Effects of Substituents on Photophysical and CO-Photoreleasing Properties of 2,6-Substituted meso-Carboxy BODIPY Derivatives †

 , ,
, ,  and
and
Abstract
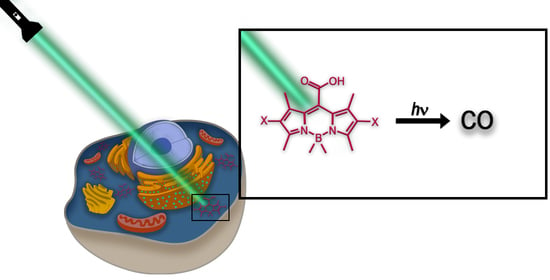
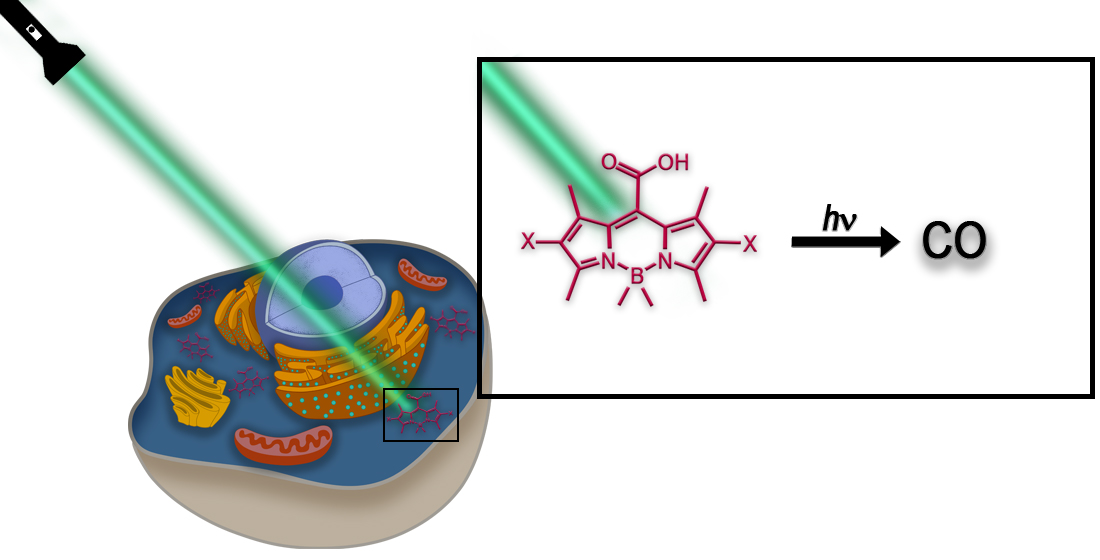
1. Introduction
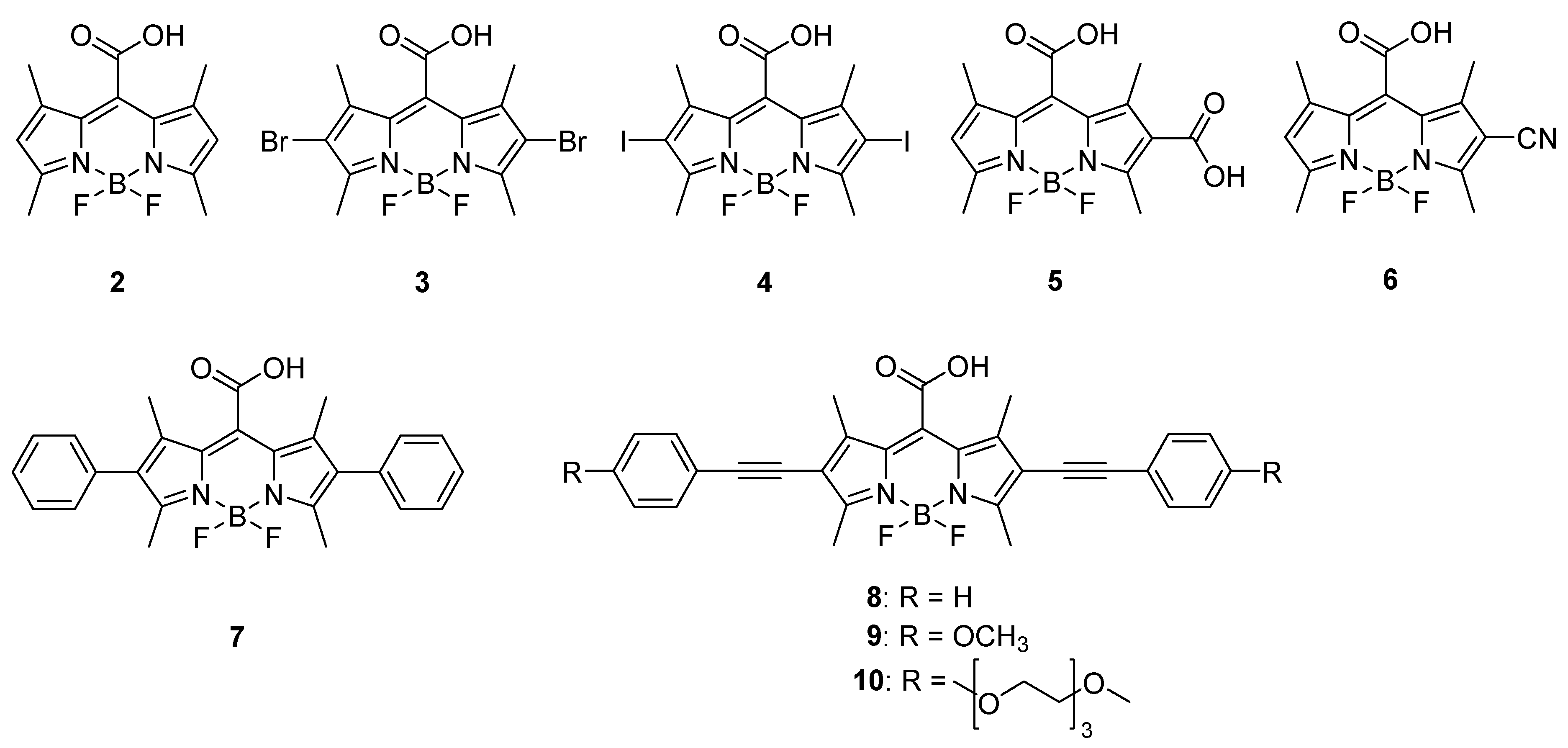
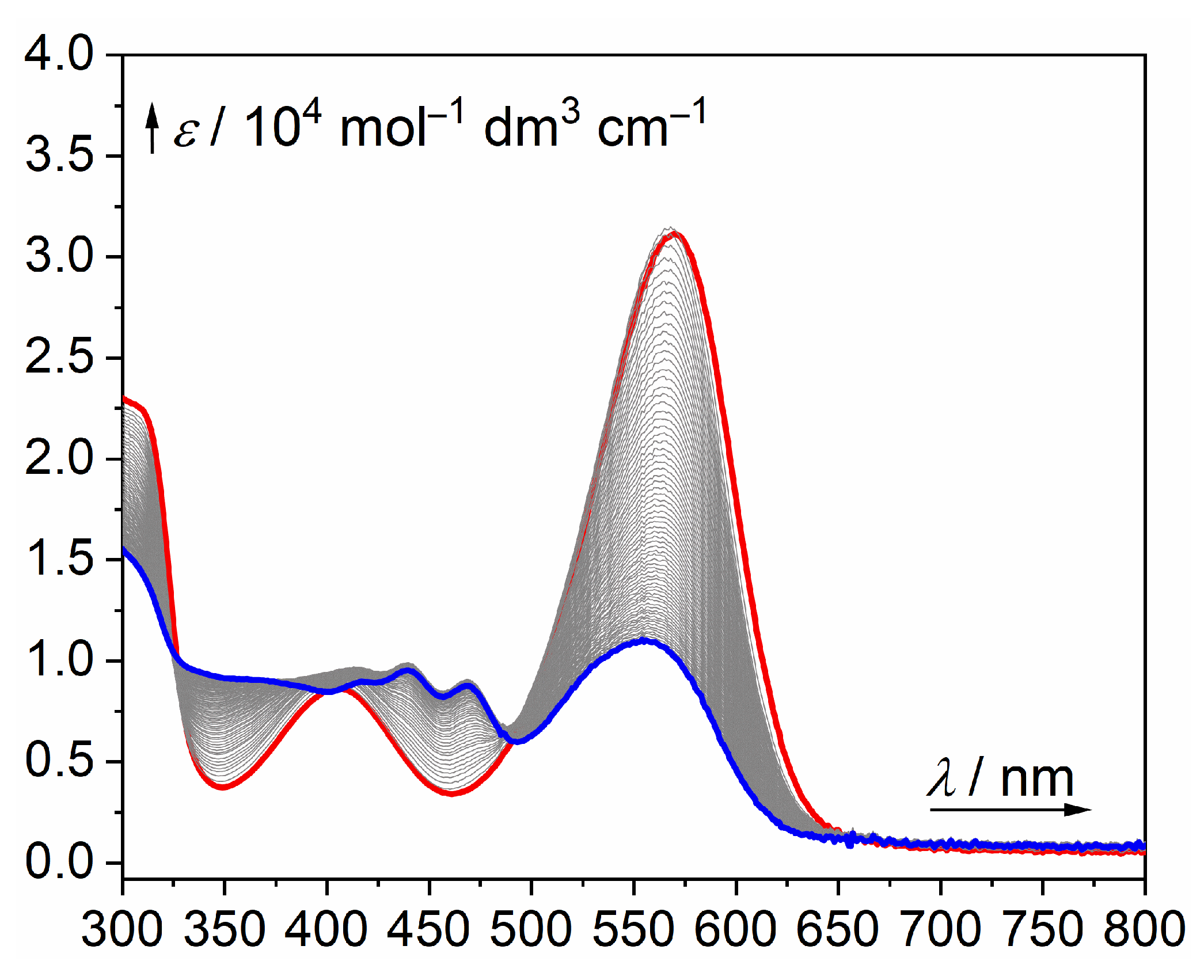
2. Results and Discussion
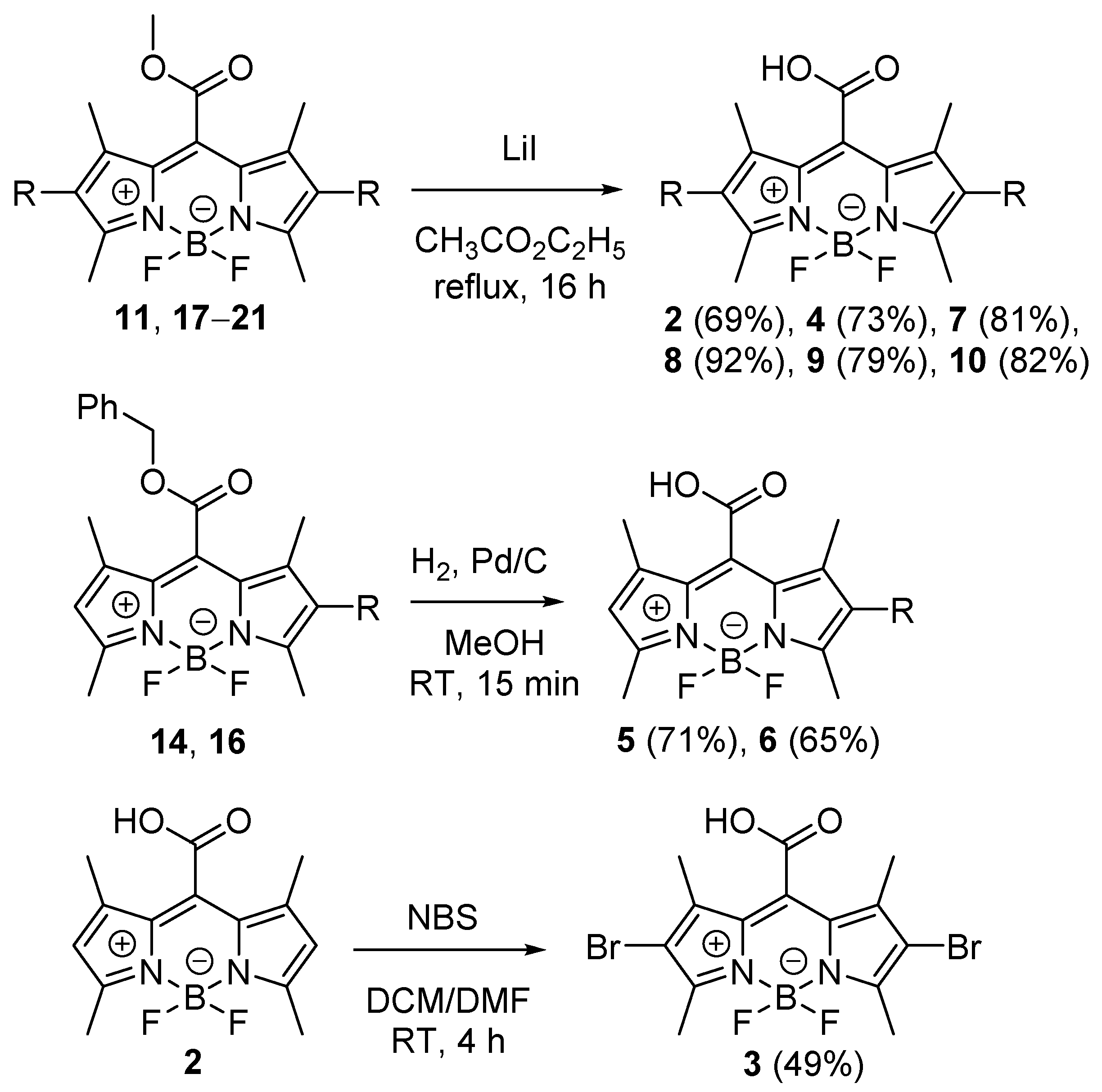
3. Experimental Section

Synthesis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wood, T.E.; Thompson, A. Advances in the chemistry of dipyrrins and their complexes. Chem. Rev. 2007, 107, 1831–1861. [Google Scholar] [CrossRef]
- Ulrich, G.; Ziessel, R.; Harriman, A. The chemistry of fluorescent BODIPY dyes: Versatility unsurpassed. Angew. Chem. Int. Ed. 2008, 47, 1184–1201. [Google Scholar] [CrossRef]
- Benniston, A.C.; Copley, G. Lighting the way ahead with boron dipyrromethene (BODIPY) dyes. Phys. Chem. Chem. Phys. 2009, 11, 4124–4131. [Google Scholar] [CrossRef] [PubMed]
- Deniz, E.; Isbasar, G.C.; Bozdemir, O.A.; Yildirim, L.T.; Siemiarczuk, A.; Akkaya, E.U. Bidirectional switching of near IR emitting boradiazaindacene fluorophores. Org. Lett. 2008, 10, 3401–3403. [Google Scholar] [CrossRef]
- Niu, L.-Y.; Guan, Y.-S.; Chen, Y.-Z.; Wu, L.-Z.; Tung, C.-H.; Yang, Q.-Z. BODIPY-based ratiometric fluorescent sensor for highly selective detection of glutathione over cysteine and homocysteine. J. Am. Chem. Soc. 2012, 134, 18928–18931. [Google Scholar] [CrossRef] [PubMed]
- Ikawa, Y.; Moriyama, S.; Furuta, H. Facile syntheses of BODIPY derivatives for fluorescent labeling of the 3′ and 5′ Ends of RNAs. Anal. Biochem. 2008, 378, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Xu, G.; Prasad, P.N. Conformationally restricted dipyrromethene boron difluoride (BODIPY) dyes: Highly fluorescent, multicolored probes for cellular imaging. Chem. Eur. J. 2008, 14, 5812–5819. [Google Scholar] [CrossRef] [PubMed]
- Kamkaew, A.; Lim, S.H.; Lee, H.B.; Kiew, L.V.; Chung, L.Y.; Burgess, K. BODIPY dyes in photodynamic therapy. Chem. Soc. Rev. 2013, 42, 77–88. [Google Scholar] [CrossRef]
- Bessette, A.; Hanan, G.S. Design, synthesis and photophysical studies of dipyrromethene-based materials: Insights into their applications in organic photovoltaic devices. Chem. Soc. Rev. 2014, 43, 3342–3405. [Google Scholar] [CrossRef]
- Boens, N.; Leen, V.; Dehaen, W. Fluorescent indicators based on BODIPY. Chem. Soc. Rev. 2012, 41, 1130–1172. [Google Scholar] [CrossRef]
- Loudet, A.; Burgess, K. BODIPY dyes and their derivatives: Syntheses and spectroscopic properties. Chem. Rev. 2007, 107, 4891–4932. [Google Scholar] [CrossRef]
- Boens, N.; Verbelen, B.; Dehaen, W. Postfunctionalization of the BODIPY core: Synthesis and spectroscopy. Eur. J. Org. Chem. 2015, 2015, 6577–6595. [Google Scholar] [CrossRef]
- DeRosa, M. Photosensitized singlet oxygen and its applications. Coord. Chem. Rev. 2002, 233, 351–371. [Google Scholar] [CrossRef]
- Al Anshori, J.; Slanina, T.; Palao, E.; Klán, P. The internal heavy-atom effect on 3-Phenylselanyl and 3-Phenyltellanyl bodipy derivatives studied by transient absorption spectroscopy. Photochem. Photobiol. Sci. 2016, 15, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Yogo, T.; Urano, Y.; Ishitsuka, Y.; Maniwa, F.; Nagano, T. Highly efficient and photostable photosensitizer based on BODIPY chromophore. J. Am. Chem. Soc. 2005, 127, 12162–12163. [Google Scholar] [CrossRef] [PubMed]
- Michl, J.; Bonačić-Koutecký, V. Electronic Aspects of Organic Photochemistry; Wiley: Chichester, UK, 1990. [Google Scholar]
- Adarsh, N.; Avirah, R.R.; Ramaiah, D. Tuning photosensitized singlet oxygen generation efficiency of Novel Aza-BODIPY dyes. Org. Lett. 2010, 12, 5720–5723. [Google Scholar] [CrossRef] [PubMed]
- Awuah, S.G.; Polreis, J.; Biradar, V.; You, Y. Singlet oxygen generation by Novel NIR BODIPY dyes. Org. Lett. 2011, 13, 3884–3887. [Google Scholar] [CrossRef]
- García-Gallego, S.; Bernardes, G.J.L. Carbon-monoxide-releasing molecules for the delivery of therapeutic CO in vivo. Angew. Chem. Int. Ed. 2014, 53, 9712–9721. [Google Scholar] [CrossRef]
- Motterlini, R.; Otterbein, L.E. The therapeutic potential of carbon monoxide. Nat. Rev. Drug Discov. 2010, 9, 728–743. [Google Scholar] [CrossRef]
- Romão, C.C.; Blättler, W.A.; Seixas, J.D.; Bernardes, G.J.L. Developing drug molecules for therapy with carbon monoxide. Chem. Soc. Rev. 2012, 41, 3571–3583. [Google Scholar] [CrossRef]
- Chakraborty, I.; Carrington, S.J.; Mascharak, P.K. Design strategies to improve the sensitivity of photoactive metal Carbonyl complexes (photoCORMs) to visible light and their potential as CO-donors to biological targets. Acc. Chem. Res. 2014, 47, 2603–2611. [Google Scholar] [CrossRef]
- Rudolf, P.; Kanal, F.; Knorr, J.; Nagel, C.; Niesel, J.; Brixner, T.; Schatzschneider, U.; Nuernberger, P. Ultrafast photochemistry of a manganese-tricarbonyl CO-releasing molecule (CORM) in aqueous solution. J. Phys. Chem. Lett. 2013, 4, 596–602. [Google Scholar] [CrossRef]
- Slanina, T.; Sebej, P. Visible-light-activated photoCORMs: Rational design of CO-releasing organic molecules absorbing in the tissue-transparent window. Photochem. Photobiol. Sci. 2018, 17, 692–710. [Google Scholar] [CrossRef] [PubMed]
- Marhenke, J.; Trevino, K.; Works, C. The chemistry, biology and design of photochemical CO releasing Molecules and the efforts to detect CO for biological applications. Coord. Chem. Rev. 2016, 306, 533–543. [Google Scholar] [CrossRef]
- Schatzschneider, U. PhotoCORMs: Light-triggered release of carbon monoxide from the coordination sphere of transition metal complexes for biological applications. Inorg. Chim. Acta 2011, 374, 19–23. [Google Scholar] [CrossRef]
- Ling, K.; Men, F.; Wang, W.C.; Zhou, Y.Q.; Zhang, H.W.; Ye, D.W. Carbon monoxide and its controlled release: Therapeutic application, detection, and development of carbon monoxide releasing molecules (CORMs). J. Med. Chem. 2018, 61, 2611–2635. [Google Scholar] [CrossRef]
- Weinstain, R.; Slanina, T.; Kand, D.; Klán, P. Visible-to-NIR-light activated release: From small molecules to nanomaterials. Chem. Rev. 2020, 120, 13135–13272. [Google Scholar] [CrossRef] [PubMed]
- Cheong, W.F.; Prahl, S.A.; Welch, A.J. A review of the optical properties of biological tissues. IEEE J. Quantum Electron. 1990, 26, 2166–2185. [Google Scholar] [CrossRef]
- Tuchin, V.V. Tissue Optics: Light Scattering Methods and Instruments for Medical Diagnostics; SPIE Press: Bellingham, WA, USA, 2015. [Google Scholar]
- Antony, L.A.P.; Slanina, T.; Šebej, P.; Šolomek, T.; Klán, P. Fluorescein analogue xanthene-9-carboxylic acid: A transition-metal-free CO releasing molecule activated by green light. Org. Lett. 2013, 15, 4552–4555. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Štacko, P.; Nachtigallová, D.; Klán, P. Mechanisms of orthogonal photodecarbonylation reactions of 3-hydroxyflavone-based acid–base forms. J. Org. Chem. 2020, 85, 3527–3537. [Google Scholar] [CrossRef]
- Štacková, L.; Russo, M.; Muchová, L.; Orel, V.; Vítek, L.; Štacko, P.; Klán, P. Cyanine-flavonol hybrids for near-infrared light-activated delivery of carbon monoxide. Chem. Eur. J. 2020, 26, 13184–13190. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, L.S.; Benninghoff, A.D.; Berreau, L.M. Development of triggerable, trackable, and targetable carbon monoxide releasing molecules. Acc. Chem. Res. 2020, 53, 2273–2285. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.N.; Richards, J.M.; Esquer, H.J.; Benninghoff, A.D.; Arif, A.M.; Berreau, L.M. A structurally-tunable 3-hydroxyflavone motif for visible light-induced carbon monoxide-releasing molecules (CORMs). ChemistryOpen 2015, 4, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Palao, E.; Slanina, T.; Muchová, L.; Šolomek, T.; Vítek, L.; Klán, P. Transition-metal-free CO-releasing BODIPY derivatives activatable by visible to NIR light as promising bioactive molecules. J. Am. Chem. Soc. 2015, 138, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Zeng, L.; Kang, N.-Y.; Huang, K.-W.; Wang, L.; Zeng, Z.; Chang, Y.-T.; Wu, J. meso-Ester and carboxylic acid substituted BODIPYs with far-red and near-infrared emission for bioimaging applications. Chem. Eur. J. 2014, 20, 2301–2310. [Google Scholar] [CrossRef]
- Orte, A.; Debroye, E.; Ruedas-Rama, M.J.; Garcia-Fernandez, E.; Robinson, D.; Crovetto, L.; Talavera, E.M.; Alvarez-Pez, J.M.; Leen, V.; Verbelen, B.; et al. Effect of the substitution position (2, 3 or 8) on the spectroscopic and photophysical properties of BODIPY dyes with a Phenyl, Styryl or Phenylethynyl group. RSC Adv. 2016, 6, 102899–102913. [Google Scholar] [CrossRef]
- Lu, H.; Mack, J.; Yang, Y.; Shen, Z. Structural modification strategies for the rational design of red/NIR region BODIPYs. Chem. Soc. Rev. 2014, 43, 4778–4823. [Google Scholar] [CrossRef]
- Lu , H.; Wang, Q.; Gai, L.; Li, Z.; Deng, Y.; Xiao, X.; Lai, G.; Shen, Z. Tuning the solid-state luminescence of BODIPY derivatives with Bulky Arylsilyl groups: Synthesis and spectroscopic properties. Chem. Eur. J. 2012, 18, 7852–7861. [Google Scholar] [CrossRef]
- Leen, V.; Leemans, T.; Boens, N.; Dehaen, W. 2-and 3-Monohalogenated BODIPY dyes and their functionalized analogues: Synthesis and spectroscopy. Eur. J. Org. Chem. 2011, 2011, 4386–4396. [Google Scholar] [CrossRef]
- Jiao, L.; Yu, C.; Wang, J.; Briggs, E.A.; Besley, N.A.; Robinson, D.; Ruedas-Rama, M.J.; Orte, A.; Crovetto, L.; Talavera, E.M.; et al. Unusual spectroscopic and photophysical properties of meso-tert-Butyl BODIPY in comparison to related alkylated BODIPY dyes. RSC Adv. 2015, 5, 89375–89388. [Google Scholar] [CrossRef]
- Prlj, A.; Fabrizio, A.; Corminboeuf, C. Rationalizing fluorescence quenching in meso-BODIPY dyes. Phys. Chem. Chem. Phys. 2016, 18, 32668–32672. [Google Scholar] [CrossRef] [PubMed]
- Prlj, A.; Vannay, L.; Corminboeuf, C. Fluorescence quenching in BODIPY dyes: The role of intramolecular interactions and charge transfer. Helv. Chim. Acta 2017, 100, e1700093. [Google Scholar] [CrossRef]
- Marfin, Y.S.; Shipalova, M.V.; Kurzin, V.O.; Ksenofontova, K.V.; Solomonov, A.V.; Rumyantsev, E.V. Fluorescent properties of BODIPY sensors based on photoinduced electron transfer. J. Fluoresc. 2016, 26, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- Ludvíková, L.; Friš, P.; Heger, D.; Šebej, P.; Wirz, J.; Klán, P. Photochemistry of rose bengal in water and acetonitrile: A comprehensive kinetic analysis. Phys. Chem. Chem. Phys. 2016, 18, 16266–16273. [Google Scholar] [CrossRef]
- Palao, E.; Duran-Sampedro, G.; de la Moya, S.; Madrid, M.; García-López, C.; Agarrabeitia, A.R.; Verbelen, B.; Dehaen, W.; Boens, N.; Ortiz, M.J. Exploring the application of the negishi reaction of HaloBODIPYs: Generality, regioselectivity, and synthetic utility in the development of BODIPY laser dyes. J. Org. Chem. 2016, 81, 3700–3710. [Google Scholar] [CrossRef]
- Montalti, M.; Murov, S.L. Handbook of Photochemistry, 3rd ed.; CRC; Taylor & Francis: Boca Raton, FL, USA, 2006; p. 650. [Google Scholar]
- Slanina, T.; Shrestha, P.; Palao, E.; Kand, D.; Peterson, J.A.; Dutton, A.S.; Rubinstein, N.; Weinstain, R.; Winter, A.H.; Klán, P. In search of the perfect photocage: Structure–reactivity relationships in meso-Methyl BODIPY photoremovable protecting groups. J. Am. Chem. Soc. 2017, 139, 15168–15175. [Google Scholar] [CrossRef] [PubMed]
- Lachish, U.; Shafferman, A.; Stein, G. Intensity dependence in laser flash photolysis experiments: Hydrated electron formation from ferrocyanide, tyrosine, and tryptophan. J. Chem. Phys. 1976, 64, 4205–4211. [Google Scholar] [CrossRef]
- Jacques, P.; Braun, A.M. Laser flash photolysis of phthalocyanines in solution and microemulsion. Helv. Chim. Acta 1981, 64, 1800–1806. [Google Scholar] [CrossRef]
- Shen, F.; Wang, T.; Yu, X.; Li, Y. Free radical oxidation reaction for selectively solvatochromic sensors with dynamic sensing ability. Chin. Chem. Lett. 2020, 31, 1919–1922. [Google Scholar] [CrossRef]
- Leen, V.; Gonzalvo, V.Z.; Deborggraeve, W.M.; Boens, N.; Dehaen, W. Direct functionalization of BODIPY dyes by oxidative nucleophilic hydrogen substitution at the 3- or 3,5-positions. Chem. Commun. 2010, 46, 4908–4910. [Google Scholar] [CrossRef]
- Kim, S.; Bouffard, J.; Kim, Y. Tailoring the solid-state fluorescence emission of BODIPY dyes by meso substitution. Chem. Eur. J. 2015, 21, 17459–17465. [Google Scholar] [CrossRef] [PubMed]
- Martínek, M.; Filipová, L.; Galeta, J.; Ludvíková, L.; Klán, P. Photochemical formation of dibenzosilacyclohept-4-yne for Cu-free click chemistry with azides and 1,2,4,5-Tetrazines. Org. Lett. 2016, 18, 4892–4895. [Google Scholar] [CrossRef] [PubMed]
- Šolomek, T.; Heger, D.; Ngoy, B.P.; Givens, R.S.; Klán, P. The pivotal role of Oxyallyl diradicals in photo-favorskii rearrangements: Transient spectroscopic and computational studies. J. Am. Chem. Soc. 2013, 135, 15209–15215. [Google Scholar] [CrossRef] [PubMed]
- Vaňková, K.; Marková, I.; Jašprová, J.; Dvořák, A.; Subhanová, I.; Zelenka, J.; Novosádová, I.; Rasl, J.; Vomastek, T.; Sobotka, R.; et al. Chlorophyll-mediated changes in the redox status of pancreatic cancer cells are associated with its anticancer effects. Oxid. Med. Cell. Longev. 2018, 2018, 4069167. [Google Scholar] [CrossRef]
- D’Aléo, A.; Picot, A.; Baldeck, P.L.; Andraud, C.; Maury, O. Design of dipicolinic acid ligands for the two-photon sensitized luminescence of europium complexes with optimized cross-sections. Inorg. Chem. 2008, 47, 10269–10279. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
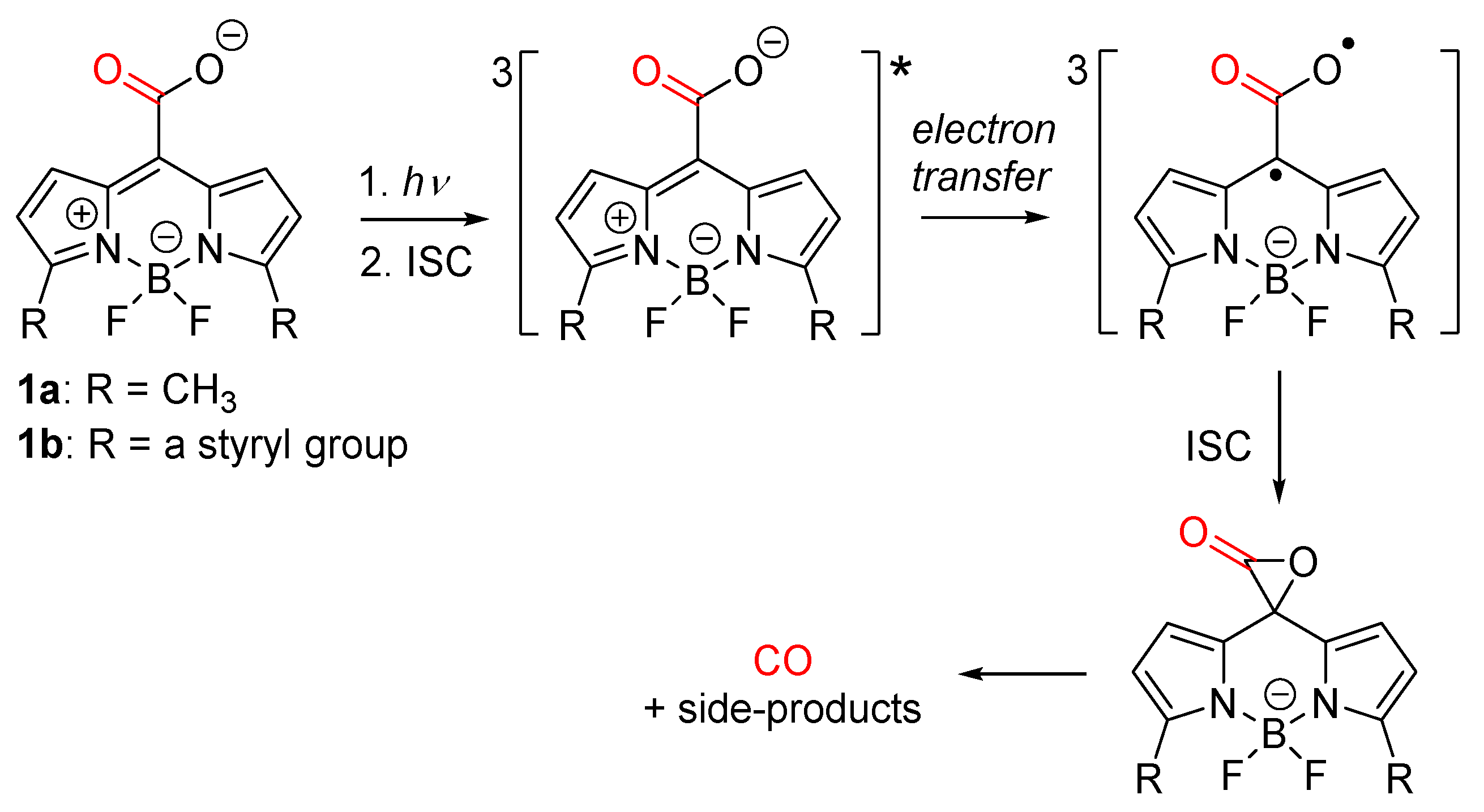{kind=link}
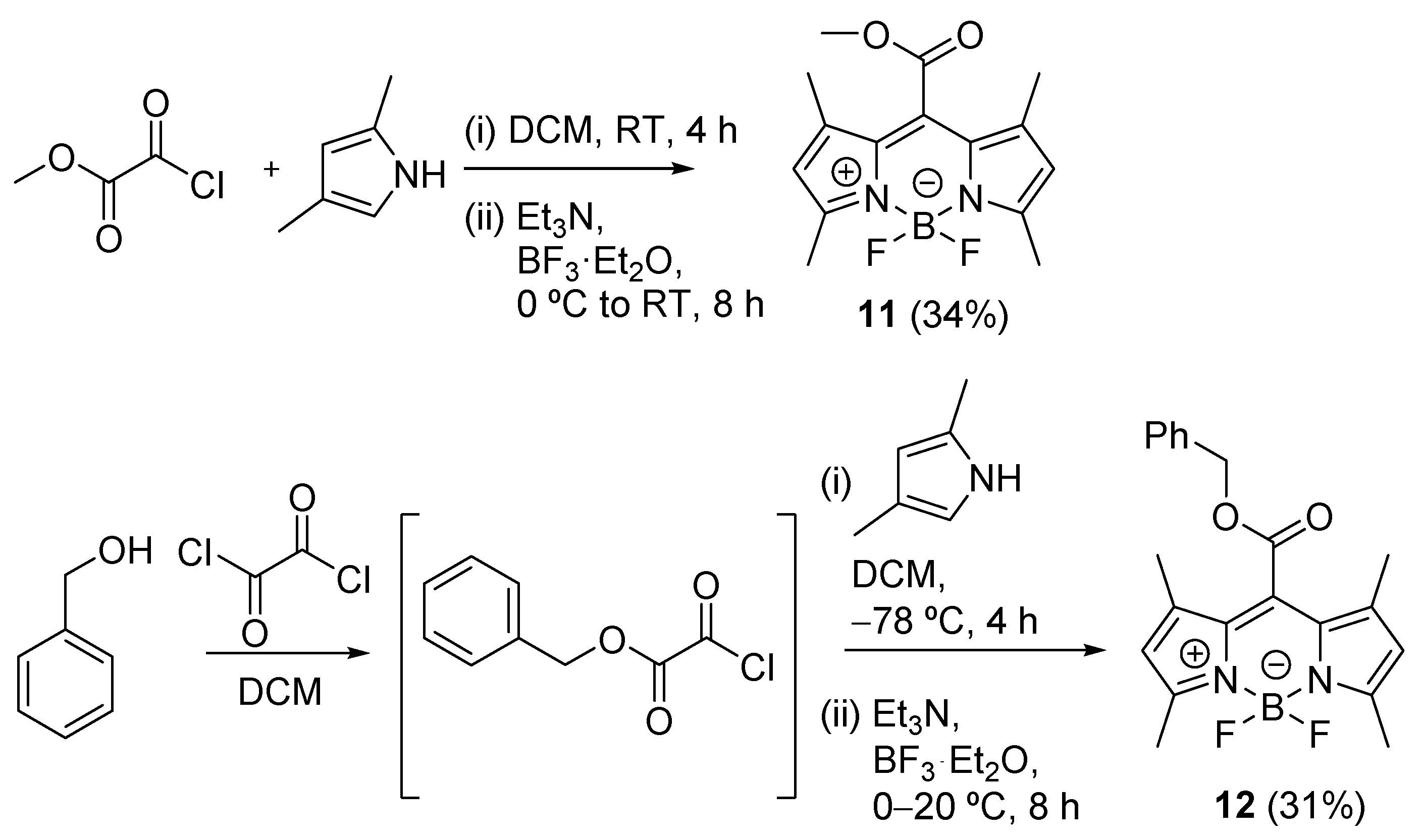{kind=link}
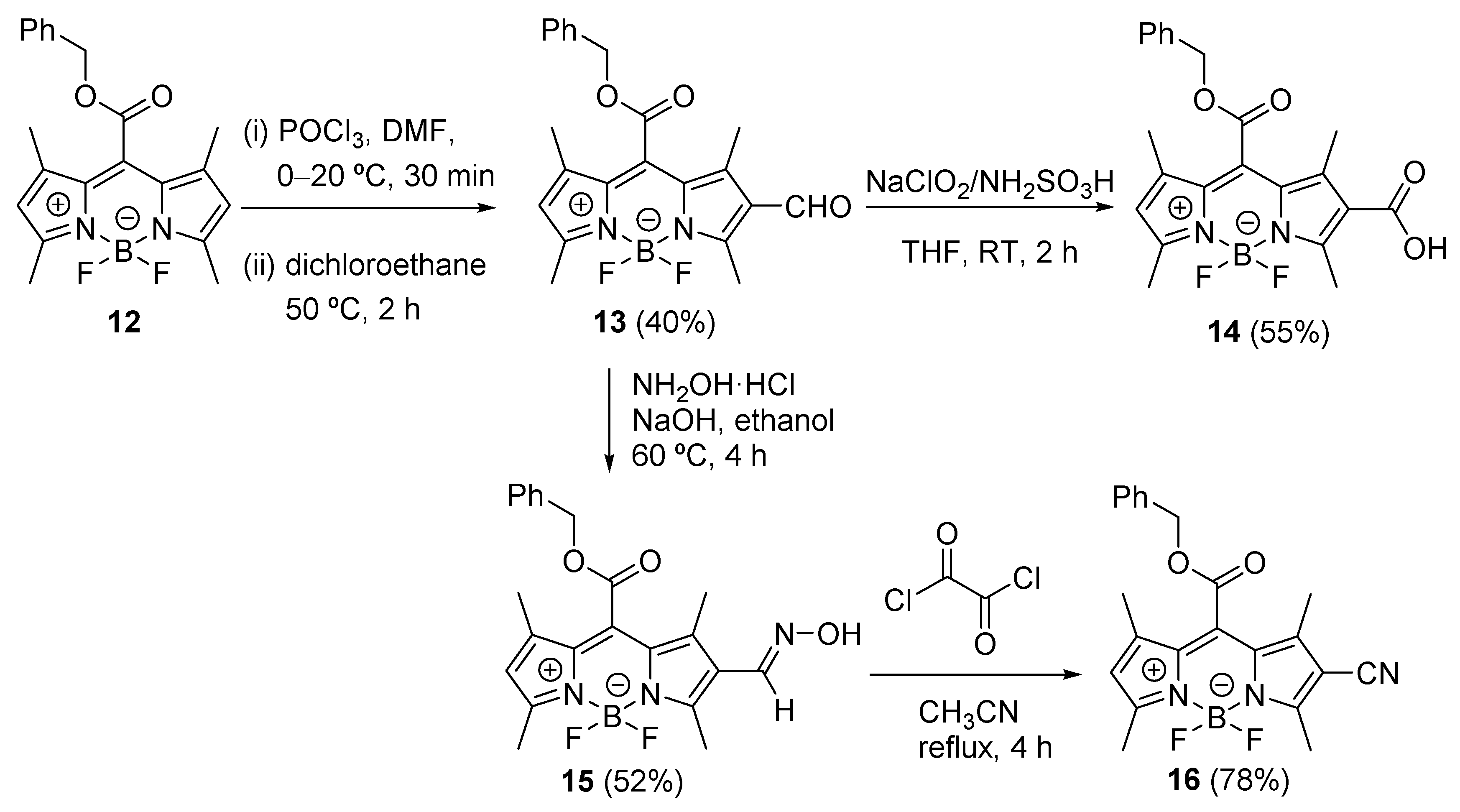{kind=link}
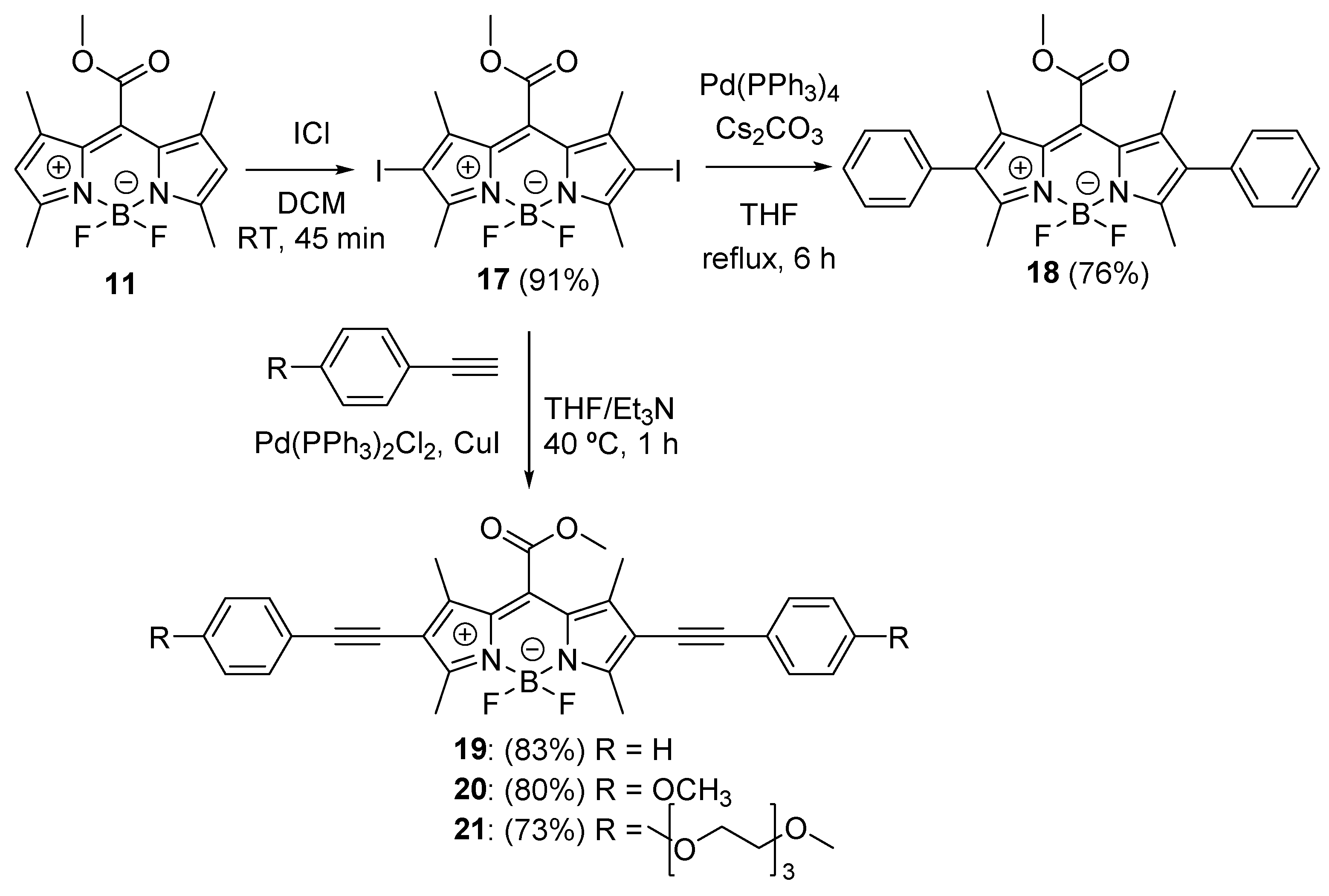{kind=link}
{kind=link}
| Compound | ε/104 M−1 cm−1 | Φfa | ΦΔb | ||
|---|---|---|---|---|---|
| 2 | 494 | 5.22 | 504 | 0.37 ± 0.03 | 0.044 ± 0.003 |
| 3 | 516 | 5.61 | 531 | 0.24 ± 0.02 | 0.59 ± 0.05 |
| 4 | 521 | 5.83 | 540 | 0.03 ± 0.002 | 0.72 ± 0.05 |
| 5 | 490 | 3.95 | 502 | 0.37 ± 0.02 | 0.055 ± 0.005 |
| 6 | 481 | 4.07 | 500 | 0.108 ± 0.003 | 0.07 ± 0.01 c |
| 7 | 518 | 3.58 | 548 | 0.53 ± 0.04 | – d |
| 8 | 554 | 4.31 | 590 | 0.41 ± 0.03 | 0.07 ± 0.01 |
| 9 | 561 | 5.06 | 616 | 0.11 ± 0.02 | 0.032 ± 0.004 |
| 10 | 562 | 5.36 | 617 | 0.15 ± 0.01 | – d |
| Compound | ε/104 M−1 cm−1 | Φfb | Φr/10−4 c | (CO)/% h | ||
|---|---|---|---|---|---|---|
| 2 | 497 | 4.35 | 507 | 0.58 ± 0.02 | 4.1 ± 0.04 d 5.2 ± 0.06 e | 7 i 15 j |
| 3 | 521 | 5.20 | 563 | 0.13 ± 0.01 | 58.4 ± 0.7 d 68.2 ± 0.05 e | 8 i 18 j |
| 4 | 528 | 5.04 | 548 | 0.012 ± 0.001 | – k | 10 i 7 j |
| 5 | 500 | 4.65 | 510 | 0.23 ± 0.01 | – k | 5 i 7 j |
| 6 | 484 | 3.82 | 500 | 0.072 ± 0.001 | – k | 13 i 8 j |
| 7 | 522 | 4.78 | 556 | 0.49 ± 0.03 | – k | 6 i 7 j |
| 8 | 558 | 4.13 | 603 | 0.13 ± 0.01 | 0.70 ± 0.02 f 1.30 ± 0.06 g | 19 i 32 j |
| 9 | 564 | 3.17 | 637 | 0.020 ± 0.002 | 0.57 ± 0.05 f 0.78 ± 0.01 g | 16 i 44 j |
| 10 | 565 | 3.95 | 633 | 0.067 ± 0.005 | 0.20 ± 0.01 f 0.80 ± 0.03 g | 24 i 43 j |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Carnerero, E.M.; Russo, M.; Jakob, A.; Muchová, L.; Vítek, L.; Klán, P. Effects of Substituents on Photophysical and CO-Photoreleasing Properties of 2,6-Substituted meso-Carboxy BODIPY Derivatives. Chemistry 2021, 3, 238-255. https://doi.org/10.3390/chemistry3010018
Sánchez-Carnerero EM, Russo M, Jakob A, Muchová L, Vítek L, Klán P. Effects of Substituents on Photophysical and CO-Photoreleasing Properties of 2,6-Substituted meso-Carboxy BODIPY Derivatives. Chemistry. 2021; 3(1):238-255. https://doi.org/10.3390/chemistry3010018
Chicago/Turabian StyleSánchez-Carnerero, Esther M., Marina Russo, Andreas Jakob, Lucie Muchová, Libor Vítek, and Petr Klán. 2021. "Effects of Substituents on Photophysical and CO-Photoreleasing Properties of 2,6-Substituted meso-Carboxy BODIPY Derivatives" Chemistry 3, no. 1: 238-255. https://doi.org/10.3390/chemistry3010018
APA StyleSánchez-Carnerero, E. M., Russo, M., Jakob, A., Muchová, L., Vítek, L., & Klán, P. (2021). Effects of Substituents on Photophysical and CO-Photoreleasing Properties of 2,6-Substituted meso-Carboxy BODIPY Derivatives. Chemistry, 3(1), 238-255. https://doi.org/10.3390/chemistry3010018