Selenium-Epoxy ‘Click’ Reaction and Se-Alkylation—Efficient Access to Organo-Selenium and Selenonium Compounds



Abstract

1. Introduction
2. Results
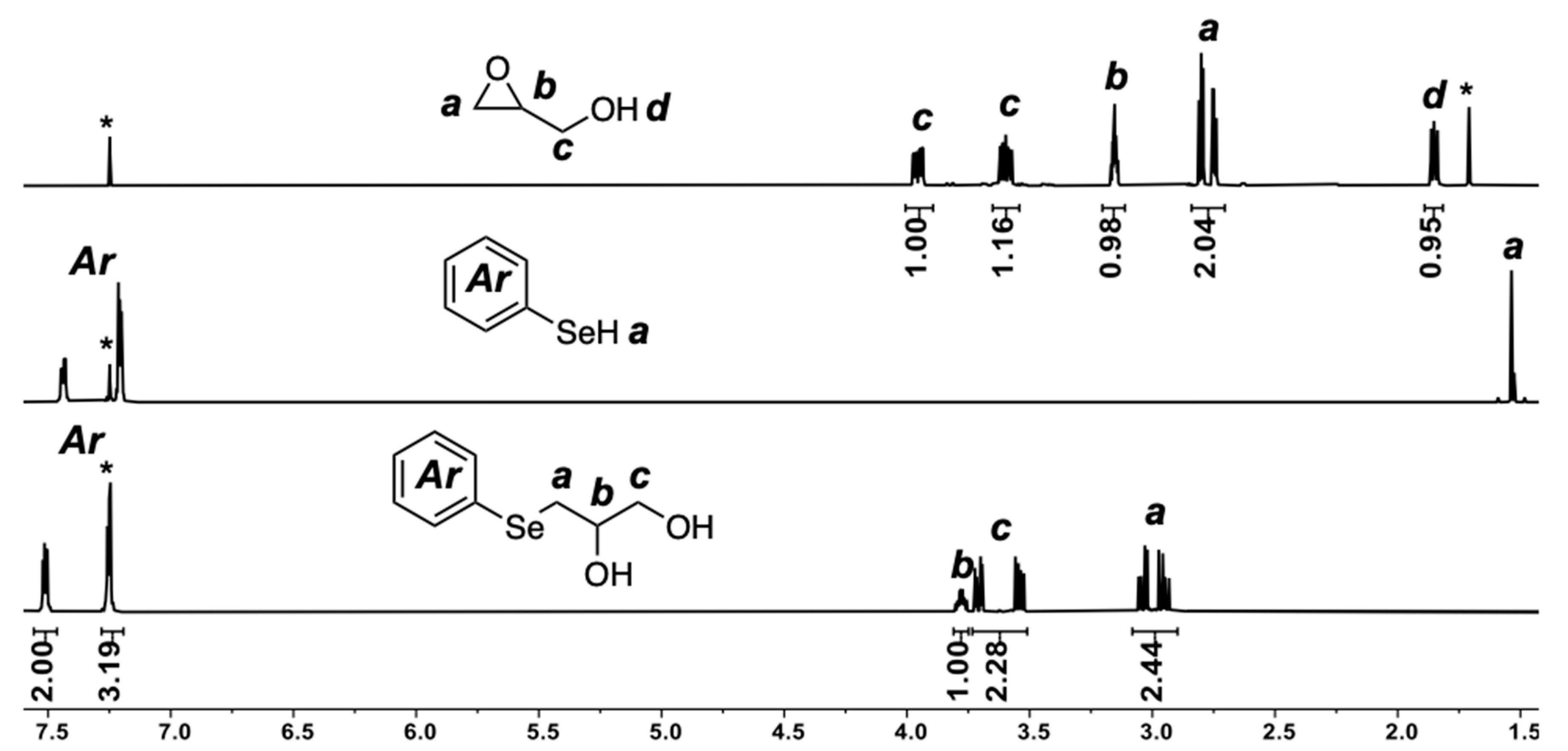
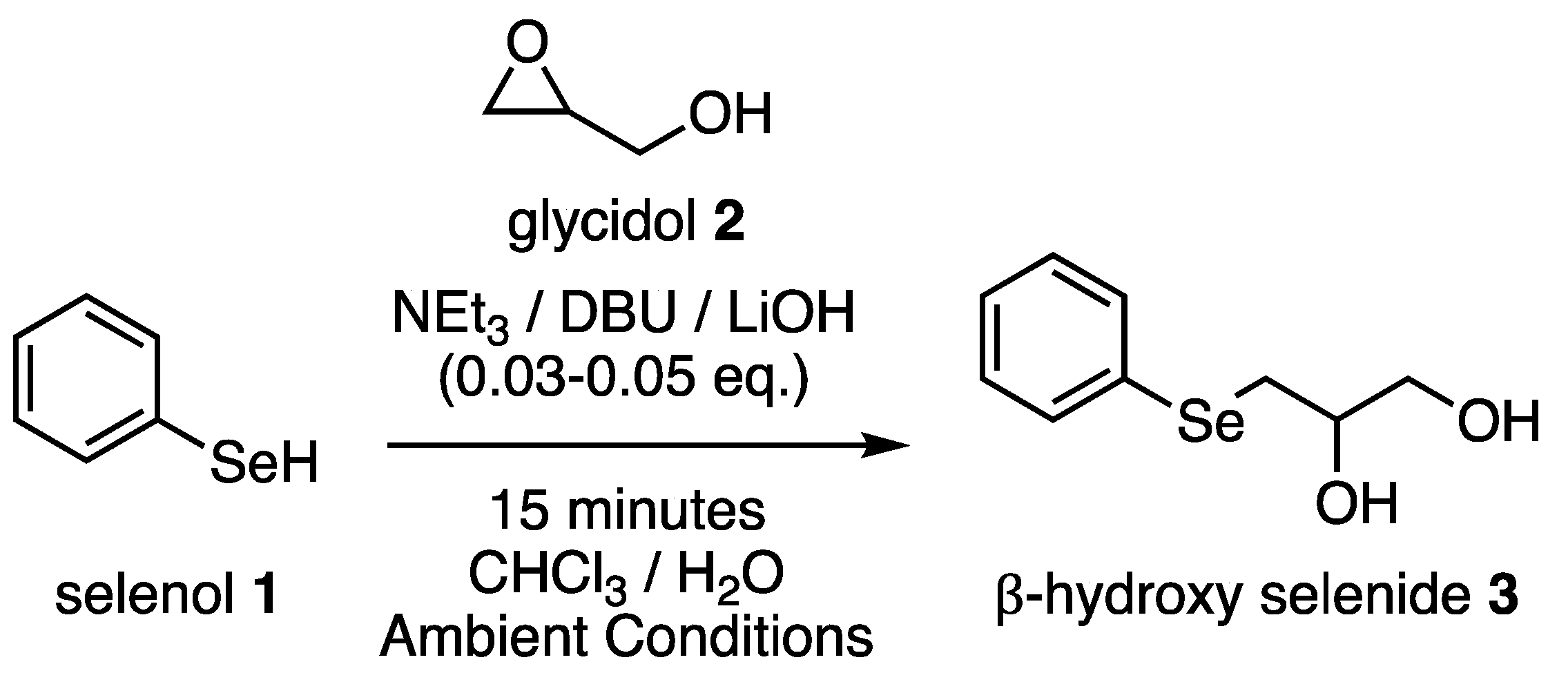
2.1. Selenol Nucleophile in Organic Medium
2.2. Selenol Nucleophile in Water
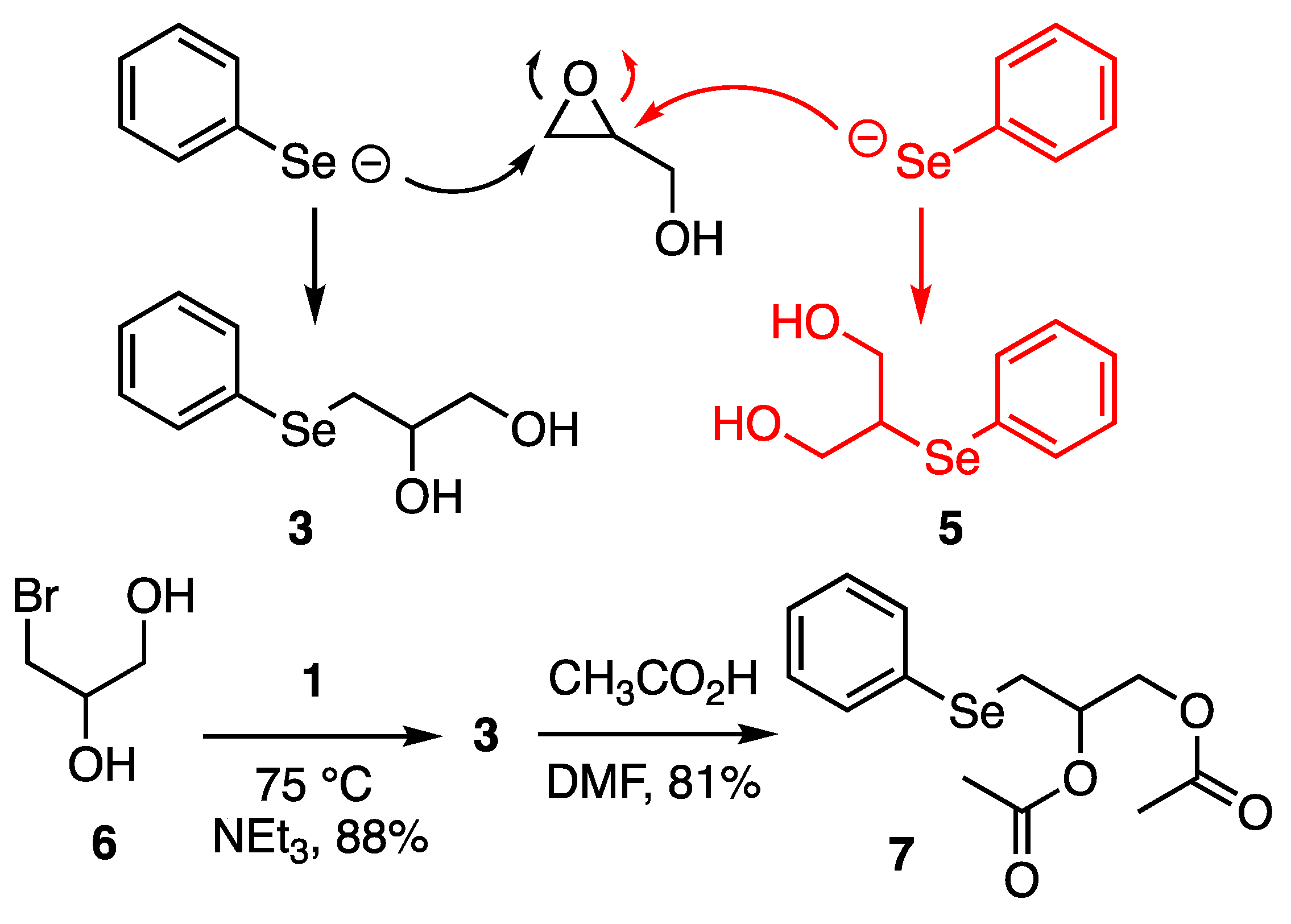
2.3. Non-Stoichiometric System
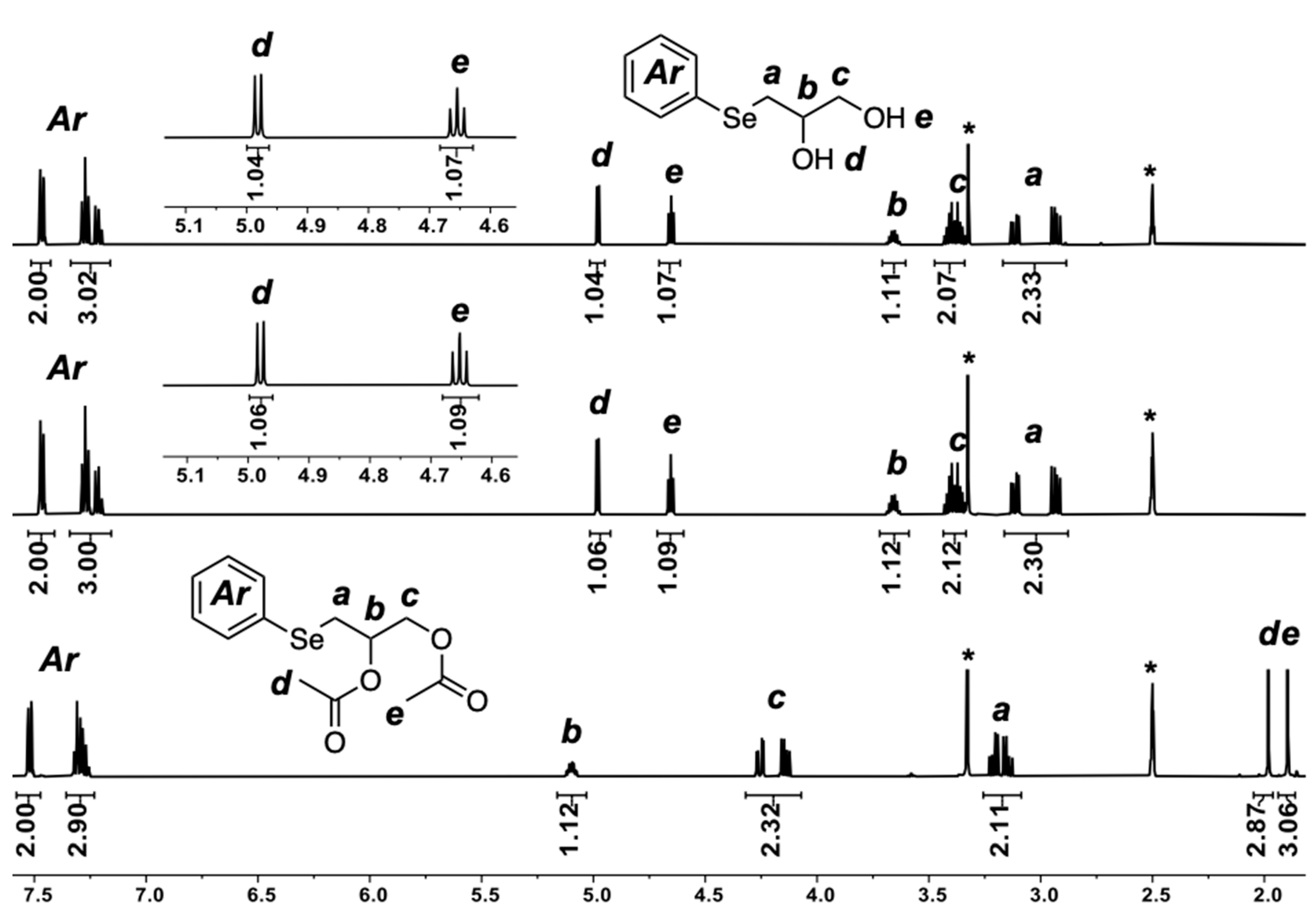
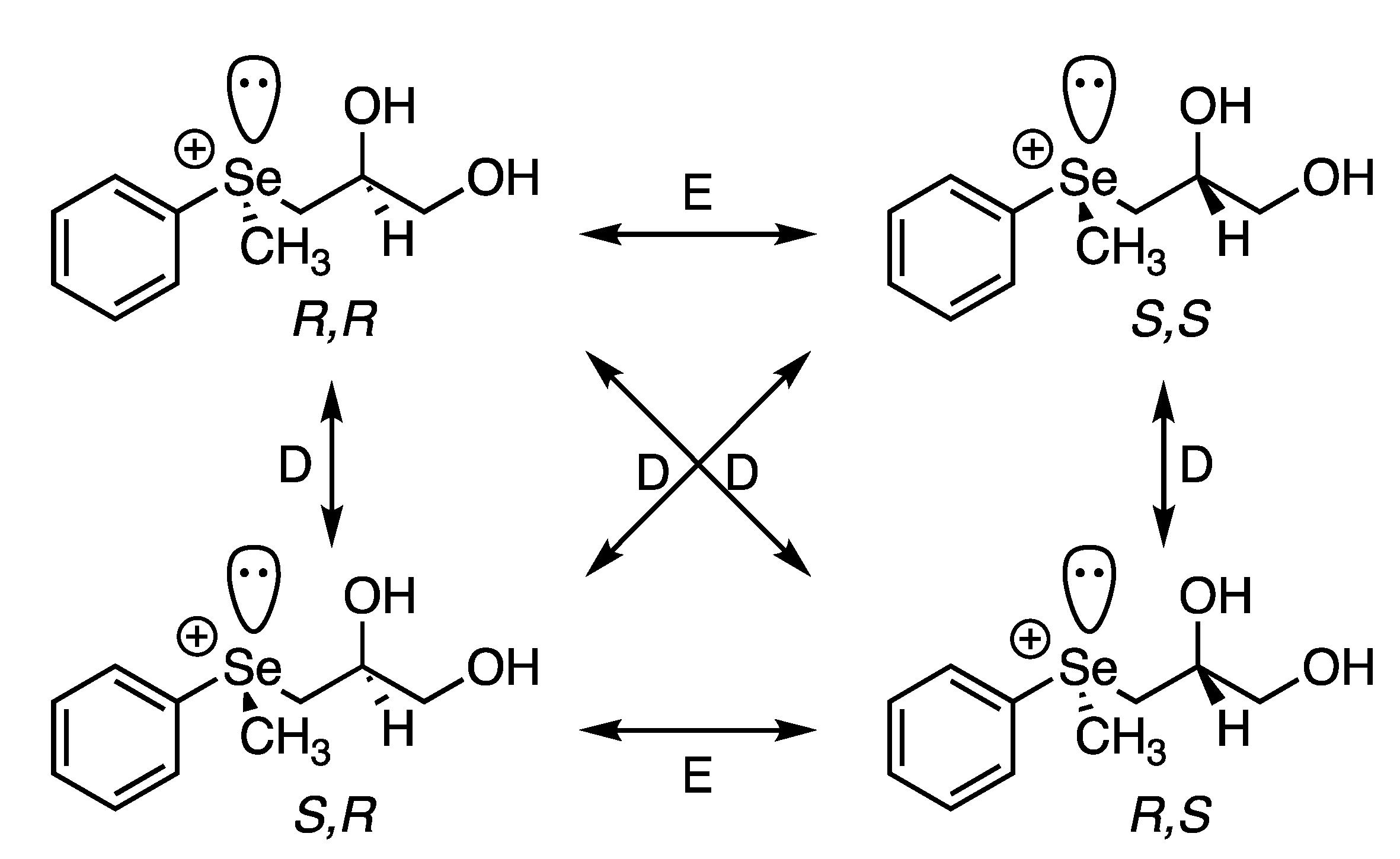
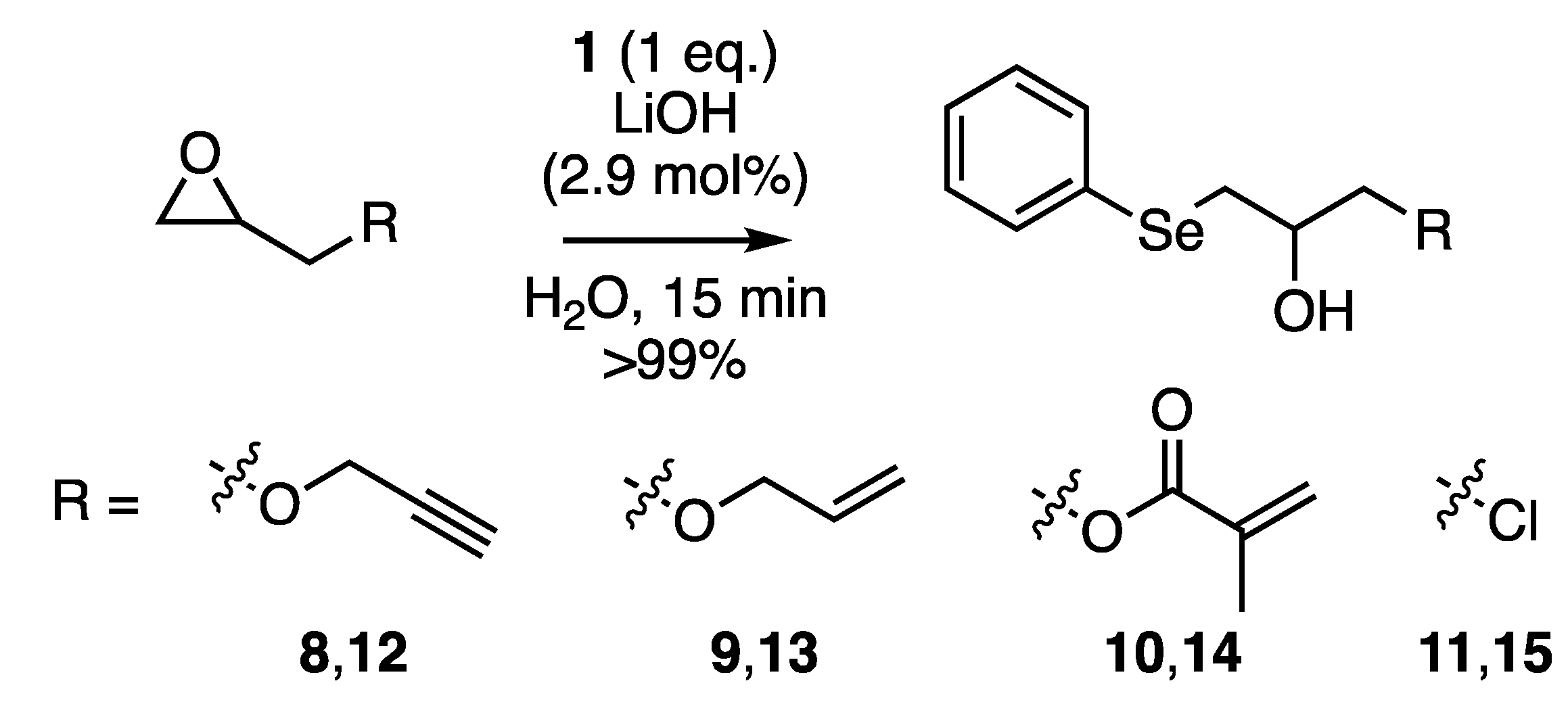
2.4. Regio-Chemistry
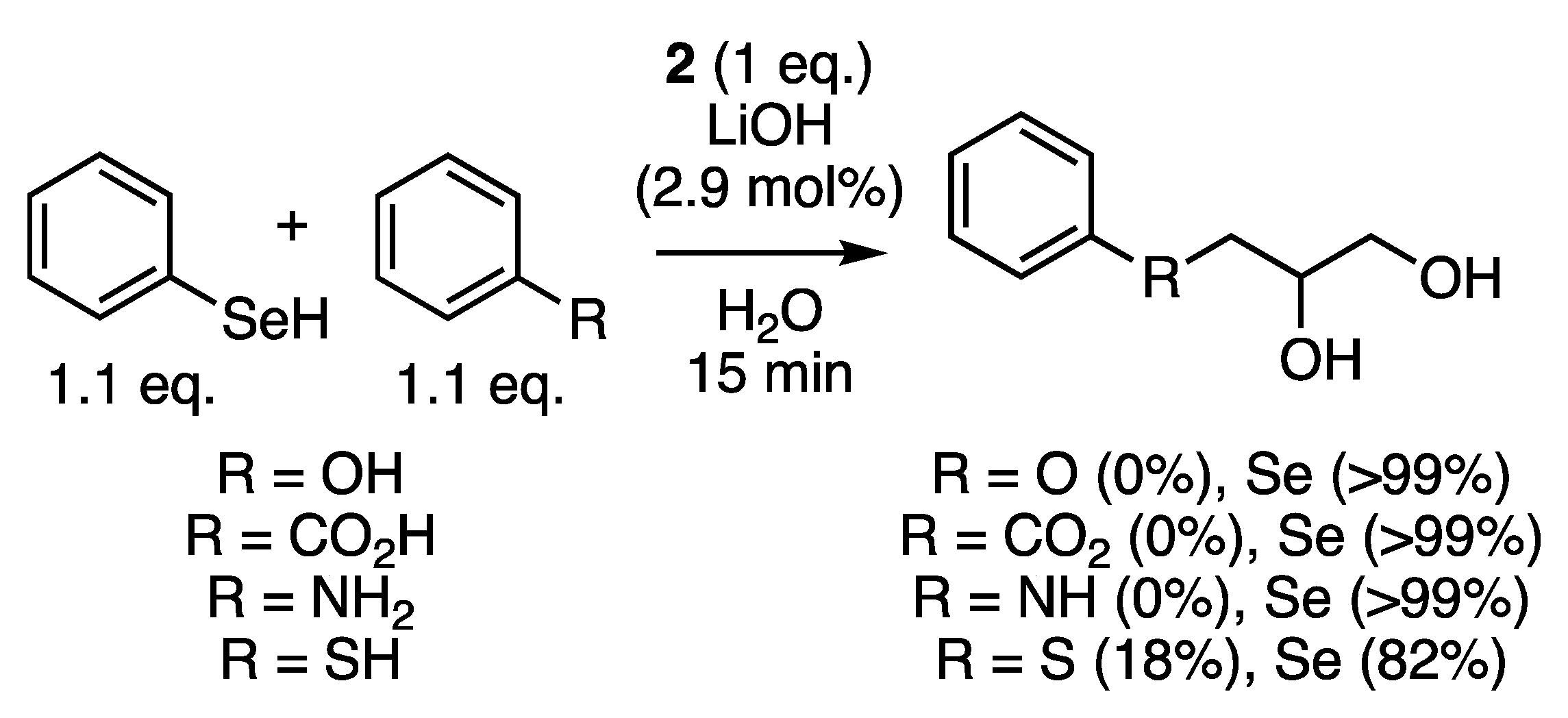
2.5. Chemo-Selectivity
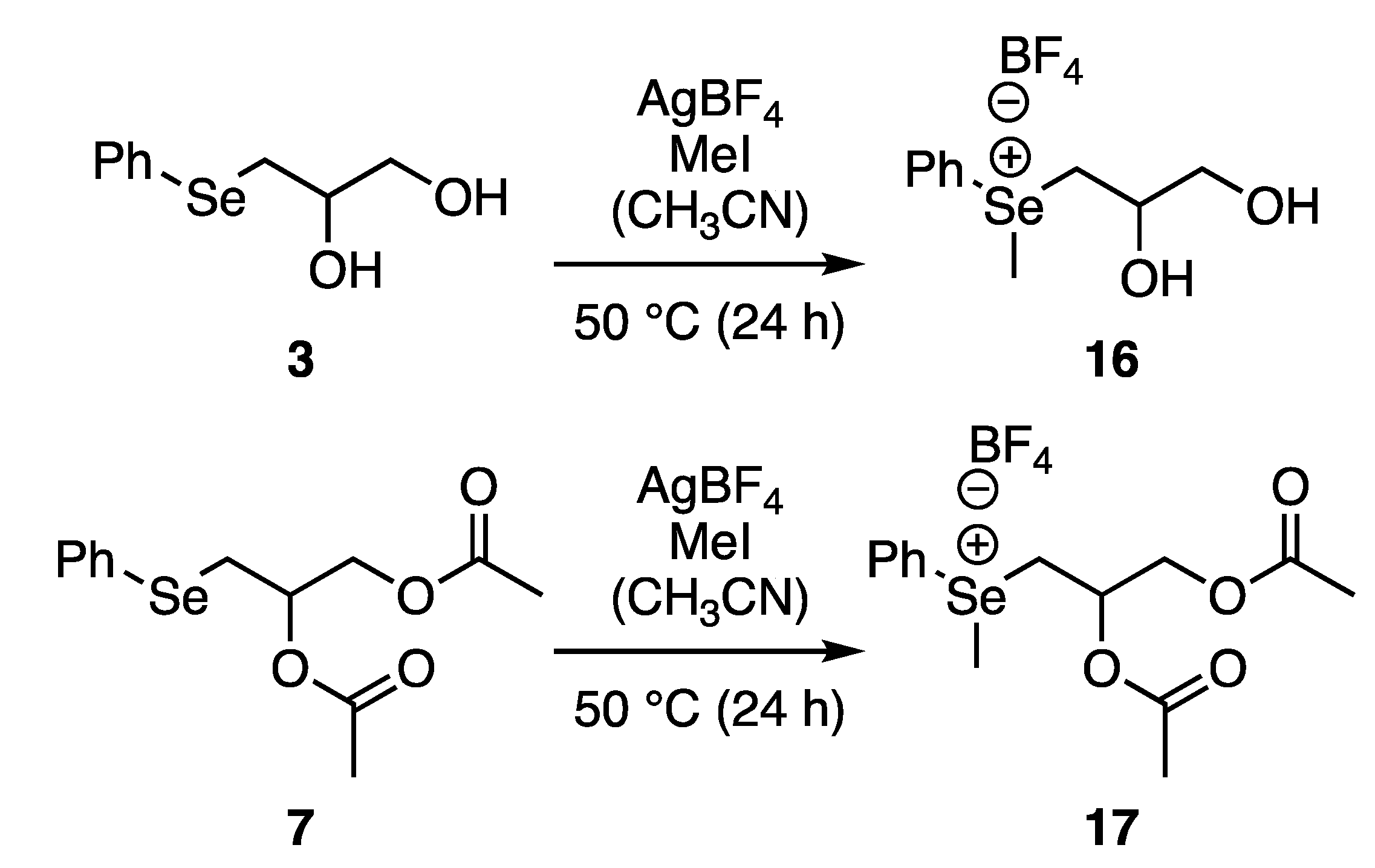
2.6. Selenium Alkylation
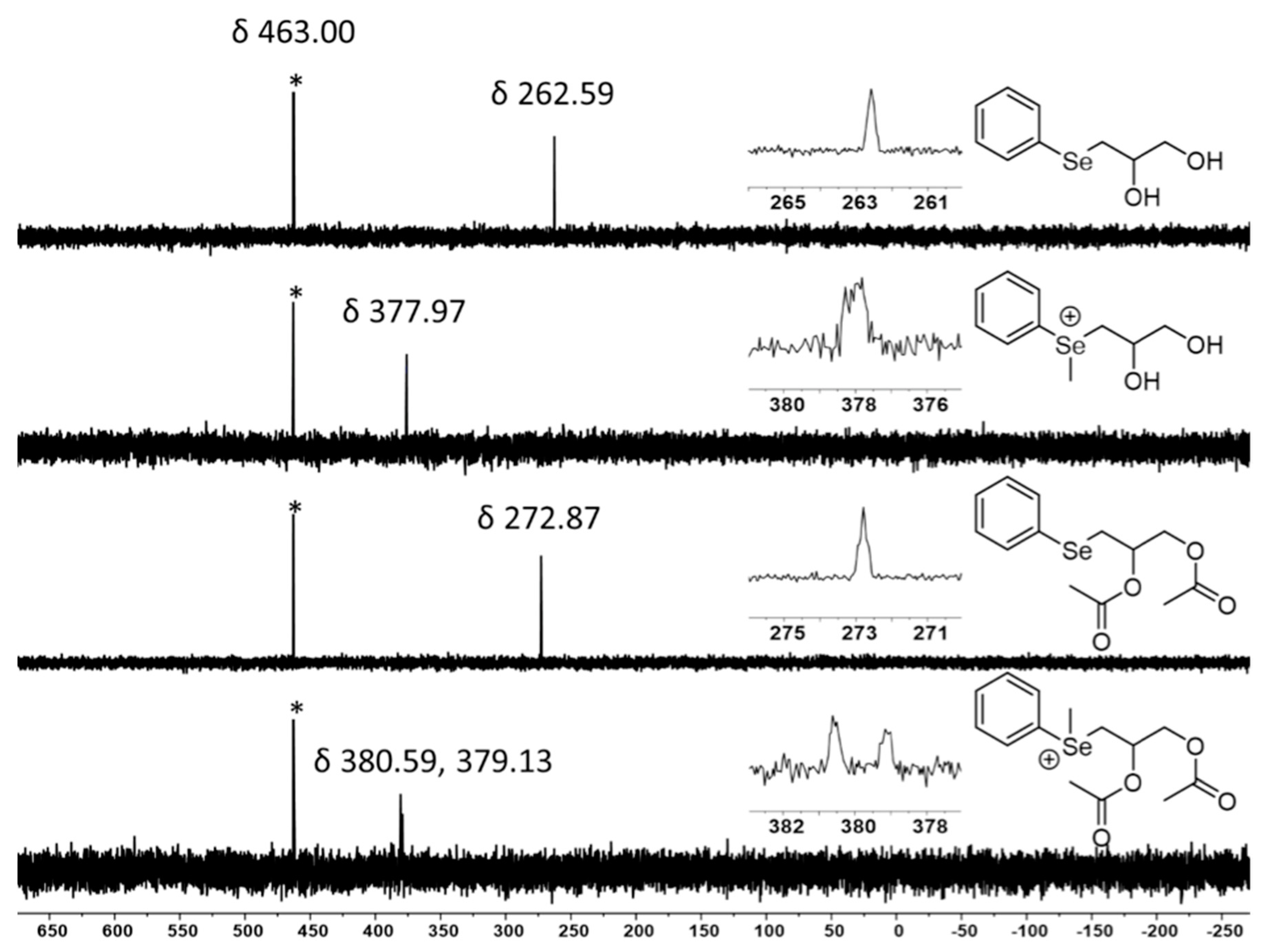
2.7. Selenium NMR
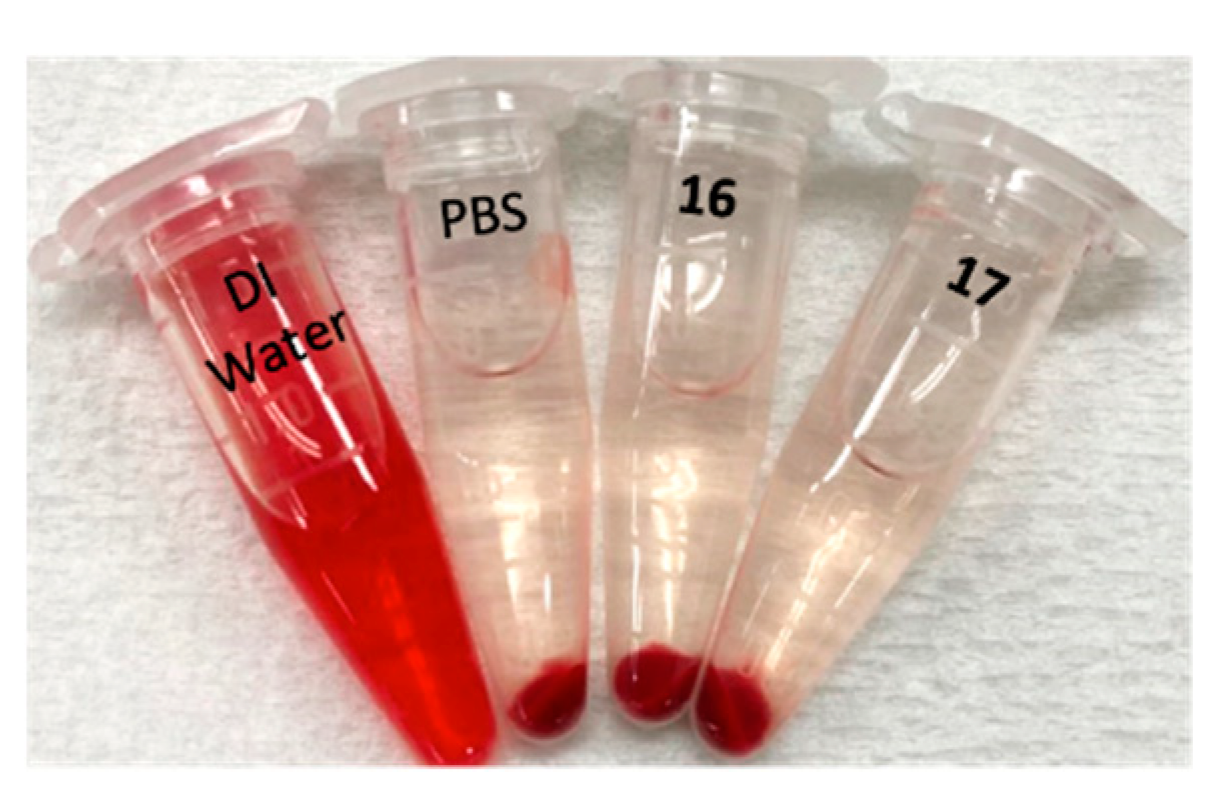
2.8. Hemolysis
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wirth, T. Organoselenium Chemistry: Synthesis and Reactions; John Wiley & Sons: Weinheim, Germany, 2012. [Google Scholar]
- Lenardao, E.J.; Santi, C.; Sancineto, L. Organoselenium compounds as reagents and catalysts to develop new green protocols. In New Frontiers in Organoselenium Compounds; Springer Science and Business Media LLC: Cham, Switzerland, 2018; pp. 1–97. [Google Scholar]
- Sharpless, K.B.; Lauer, R.F. Mild procedure for the conversion of epoxides to allylic alcohols. First organoselenium reagent. J. Am. Chem. Soc. 1973, 95, 2697–2699. [Google Scholar] [CrossRef]
- Nogueira, C.W.; Zeni, G.; Da Rocha, J.B.T. Organoselenium and organotellurium compounds: Toxicology and pharmacology. Chem. Rev. 2004, 104, 6255–6286. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Hawker, C.J.; Fokin, V.V.; Finn, M.G.; Sharpless, K.B. Bringing efficiency to materials synthesis: The philosophy of click chemistry. Aust. J. Chem. 2007, 60, 381–383. [Google Scholar] [CrossRef]
- Iha, R.K.; Wooley, K.L.; Nyström, A.M.; Burke, D.J.; Kade, M.J.; Hawker, C.J. Applications of orthogonal “click” chemistries in the synthesis of functional soft materials. Chem. Rev. 2009, 109, 5620–5686. [Google Scholar] [CrossRef]
- Espeel, P.; Du Prez, F.E. “Click”-inspired chemistry in macromolecular science: Matching recent progress and user expectations. Macromolecules 2014, 48, 2–14. [Google Scholar] [CrossRef]
- Sumerlin, B.S.; Vogt, A.P. Macromolecular engineering through click chemistry and other efficient transformations. Macromolecules 2010, 43, 1–13. [Google Scholar] [CrossRef]
- Durmaz, H.; Sanyal, A.; Hizal, G.; Tunca, U. Double click reaction strategies for polymer conjugation and post-functionalization of polymers. Polym. Chem. 2012, 3, 825–835. [Google Scholar] [CrossRef]
- Becer, C.R.; Hoogenboom, R.; Schubert, U.S. Click chemistry beyond metal-catalyzed cycloaddition. Angew. Chem. Int. Ed. 2009, 48, 4900–4908. [Google Scholar] [CrossRef]
- Saha, A.; De, S.; Stuparu, M.C.; Khan, A. Facile and general preparation of multifunctional main-chain cationic polymers through application of robust, efficient and orthogonal click chemistries. J. Am. Chem. Soc. 2012, 134, 17291–17297. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Lee, D.G.; Yeo, H.; Rao, J.; Zhu, Z.; Shin, J.; Jeong, K.; Kim, S.; Jung, H.W.; Khan, A. Proton transfer hydrogels: Versatility and applications. J. Am. Chem. Soc. 2018, 140, 6700–6709. [Google Scholar] [CrossRef] [PubMed]
- Yeo, H.; Khan, A. Photoinduced Proton-Transfer Polymerization: A practical synthetic tool for soft lithography applications. J. Am. Chem. Soc. 2020, 142, 3479–3488. [Google Scholar] [CrossRef] [PubMed]
- Tanini, D.; Capperucci, A. Ring opening reactions of heterocycles with selenium and tellurium nucleophiles. N. J. Chem. 2019, 43, 11451–11468. [Google Scholar] [CrossRef]
- Movassagh, B.; Shamsipoor, M. Stereo and regioselective zinc-mediated ring-opening of epoxides with diselenides. Synlett 2005, 8, 1316–1318. [Google Scholar] [CrossRef]
- Tiecco, M.; Testaferri, L.; Marini, F.; Sternativo, S.; Del Verme, F.; Santi, C.; Bagnoli, L.; Temperini, A. Synthesis of enantiomerically enriched β-hydroxy selenides by catalytic asymmetric ring opening of meso-epoxides with (phenylseleno) silanes. Tetrahedron 2008, 64, 3337–3342. [Google Scholar] [CrossRef]
- Santi, C.; Santoro, S.; Battistelli, B.; Testaferri, L.; Tiecco, M. Preparation of the first bench-stable phenyl selenolate: An interesting “on water” nucleophilic reagent. Eur. J. Org. Chem. 2008, 2008, 5387–5390. [Google Scholar] [CrossRef]
- Santi, C.; Santoro, S.; Testaferri, L.; Tiecco, M. A Simple zinc-mediated preparation of selenols. Synlett 2008, 2008, 1471–1474. [Google Scholar] [CrossRef]
- Huber, R.; Criddle, R. Comparison of the chemical properties of selenocysteine and selenocystine with their sulfur analogs. Arch. Biochem. Biophys. 1967, 122, 164–173. [Google Scholar] [CrossRef]
- Sridhar, R.; Srinivas, B.; Surendra, K.; Krishnaveni, N.S.; Rao, K.R. Synthesis of β-hydroxy selenides using benzeneselenol and oxiranes under supramolecular catalysis in the presence of β-cyclodextrin in water. Tetrahedron Lett. 2005, 46, 8837–8839. [Google Scholar] [CrossRef]
- Yang, M.-H.; Yan, G.-B.; Zheng, Y.-F. Regioselective ring-opening reactions of 1,2-epoxides with thiols and arylselenols directly promoted by [Bmim]BF4. Tetrahedron Lett. 2008, 49, 6471–6474. [Google Scholar] [CrossRef]
- Yang, M.; Zhu, C.; Yuan, F.; Huang, Y.; Pan, Y. Enantioselective ring-opening reaction of meso-epoxides with ArSeH catalyzed by heterometallic Ti−Ga−salen system. Org. Lett. 2005, 7, 1927–1930. [Google Scholar] [CrossRef] [PubMed]
- Tanini, D.; Tiberi, C.; Gellini, C.; Salvi, P.R.; Capperucci, A. A straightforward access to stable β-functionalized alkyl selenols. Adv. Synth. Catal. 2018, 360, 3367–3375. [Google Scholar] [CrossRef]
- Freudendahl, D.M.; Santoro, S.; Shahzad, S.A.; Santi, C.; Wirth, T. Green chemistry with selenium reagents: Development of efficient catalytic reactions. Angew. Chem. Int. Ed. 2009, 48, 8409–8411. [Google Scholar] [CrossRef] [PubMed]
- Santi, C.; Jacob, R.G.; Monti, B.; Bagnoli, L.; Sancineto, L.; Lenardao, E.J. Water and aqueous mixtures as convenient alternative media for organoselenium chemistry. Molecules 2016, 21, 1482. [Google Scholar] [CrossRef]
- Muzammil, E.M.; Khan, A.; Stuparu, M.C. Post-polymerization modification reactions of poly(glycidyl methacrylate)s. RSC Adv. 2017, 7, 55874–55884. [Google Scholar] [CrossRef]
- Kade, M.J.; Burke, D.J.; Hawker, C.J. The power of thiol-ene chemistry. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 743–750. [Google Scholar] [CrossRef]
- Lowe, A.B. Thiol-yne ‘click’/coupling chemistry and recent applications in polymer and materials synthesis and modification. Polymer 2014, 55, 5517–5549. [Google Scholar] [CrossRef]
- Daglar, O.; Gunay, U.S.; Hizal, G.; Tunca, U.; Durmaz, H. Extremely rapid polythioether synthesis in the presence of TBD. Macromolecules 2019, 52, 3558–3572. [Google Scholar] [CrossRef]
- Gunay, U.S.; Cetin, M.; Daglar, O.; Hizal, G.; Tunca, U.; Durmaz, H.; Butun, M. Ultrafast and efficient aza- and thiol-Michael reactions on a polyester scaffold with internal electron deficient triple bonds. Polym. Chem. 2018, 9, 3037–3054. [Google Scholar] [CrossRef]
- Daglar, O.; Ozcan, B.; Gunay, U.S.; Hizal, G.; Tunca, U.; Durmaz, H. Extremely rapid postfunctionalization of maleate and fumarate main chain polyesters in the presence of TBD. Polymer 2019, 182, 121844. [Google Scholar] [CrossRef]
- Nacca, F.G.; Monti, B.; Lenardao, E.J.; Evans, P.; Santi, C. A Simple zinc-mediated method for selenium addition to michael acceptors. Molecules 2020, 25, 2018. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Wang, X.; Tse, Y.-L.S.; Ke, Z.; Yeung, Y.-Y. Applications of selenonium cations as lewis acids in organocatalytic reactions. Angew. Chem. Int. Ed. 2018, 57, 12869–12873. [Google Scholar] [CrossRef] [PubMed]
- Duddeck, H. Selenium-77 nuclear magnetic resonance spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc. 1995, 27, 1–323. [Google Scholar] [CrossRef]
- Goodhead, L.K.; Macmillan, F.M. Measuring osmosis and hemolysis of red blood cells. Adv. Physiol. Educ. 2017, 41, 298–305. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Solvent | Selenol (1) | Epoxide (2) | Catalyst | Catalyst (mol%) | Catalyst (eq./SeH) | Time (min) | Ring Opening (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | CHCl3 | 1 | 1 | TEA | 0.99 | 0.01 | 15 | 75 |
| 2 | CHCl3 | 1 | 1 | TEA | 0.99 | 0.01 | 30 | 77 |
| 3 | CHCl3 | 1 | 1 | TEA | 2.91 | 0.03 | 15 | 81 |
| 4 | CHCl3 | 1 | 1 | TEA | 2.91 | 0.03 | 30 | 83 |
| 5 | CHCl3 | 1 | 1 | TEA | 4.76 | 0.05 | 15 | 87 |
| 6 | CHCl3 | 1 | 1 | TEA | 4.76 | 0.05 | 30 | 87 |
| 7 | CHCl3 | 1 | 1 | DBU | 0.99 | 0.01 | 15 | 88 |
| 8 | CHCl3 | 1 | 1 | DBU | 0.99 | 0.01 | 30 | 89 |
| 9 | CHCl3 | 1 | 1 | DBU | 2.91 | 0.03 | 15 | 89 |
| 10 | CHCl3 | 1 | 1 | DBU | 2.91 | 0.03 | 30 | 90 |
| 11 | CHCl3 | 1 | 1 | DBU | 4.76 | 0.05 | 15 | 94 |
| 12 | CHCl3 | 1 | 1 | DBU | 4.76 | 0.05 | 30 | 94 |
| 13 | CHCl3 | 1 | 1 | LiOH | 0.99 | 0.01 | 15 | 94 |
| 14 | CHCl3 | 1 | 1 | LiOH | 0.99 | 0.01 | 30 | 94 |
| 15 | CHCl3 | 1 | 1 | LiOH | 2.91 | 0.03 | 15 | 95 |
| 16 | CHCl3 | 1 | 1 | LiOH | 2.91 | 0.03 | 30 | 96 |
| 17 | CHCl3 | 1 | 1 | LiOH | 4.76 | 0.05 | 15 | 97 |
| 18 | CHCl3 | 1 | 1 | LiOH | 4.76 | 0.05 | 30 | 97 |
| 19 | CHCl3 | 1.1 | 1 | TEA | 3.19 | 0.03 | 15 | 91 |
| 20 | CHCl3 | 1.1 | 1 | DBU | 3.19 | 0.03 | 15 | >99 |
| 21 | CHCl3 | 1.1 | 1 | LiOH | 3.19 | 0.03 | 15 | >99 |
| Entry | Solvent | Selenol (1) | Epoxide (2) | Catalyst | Catalyst (mol%) | Catalyst (eq./SeH) | Time (min) | Ring Opening (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | H2O | 1 | 1 | TEA | 0.99 | 0.01 | 15 | 94 |
| 2 | H2O | 1 | 1 | TEA | 0.99 | 0.01 | 30 | 95 |
| 3 | H2O | 1 | 1 | TEA | 2.91 | 0.03 | 15 | >99 |
| 4 | H2O | 1 | 1 | TEA | 2.91 | 0.03 | 30 | >99 |
| 5 | H2O | 1 | 1 | TEA | 4.76 | 0.05 | 15 | >99 |
| 6 | H2O | 1 | 1 | TEA | 4.76 | 0.05 | 30 | >99 |
| 7 | H2O | 1 | 1 | DBU | 0.99 | 0.01 | 15 | 94 |
| 8 | H2O | 1 | 1 | DBU | 0.99 | 0.01 | 30 | 95 |
| 9 | H2O | 1 | 1 | DBU | 2.91 | 0.03 | 15 | >99 |
| 10 | H2O | 1 | 1 | DBU | 2.91 | 0.03 | 30 | >99 |
| 11 | H2O | 1 | 1 | DBU | 4.76 | 0.05 | 15 | >99 |
| 12 | H2O | 1 | 1 | DBU | 4.76 | 0.05 | 30 | >99 |
| 13 | H2O | 1 | 1 | LiOH | 0.99 | 0.01 | 15 | >99 |
| 14 | H2O | 1 | 1 | LiOH | 0.99 | 0.01 | 30 | >99 |
| 15 | H2O | 1 | 1 | LiOH | 2.91 | 0.03 | 15 | >99 |
| 16 | H2O | 1 | 1 | LiOH | 2.91 | 0.03 | 30 | >99 |
| 17 | H2O | 1 | 1 | LiOH | 4.76 | 0.05 | 15 | >99 |
| 18 | H2O | 1 | 1 | LiOH | 4.76 | 0.05 | 30 | >99 |
| 19 | H2O | 1.1 | 1 | TEA | 3.19 | 0.03 | 15 | >99 |
| 20 | H2O | 1.1 | 1 | DBU | 3.19 | 0.03 | 15 | >99 |
| 21 | H2O | 1.1 | 1 | LiOH | 3.19 | 0.03 | 15 | >99 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eom, T.; Khan, A. Selenium-Epoxy ‘Click’ Reaction and Se-Alkylation—Efficient Access to Organo-Selenium and Selenonium Compounds. Chemistry 2020, 2, 827-836. https://doi.org/10.3390/chemistry2040054
Eom T, Khan A. Selenium-Epoxy ‘Click’ Reaction and Se-Alkylation—Efficient Access to Organo-Selenium and Selenonium Compounds. Chemistry. 2020; 2(4):827-836. https://doi.org/10.3390/chemistry2040054
Chicago/Turabian StyleEom, Taejun, and Anzar Khan. 2020. "Selenium-Epoxy ‘Click’ Reaction and Se-Alkylation—Efficient Access to Organo-Selenium and Selenonium Compounds" Chemistry 2, no. 4: 827-836. https://doi.org/10.3390/chemistry2040054
APA StyleEom, T., & Khan, A. (2020). Selenium-Epoxy ‘Click’ Reaction and Se-Alkylation—Efficient Access to Organo-Selenium and Selenonium Compounds. Chemistry, 2(4), 827-836. https://doi.org/10.3390/chemistry2040054