst, L.; Trofymchuk, K.; Steiner, F.; Glembockyte, V.; Tinnefeld, P. Fluorophore photostability and saturation in the hotspot of DNA origami nanoantennas. Methods Appl. Fluoresc. 2020, 8, 024003. [Google Scholar] [CrossRef]
- Widengren, J.; Chmyrov, A.; Eggeling, C.; Löfdahl, P.-Å.; Seidel, C.A.M. Strategies to Improve Photostabilities in Ultrasensitive Fluorescence Spectroscopy. J. Phys. Chem. A 2007, 111, 429–440. [Google Scholar] [CrossRef]
- Liphardt, B.; Lüttke, W. Laser-Dyes. I. Bifluorophoric Laser-Dyes for Increase of the Efficiency of Dye-Lasers. Liebigs Ann. Chem. 1981, 1118–1138. [Google Scholar] [CrossRef]
- Liphardt, B.; Liphardt, B.; Lüttke, W. Laser dyes with intramolecular triplet quenching. Opt. Commun. 1981, 38, 207–210. [Google Scholar] [CrossRef]
- Schäfer, F.P.; Zhang, F.G.; Jethwa, J. Intramolecular TT-energy transfer in bifluorophoric laser dyes. Appl. Phys. B 1982, 28, 37–41. [Google Scholar] [CrossRef]
- Altman, R.B.; Terry, D.S.; Zhou, Z.; Zheng, Q.; Geggier, P.; Kolster, R.A.; Zhao, Y.; Javitch, J.A.; Warren, J.D.; Blanchard, S.C. Cyanine fluorophore derivatives with enhanced photostability. Nat. Methods 2012, 9, 68–71. [Google Scholar] [CrossRef]
- Tinnefeld, P.; Cordes, T. ‘Self-healing’ dyes: Intramolecular stabilization of organic fluorophores. Nat. Methods 2012, 9, 426–427. [Google Scholar] [CrossRef]
- Altman, R.B.; Zheng, Q.; Zhou, Z.; Terry, D.S.; Warren, J.D.; Blanchard, S.C. Enhanced photostability of cyanine fluorophores across the visible spectrum. Nat. Methods 2012, 9, 428–429. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Jockusch, S.; Zhou, Z.; Altman, R.B.; Warren, J.D.; Turro, N.J.; Blanchard, S.C. On the Mechanisms of Cyanine Fluorophore Photostabilization. J. Phys. Chem. Lett. 2012, 3, 2200–2203. [Google Scholar] [CrossRef] [PubMed]
- Van der Velde, J.H.M.; Ploetz, E.; Hiermaier, M.; Oelerich, J.; de Vries, J.W.; Roelfes, G.; Cordes, T. Mechanism of Intramolecular Photostabilization in Self-Healing Cyanine Fluorophores. ChemPhysChem 2013, 14, 4084–4093. [Google Scholar] [CrossRef] [PubMed]
- Van der Velde, J.H.M.; Oelerich, J.; Huang, J.Y.; Smit, J.H.; Hiermaier, M.; Ploetz, E.; Herrmann, A.; Roelfes, G.; Cordes, T. The Power of Two: Covalent Coupling of Photostabilizers for Fluorescence Applications. J. Phys. Chem. Lett. 2014, 5, 3792–3798. [Google Scholar] [CrossRef]
- Zheng, Q.; Juette, M.F.; Jockusch, S.; Wasserman, M.R.; Zhou, Z.; Altman, R.B.; Blanchard, S.C. Ultra-stable organic fluorophores for single-molecule research. Chem. Soc. Rev. 2014, 43, 1044–1056. [Google Scholar] [CrossRef]
- Zheng, Q.; Jockusch, S.; Zhou, Z.; Blanchard, S.C. The Contribution of Reactive Oxygen Species to the Photobleaching of Organic Fluorophores. Photochem. Photobiol. 2014, 90, 448–454. [Google Scholar] [CrossRef]
- Juette, M.F.; Terry, D.S.; Wasserman, M.R.; Zhou, Z.; Altman, R.B.; Zheng, Q.; Blanchard, S.C. The bright future of single-molecule fluorescence imaging. Curr. Opin. Chem. Biol. 2014, 20, 103–111. [Google Scholar] [CrossRef]
- Van der Velde, J.H.M.; Uusitalo, J.J.; Ugen, L.J.; Warszawik, E.M.; Herrmann, A.; Marrink, S.J.; Cordes, T. Intramolecular photostabilization via triplet-state quenching: Design principles to make organic fluorophores “self-healing”. Faraday Discuss. 2015, 184, 221–235. [Google Scholar] [CrossRef]
- Zheng, Q.S.; Jockusch, S.; Rodriguez-Calero, G.G.; Zhou, Z.; Zhao, H.; Altman, R.B.; Abruna, H.D.; Blanchard, S.C. Intra-molecular triplet energy transfer is a general approach to improve organic fluorophore photostability. Photochem. Photobiol. Sci. 2016, 15, 196–203. [Google Scholar] [CrossRef]
- Van der Velde, J.H.M.; Oelerich, J.; Huang, J.; Smit, J.H.; Aminian Jazi, A.; Galiani, S.; Kolmakov, K.; Gouridis, G.; Eggeling, C.; Herrmann, A.; et al. A simple and versatile design concept for fluorophore derivatives with intramolecular photostabilization. Nature Comm. 2016, 7, 10144. [Google Scholar] [CrossRef]
- Zheng, Q.; Jockusch, S.; Zhou, Z.; Altman, R.B.; Zhao, H.; Asher, W.; Holsey, M.; Mathiasen, S.; Geggier, P.; Javitch, J.A.; et al. Electronic tuning of self-healing fluorophores for live-cell and single-molecule imaging. Chem. Sci. 2017, 8, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Lavis, L.D. Development of photostable fluorophores for molecular imaging. Curr. Opin. Chem. Biol. 2017, 39, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Minoshima, M.; Kikuchi, K. Photostable and photoswitching fluorescent dyes for super-resolution imaging. JBIC J. Biol. Inorg. Chem. 2017, 22, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Van der Velde, J.H.M.; Smit, J.H.; Hebisch, E.; Punter, M.; Cordes, T. Self-healing dyes for super-resolution fluorescence microscopy. J. Phys. D Appl. Phys. 2018, 52, 034001. [Google Scholar] [CrossRef]
- Glembockyte, V.; Wieneke, R.; Gatterdam, K.; Gidi, Y.; Tampé, R.; Cosa, G. Tris-N-Nitrilotriacetic Acid Fluorophore as a Self-Healing Dye for Single-Molecule Fluorescence Imaging. J. Am. Chem. Soc. 2018, 140, 11006–11012. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Liu, L.; Jing, T.; Ruan, Z.; Yuan, P.; Yan, L. Self-Healing Organic Fluorophore of Cyanine-Conjugated Amphiphilic Polypeptide for Near-Infrared Photostable Bioimaging. ACS Appl. Mater. Interfaces 2018, 10, 14517–14530. [Google Scholar] [CrossRef]
- Gong, W.; Das, P.; Samanta, S.; Xiong, J.; Pan, W.; Gu, Z.; Zhang, J.; Qu, J.; Yang, Z. Redefining the photo-stability of common fluorophores with triplet state quenchers: Mechanistic insights and recent updates. Chem. Comm. 2019, 55, 8695–8704. [Google Scholar] [CrossRef]
- Smit, J.H.; van der Velde, J.H.M.; Huang, J.; Trauschke, V.; Henrikus, S.S.; Chen, S.; Eleftheriadis, N.; Warszawik, E.M.; Herrmann, A.; Cordes, T. On the impact of competing intra- and intermolecular triplet-state quenching on photobleaching and photoswitching kinetics of organic fluorophores. Phys. Chem. Chem. Phys. 2019, 21, 3721–3733. [Google Scholar] [CrossRef]
- Isselstein, M.; Zhang, L.; Glembockyte, V.; Brix, O.; Cosa, G.; Tinnefeld, P.; Cordes, T. Self-Healing Dyes—Keeping the Promise? J. Phys. Chem. Lett. 2020, 11, 4462–4480. [Google Scholar] [CrossRef]
- Pati, A.K.; El Bakouri, O.; Jockusch, S.; Zhou, Z.; Altman, R.B.; Fitzgerald, G.A.; Asher, W.B.; Terry, D.S.; Borgia, A.; Holsey, M.D.; et al. Tuning the Baird aromatic triplet-state energy of cyclooctatetraene to maximize the self-healing mechanism in organic fluorophores. Proc. Natl. Acad. Sci. USA 2020, 117, 24305–24315. [Google Scholar] [CrossRef]
- Yang, Z.; Li, L.; Ling, J.; Liu, T.; Huang, X.; Ying, Y.; Zhao, Y.; Zhao, Y.; Lei, K.; Chen, L.; et al. Cyclooctatetraene-conjugated cyanine mitochondrial probes minimize phototoxicity in fluorescence and nanoscopic imaging. Chem. Sci. 2020, 11, 8506–8516. [Google Scholar] [CrossRef]
- Ostapko, J.; Gorski, A.; Buczyńska, J.; Golec, B.; Nawara, K.; Kharchenko, A.; Listkowski, A.; Ceborska, M.; Pietrzak, M.; Waluk, J. Towards More Photostable, Brighter, and Less Phototoxic Chromophores: Synthesis and Properties of Porphyrins Functionalized with Cyclooctatetraene. Chem. Eur. J. 2020, 70, 16666–16675. [Google Scholar] [CrossRef] [PubMed]
- Pineiro, M.; Carvalho, A.L.; Pereira, M.M.; Gonsalves, A.M.A.R.; Arnaut, L.G.; Formosinho, S.J. Photoacoustic measurements of porphyrin triplet-state quantum yields and singlet-oxygen efficiencies. Chem. Eur. J. 1998, 11, 2299–2307. [Google Scholar] [CrossRef]
- Schmidt, R.; Tanielian, C.; Dunsbach, R.; Wolff, C. Phenalenone, a universal reference compound for the determination of quantum yields of singlet oxygen O2 (1Δg) sensitization. J. Photochem. Photobiol. A Chem. 1994, 79, 11–17. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Cavaleiro, J.A.S.; Görner, H.; Lacerda, P.S.S.; MacDonald, J.G.; Mark, G.; Neves, M.G.P.M.S.; Nohr, R.S.; Schuchmann, H.P.; van Sonntag, C.; Tome, A.C. Singlet oxygen formation and photostability of meso-tetraarylporphyrin derivatives and their copper complexes. J. Photochem. Photobiol. A Chem. 2001, 144, 131–140. [Google Scholar] [CrossRef]
- Cavaleiro, J.A.S.; Hewlins, M.J.E.; Jackson, A.H.; Neves, G.P.M.S. Structures of the Ring-Opened Oxidation Products from meso-Tetraphenylporphyrins. J. Chem. Soc. Chem. Comm. 1986, 142–144. [Google Scholar] [CrossRef]
- Cavaleiro, J.A.S.; Neves, M.G.P.S.; Hewlins, M.J.E.; Jackson, A.H. The Photooxidation of Meso-Tetraphenylporphyrins. J. Chem. Soc. Perkin Trans. 1 1990, 7, 1937–1943. [Google Scholar] [CrossRef]
- Smith, K.M.; Brown, S.B.; Troxler, R.F.; Lai, J.J. Mechanism of photo-oxygenation of meso-tetraphenylporphyrin metal complexes. Tetrahedron Lett. 1980, 21, 2763–2766. [Google Scholar] [CrossRef]
- Smith, K.M.; Brown, S.B.; Troxler, R.F.; Lai, J.J. Photooxygenation of meso-tetraphenylporphyrin complexes. Photochem. Photobiol. 1982, 36, 147–152. [Google Scholar] [CrossRef]
- Wojaczyński, J.; Popiel, M.; Szterenberg, L.; Latos-Grażyński, L. Common Origin, Common Fate: Regular Porphyrin and N-Confused Porphyrin Yield an Identical Tetrapyrrolic Degradation Product. J. Org. Chem. 2011, 76, 9956–9961. [Google Scholar] [CrossRef]
- Silva, A.M.S.; Neves, M.G.P.M.S.; Martins, R.R.L.; Cavaleiro, J.A.S.; Boschi, T.; Tagliatesta, P. Photo-oxygenation of meso-tetraphenylporphyrin derivatives: The influence of the substitution pattern and characterization of the reaction products. J. Porphyr. Phthalocyanines 1998, 2, 45–51. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
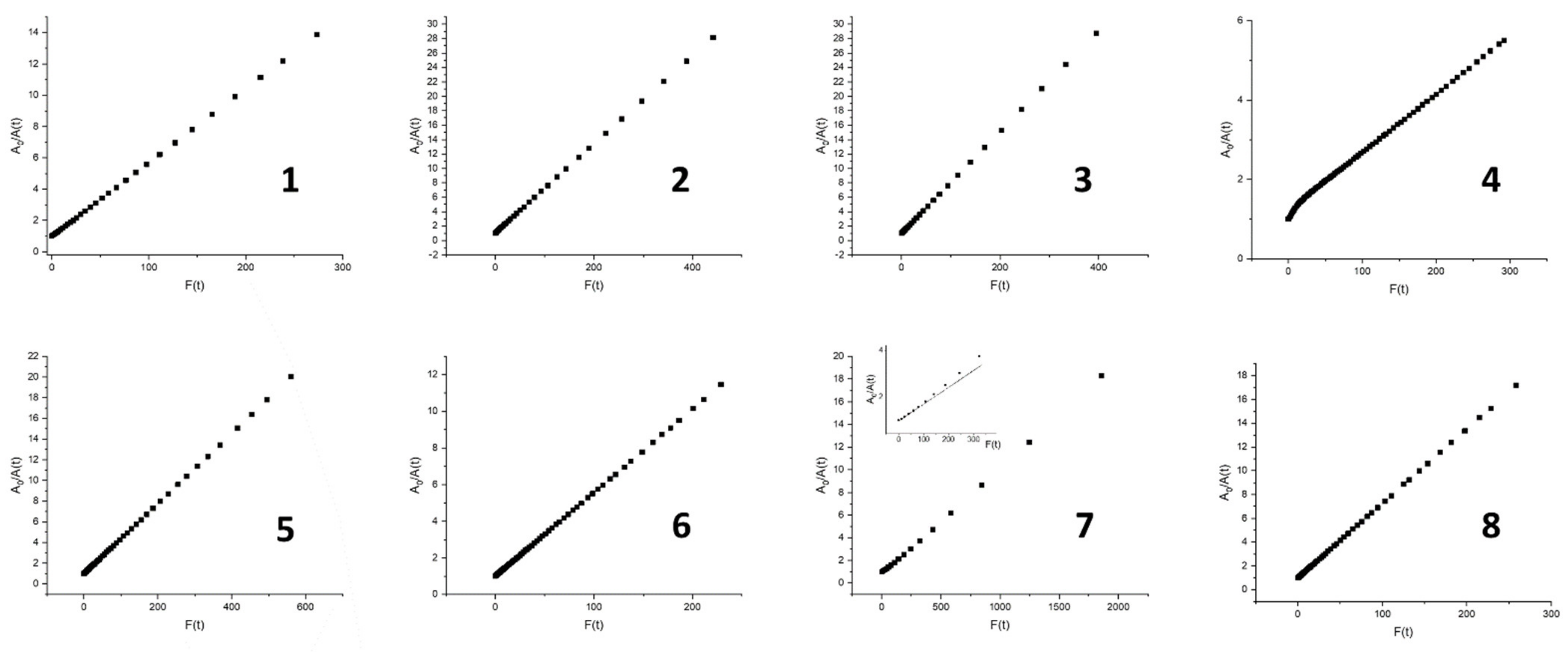
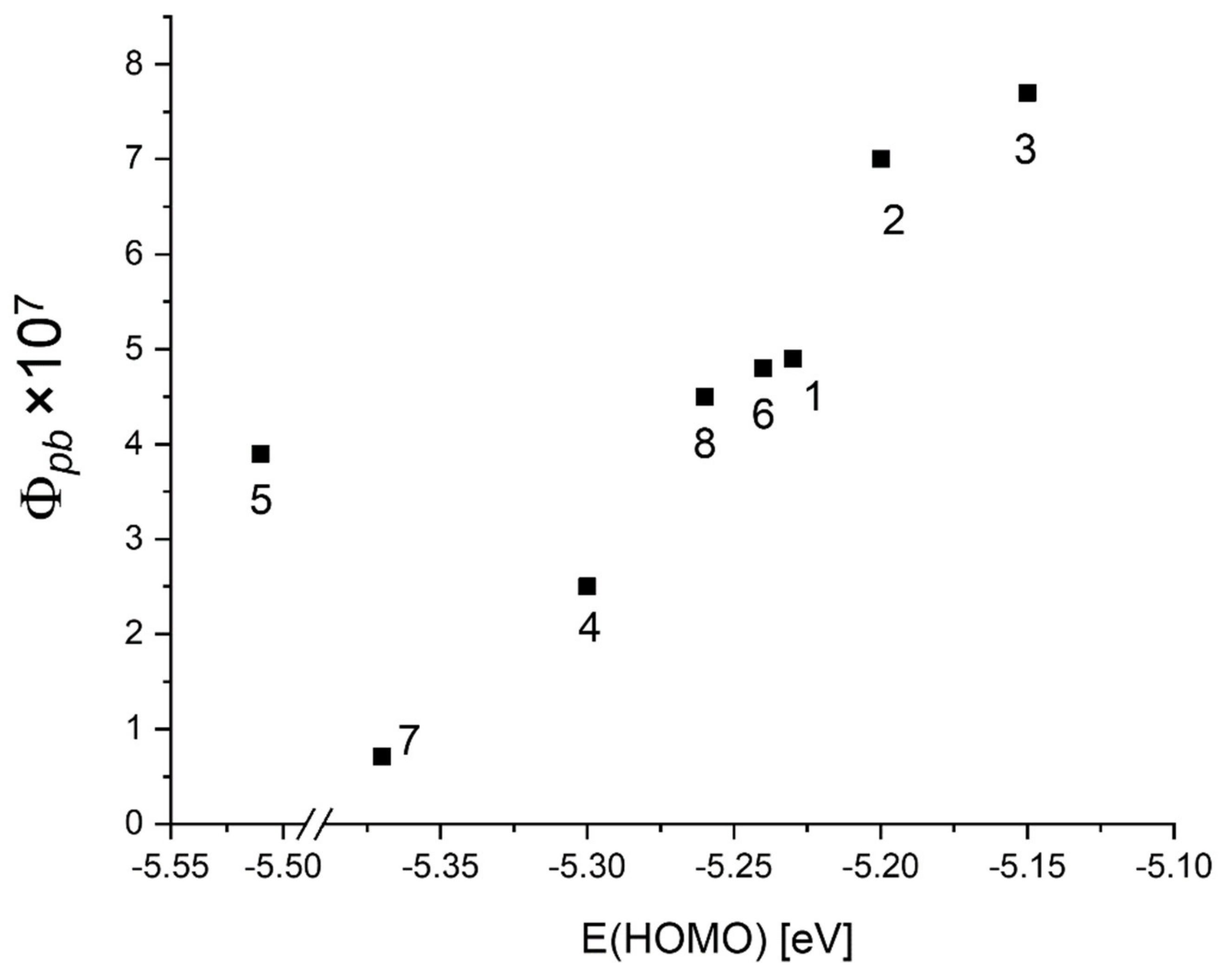
| Abs. Maxima [nm] | Abs. Coeff [M−1 cm−1] | Φfl | S1 Lifetime [ns] | T1 Lifetime Aerated Degassed [ns] [μs] | ΦΔ | Φpb | |
|---|---|---|---|---|---|---|---|
| 1 | 421 516 551.5 593.5 649.5 | 416,860 17,558 9056 5016 3901 | 0.12 | 9.7 | 293 458 | 0.78 | 4.9 × 10−7 |
| 2 | 420.5 516 551 592.5 648.5 | 440,270 17,921 9077 5035 4014 | 0.12 | 9.6 | 297 14.7 | 0.7 | 7.0 × 10−7 |
| 3 | 421.5 517 552 594 649.5 | 389,104 15,590 8793 4486 3872 | 0.12 | 9.5 | 298 112 | 0.75 | 7.7 × 10−7 |
| 4 | 420 514.5 548 591.5 650 | 426,346 18,376 7350 5052 4380 | 0.13 | 10.3 | 386 9.3 | 0.72 | 2.5 × 10−7 |
| 5 | 422 516 550.5 593 649 | 383,662 15,931 7171 4281 2949 | 0.13 | 10.2 | 329 257 | 0.66 | 3.9 × 10−7 |
| 6 | 421 516 551 593.5 649.5 | 401,207 16,709 8627 4795 3735 | 0.12 | 9.6 | 297 645 | 0.78 | 4.8 × 10−7 |
| 7 | 420.5 515 548.5 592.5 650.5 | 431,582 20,127 8252 5515 4621 | 0.14 | 10.4 | 509 1070 | 0.78 | 7.1 × 10−8 |
| 8 | 421.5 517 552.5 593.5 650.5 | 449,392 15,922 8839 4545 3849 | 0.13 | 9.5 | 294 820 | 0.79 | 4.5 × 10−7 |
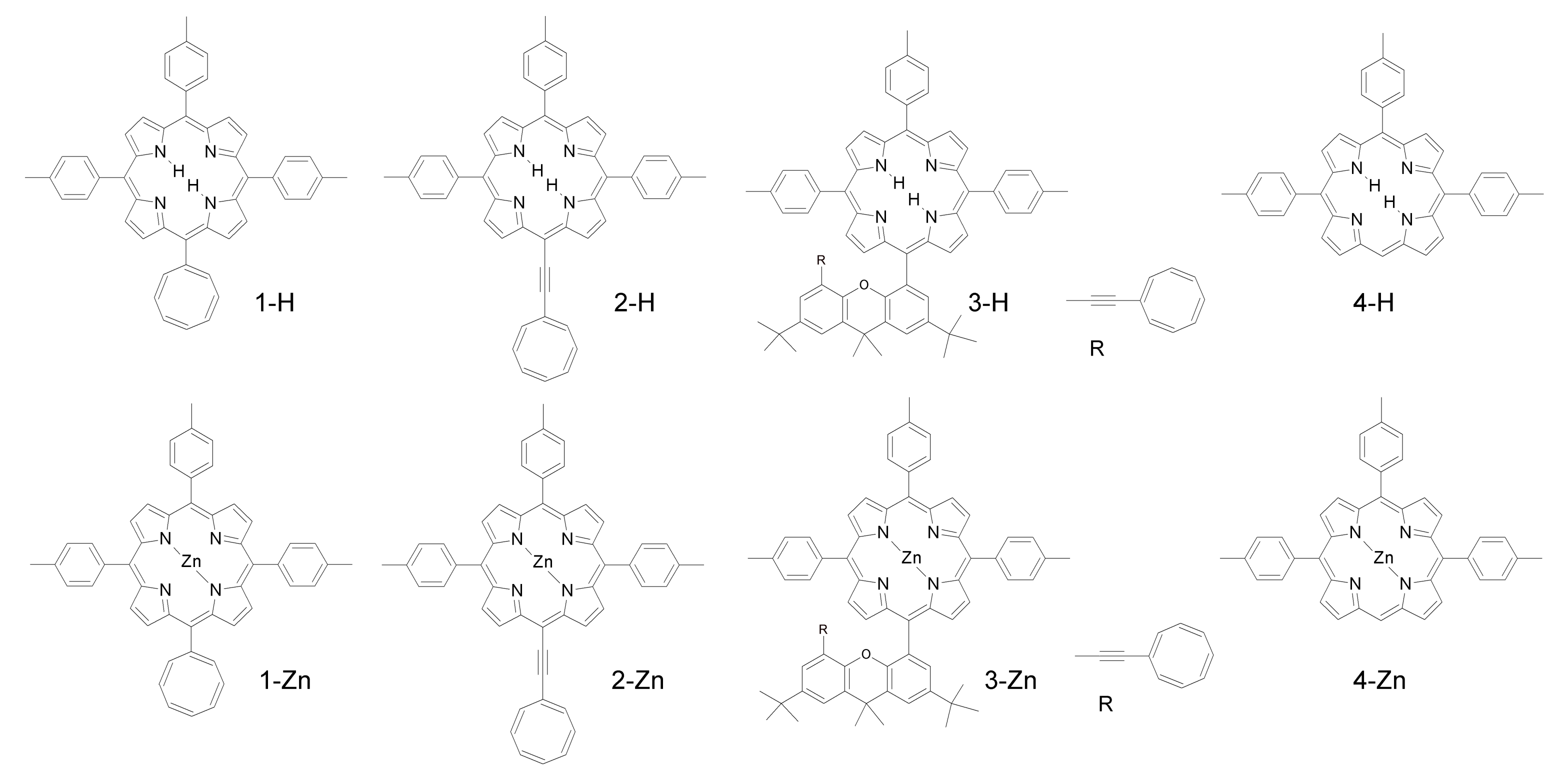
| Compound | kintr [106 s−1] | kq[O2] [106 s−1] |
|---|---|---|
| 1-H | 5.6 | 3.8 |
| 2-H | 5.4 | 7.6 |
| 3-H | 5.2 | 6.8 |
| 4-H | 0.0048 | 4 |
| 1-Zn | 5.6 | 4.1 |
| 2-Zn | 5.2 | 7.8 |
| 3-Zn | 17 | 4 |
| 4-Zn | 0.0038 | 2.8 |
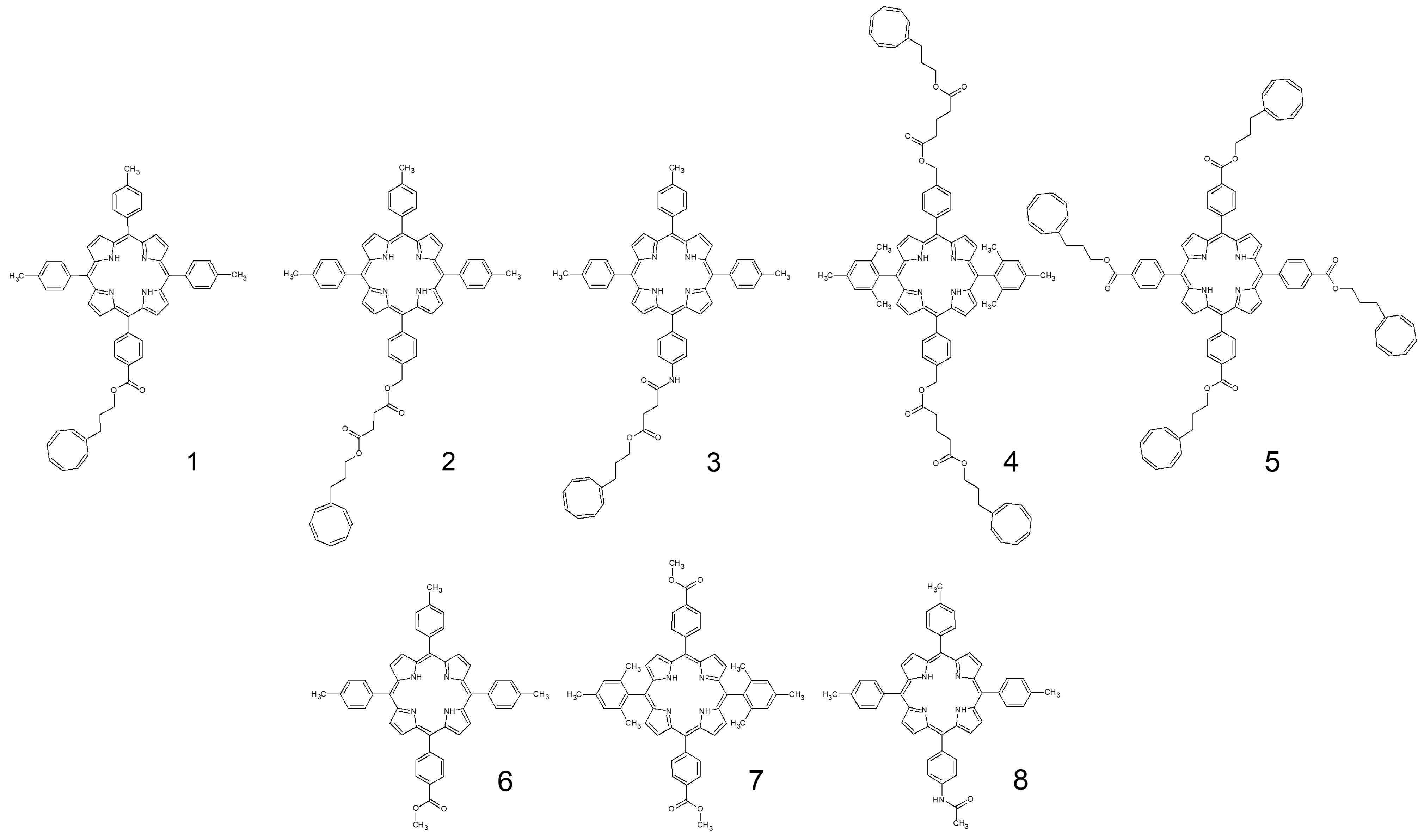
| 1 | 0.0022 | 3.4 |
| 2 | 0.068 | 3.4 |
| 3 | 0.0089 | 3.4 |
| 4 | 0.11 | 2.5 |
| 5 | 0.0039 | 3 |
| 6 | 0.0016 | 3.4 |
| 7 | 0.00093 | 2 |
| 8 | 0.0012 | 3.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buczyńska, J.; Gajewska, A.; Gorski, A.; Golec, B.; Nawara, K.; Rybakiewicz, R.; Waluk, J. Synthesis and Photostability of Cyclooctatetraene-Substituted Free Base Porphyrins. Chemistry 2021, 3, 104-115. https://doi.org/10.3390/chemistry3010008
Buczyńska J, Gajewska A, Gorski A, Golec B, Nawara K, Rybakiewicz R, Waluk J. Synthesis and Photostability of Cyclooctatetraene-Substituted Free Base Porphyrins. Chemistry. 2021; 3(1):104-115. https://doi.org/10.3390/chemistry3010008
Chicago/Turabian StyleBuczyńska, Joanna, Agnieszka Gajewska, Aleksander Gorski, Barbara Golec, Krzysztof Nawara, Renata Rybakiewicz, and Jacek Waluk. 2021. "Synthesis and Photostability of Cyclooctatetraene-Substituted Free Base Porphyrins" Chemistry 3, no. 1: 104-115. https://doi.org/10.3390/chemistry3010008
APA StyleBuczyńska, J., Gajewska, A., Gorski, A., Golec, B., Nawara, K., Rybakiewicz, R., & Waluk, J. (2021). Synthesis and Photostability of Cyclooctatetraene-Substituted Free Base Porphyrins. Chemistry, 3(1), 104-115. https://doi.org/10.3390/chemistry3010008