Abstract
Pyrimidine cyclobutane dimers are hazardous DNA lesions formed upon exposure of DNA to UV light, which can be repaired through oxidative electron transfer (ET). Laser flash photolysis and computational studies were performed to explore the role of configuration and constitution at the cyclobutane ring on the oxidative repair process, using the nitrate radical (NO3•) as oxidant. The rate coefficients of 8–280 × 107 M−1 s−1 in acetonitrile revealed a very high reactivity of the cyclobutane dimers of N,N’-dimethylated uracil (DMU), thymine (DMT), and 6-methyluracil (DMU6-Me) towards NO3•, which likely proceeds via ET at N(1) as a major pathway. The overall rate of NO3• consumption was determined by (i) the redox potential, which was lower for the syn- than for the anti-configured dimers, and (ii) the accessibility of the reaction site for NO3•. In the trans dimers, both N(1) atoms could be approached from above and below the molecular plane, whereas in the cis dimers, only the convex side was readily accessible for NO3•. The higher reactivity of the DMT dimers compared with isomeric DMU dimers was due to the electron-donating methyl groups on the cyclobutane ring, which increased their susceptibility to oxidation. On the other hand, the approach of NO3• to the dimers of DMU6-Me was hindered by the methyl substituents adjacent to N(1), making these dimers the least reactive in this series.
1. Introduction
Exposure to UV light from the sun (λ = 180–400 nm) causes hazardous DNA lesions in the human body, which can lead to melanoma, basal cell, and squamous cell skin cancer [1]. The primary photo adducts implicated in skin cancer are pyrimidine cyclobutane dimers, which are formed through [2 + 2] cycloaddition between adjacent thymine nucleobases in the same oligonucleotide strand (Scheme 1, path A). A minor pathway (ca. 25%) proceeds through [2 + 2] photocycloaddition between the alkene and carbonyl moiety to give an oxetane intermediate that undergoes ring-opening to the 6–4 adduct (Scheme 1, path B) [2]. Cytosine forms the same type of dimers as thymine, but deamination occurs within only a few hours to yield the highly mutagenic uracil dimer (not shown) [3]. In general, pyrimidine cyclobutane dimers can exist in four isomeric structures with head-to-head (syn, s), head-to-tail (anti, a), as well as cis (c) and trans (t) configuration at the cyclobutane ring. However, because of geometrical constraints, only the cis,syn dimer is formed in DNA [4]. Such pyrimidine dimers can prevent DNA polymerases from transcribing DNA, thereby promoting the accumulation of errors [5].
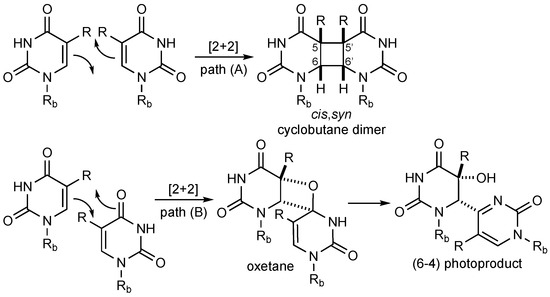
Scheme 1.
Light-induced dimerization of pyrimidines. The deoxyribose phosphate backbone of DNA is abbreviated as Rb; thymine (R = Me), uracil (R = H).
The endogenous repair mechanism of damaged DNA nucleotides in placental mammals involves excision and replacement of the affected base or strand cluster that is orchestrated by a large system of reparative enzymes [6,7]. Other species, such as plants or fish, employ the enzyme photolyase and its reducing cofactor FADH (reduced flavine adenine dinucleotide) to repair dimer lesions through a reductive one-electron transfer (ET) process [8,9].
In addition to repair by excision or enzymatic reduction, pyrimidine cyclobutane dimers can also be oxidatively cleaved into their monomers, for example, by oxidizing radicals and radical ions [10]. Heelis et al. showed that the reaction of the thymine cyclobutane dimer with sulfate radical anions (SO4•−) or hydroxyl radicals (•OH, this radical is commonly formed in vivo and can lead to oxidative damage in biomolecules [11]) led to the recovery of the monomeric pyrimidine and suggested a mechanism initiated by the abstraction of a hydrogen atom at C(6) (see below) [12]. In experiments involving the cis,syn-configured N,N’-dimethyluracil cyclobutane dimer (c,s-DMU<>DMU), Yan et al. showed that oxidative repair and formation of N,N’-dimethyluracil (DMU) occurred by reaction with •OH (possibly through initial hydrogen abstraction) and bromide radical anions (Br2•−, presumably through initial oxidation) but not with the azide radical (N3•) [13], which could be due to the only moderately high oxidizing ability of this radical (E0 (N3•/N3−) = 1.35 V vs. NHE [14])). Comparable results were obtained by Ito et al., who found in the reaction of the C(5)–C(5′) linked thymine dimer with •OH, SO4•− and N3• that repair only occurred in the case of the former two radicals [15].
In previous published and unpublished work, we performed product studies of the oxidative repair of pyrimidine cyclobutane dimers by nitrate radicals (NO3•), which are important oxidants in the nighttime troposphere. NO3• was generated through photo-induced ET from cerium(IV) ammonium nitrate (CAN) at λ = 350 nm in acetonitrile in the presence of various stereoisomeric N,N’-dimethylated cyclobutane dimers of uracil (DMU), thymine (DMT), and the unnatural pyrimidine 6-methyl uracil (DMU6-Me), according to Reaction (1) [16,17]:
(NH4)2Ce(NO3)6 + hν (350 nm) → NO3• + (NH4)2Ce(NO3)5
Table 1 shows the major products formed after a reaction time of two hours with a reactant ratio of [dimer]:[CAN] = 5. The data were obtained by gas chromatographic (GC) analysis of the reaction mixture from the relative peak areas [17] and, therefore, provided only a qualitative picture. In the case of c,s-DMU<>DMU, the major products were the monomer DMU and the partially cleaved dimer 1, whereas only small amounts of the intact dimer remained (entry 1). Cyclobutane cleavage was considerably less efficient with t,s-DMU<>DMU, where mainly 1 and smaller amounts of DMU were obtained (entry 2). Likewise, repair of c,a-DMU<>DMU, t,a-DMU<>DMU, c,s-DMT<>DMT, and c,a-DMT<>DMT was also incomplete with c,a-DMT<>DMT being the least efficiently cleaved dimer under these conditions (entries 3–6). Repair of the cis,syn configured dimer of DMU6-Me, on the other hand, appeared to be more efficient with only 22% of c,s-DMU6-Me<>DMU6-Me still present after the reaction (entry 7).

Table 1.
Product and competition studies of the reaction of NO3• with isomeric pyrimidine cyclobutane dimers of DMU, DMT, and DMU6-Me.1
Since NO3• is a strong one-electron oxidant (E0 (NO3•/NO3−) = 2.3–2.5 V vs. NHE [19]), these data suggested that NO3• initiated the cleavage process by oxidative ET to form the dimer radical cation, as shown in Scheme 2a for c,s-DMU<>DMU. Stepwise scission of the C(6)–C(6′) bond followed by the C(5)–C(5′) bond gives the repaired monomer DMU and its radical cation DMU•+ [17,18]. Similarly, oxidative repair of c,a-DMU<>DMU should proceed through the first scission of the C(6)–C(5′) and then the C(5)–C(6′) bond (Scheme 2b). However, according to the standard potentials E0 available for some of the dimers [18], the rate of oxidation should not be significantly different for dimers with a cis or a trans configuration at the cyclobutane ring and whether or not a methyl group at the C(5) and C(5′) position is present (entries 1, 2, and 5) [13], which contradicts some of the experimental findings from the product studies. On the other hand, the observed lower efficiency of the oxidative repair of the anti-configured dimers by NO3• confirmed previous findings with other oxidizing species [20] and could be rationalized by their higher E0 value (see entry 3 as an example) [18,21]. It should be noted that in many cases, the extent of dimer consumption was larger than would be expected on the basis of the reactant ratio, suggesting that the monomer radical cation, such as DMU•+, could subsequently oxidize another dimer molecule, thereby propagating a radical chain process [17].
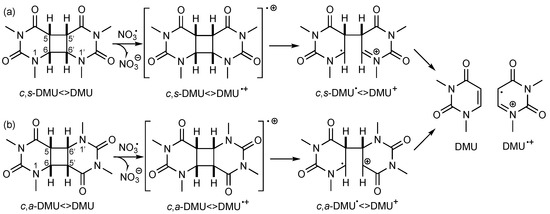
Scheme 2.
Suggested cleavage mechanism of (a) c,s-DMU<>DMU and (b) c,a-DMU<>DMU following one-electron oxidation by NO3•.
The discrepancies become even more apparent in competition studies, where equal amounts of two isomeric dimers were irradiated in the presence of one equivalent of CAN (entries 8–13). For example, a mixture of c,s- and t,s-DMU<>DMU revealed a much more efficient cleavage of the trans,syn-configured isomer (entry 8), whereas in a mixture of c,s-DMU<>DMU and c,s-DMT<>DMT, preferential cleavage of the former dimer occurred (entry 11) [22]. However, as mentioned above, it is highly likely that, apart from NO3•-induced dimer cleavage, additional oxidation of both dimers could also occur by the monomer radical cations formed. Such competition experiments can, therefore, only provide qualitative information about the susceptibility of particular dimers for radical-induced oxidative repair. To obtain quantitative data whether and how the configuration and constitution at the cyclobutane ring affect the rate of the initial step of the oxidative repair, we performed kinetic studies to determine the absolute rate coefficients for the reaction of a series of stereo- and regioisomeric pyrimidine cyclobutane dimers with NO3•, using laser flash photolysis techniques. Apart from being an important environmental oxidant, a major advantage of NO3• is that this radical allows investigating irreversible oxidation processes in biological molecules, since the back-electron transfer, which often hampers studies where the oxidant is produced through photo-induced ET in donor-acceptor pairs, cannot occur. The experimental studies were augmented with density functional theory (DFT) calculations to provide further mechanistic insight.
2. Materials and Methods
2.1. Synthesis of the Pyrimidine Cyclobutane Dimers
2.1.1. General
The starting materials for the synthesis were purchased from commercial suppliers (Sigma-Aldrich, Castle Hill, Australia) and used without purification. Thin-layer chromatography (TLC) was performed to monitor the reactions using aluminum plates coated with silica gel 60 F254 (Merck, ASIS Scientific, Hindmarsh, Australia). UV light at λ = 254 nm, potassium permanganate (KMnO4) stain followed by heating, or iodine ground in Davisil Chromatography Silica Gel LC60A (40–63 microns, 230–400 mesh) were used to visualize TLC plates. The crude products were purified by recrystallization from hot solvent or silica column chromatography with approximately 30–50 g of dry silica (Davisil Chromatography Silica Gel LC60A, 40–63 microns, 230–400 mesh) per 1 g of the crude product mixture. The eluting solvent consisted of a mixture of chloroform and acetone. Solvents were removed under reduced pressure and elevated temperature using a rotary evaporator (Büchi, In Vitro Technologies, Melbourne, Australia). The purity was assessed by an Agilent 1100 reversed-phase HPLC on a Phenomenex Aeris XB-C18 column 250 mm × 4.5 mm × 3.6 µm (Gradient: 100% water buffered with 0.1% trifluoroacetic acid (TFA) to 100% acetonitrile buffered with 0.1% TFA over 25 min, 4% min−1, flow rate: 1 mL min−1).
1H and 13C-NMR spectra were recorded on an Agilent MR 400 MHz NMR spectrometer or an Agilent DD2 500 MHz NMR spectrometer in deuterated dimethyl sulfoxide (DMSO-d6) or in deuterated chloroform (CDCl3) at 25 °C. Chemical shifts were expressed in parts per million (ppm, δ) relative to either DMSO-d6 (1H δ = 2.50 ppm, 13C δ = 39.5 ppm) or CDCl3 (1H δ = 7.26 ppm, 13C δ = 77.2 ppm). High-resolution mass spectrometry (HRMS) was performed by ionizing the sample using electrospray ionization (ESI) into a Thermo Scientific Exactive Plus Orbitrap mass spectrometer (Thermo Scientific Australia, Scoresby, Australia). The detected molecular ions were formed in the positive ion mode and were expressed as [M + H]+. The 1H and 13C-NMR spectra of the pyrimidines and pyrimidine cyclobutane dimers prepared in this study are provided in the Supplementary Material.
2.1.2. Synthesis of N-Methylated Pyrimidines
Method 1. The pyrimidine was stirred in a solution of powdered potassium hydroxide (8 equiv.) and dimethyl sulfoxide (1 mL per mmol of KOH) at 0 °C for 10 min under an inert atmosphere. Methyl iodide (16 equiv.) was then added dropwise, and the mixture was stirred overnight at room temperature. The solution was diluted with cold water and extracted three times with dichloromethane. The combined organic layers were washed with 2 M aqueous sodium hydroxide solution and brine, dried over magnesium sulfate, and the solvent was removed in vacuo. The crude product was purified by recrystallization from ethanol.
Method 2. The pyrimidine and potassium carbonate (3.5 equiv.) were refluxed in acetone (2 mL per mmol of pyrimidine) for 10 min under an inert atmosphere. Methyl iodide (8 equiv.) was then added dropwise, and the reaction mixture was stirred overnight. The reaction was quenched with water (2 mL per mL of acetone) and extracted three times with dichloromethane. The combined organic layers were washed with 2 M aqueous sodium hydroxide solution and brine, dried over magnesium sulfate, and the solvent was removed in vacuo. The crude solid was purified by recrystallization from ethanol.
1,3-Dimethyluracil (DMU)
Prepared from uracil using both methods 1 and 2. 1H-NMR (400 MHz, DMSO-d6): δ 7.66 (d, J = 7.8 Hz, 1H), 5.66 (d, J = 7.8 Hz, 1H), 3.29 (s, 3H), 3.15 ppm (s, 3H). 13C {1H}-NMR (101 MHz, DMSO-d6): δ 162.8, 151.5, 144.6, 99.6, 36.4, 27.2 ppm. HRMS (ESI) m/z calcd. for [C6H9N2O2]+: 141.0664 [M + H]+, found 141.0660.
1,3-Dimethylthymine (DMT)
Prepared from thymine using both methods 1 and 2. 1H-NMR (400 MHz, DMSO-d6): δ 7.56 (s, 1H), 3.26 (s, 3H), 3.16 (s, 3H), 1.79 ppm (s, 3H). 13C {1H}-NMR (101 MHz, DMSO-d6): δ 163.4, 151.3, 140.7, 107.1, 36.1, 27.4, 12.5 ppm. HRMS (ESI) m/z calcd. for [C7H11N2O2]+: 155.0821 [M + H]+, found 155.0815.
1,3.6-Trimethyluracil (DMU6-Me)
N,N’-Dimethylurea (8.81 g, 100 mmol), 4-dimethylamino pyridine (12.2 g, 100 mmol), and acetic anhydride (31.2 mL, 330 mmol) were stirred in anhydrous pyridine (150 mL) under an inert atmosphere at room temperature for 2 h. The resulting red solution was concentrated in vacuo; the residue was diluted with dichloromethane (700 mL) and washed sequentially with 2 M aqueous hydrochloric acid (100 mL), 2 M aqueous sodium bicarbonate solution (100 mL), and 2 M aqueous copper(II) sulfate solution (100 mL). The organic layer was dried over magnesium sulfate. The solvent was removed in vacuo. The crude pale-yellow solid was recrystallized from ethyl acetate and washed repeatedly with cold ethyl acetate until the yellow color had disappeared. White crystals (10.9 g, 70%). 1H-NMR (400 MHz, CDCl3): δ 5.61 (s, 1H), 3.39 (s, 3H), 3.32 (s, 3H), 2.23 ppm (s, 3H). 13C {1H}-NMR (101 MHz, CDCl3): δ 162.4, 152.6, 151.4, 101.2, 31.7, 27.9, 20.2 ppm. HRMS (ESI) m/z calcd. for [C7H11N2O2]+: 155.0821 [M + H]+, found 155.0817.
2.1.3. Dimerization of N-Methylated Pyrimidines
The pyrimidine was dissolved in UV-grade acetone and degassed under an inert atmosphere with sonication for 30 min. The solution was then added to a Pyrex reactor and diluted to 300 mL with UV-grade acetone. Under an inert atmosphere, the solution was stirred for 10 min before exposure to a medium-pressure mercury lamp. Reaction progress was monitored using TLC (KMnO4 stain) until complete consumption of starting material was observed. The solvent was removed in vacuo to afford a mixture of the dimerized substrates as a white solid, which were separated by column chromatography (SiO2, a mixture of CHCl3/acetone). The separated dimers were further purified by recrystallization from ethyl acetate or ethanol and identified by X-ray crystallography. The X-ray structures are given in the Supplementary Material.
Synthesis of DMU<>DMU Isomers
DMU (14.00 g, 100.00 mmol) was dimerized with an overall yield of 25% (3.57 g, 12.70 mmol).
- (a)
- t,a-DMU<>DMU: White crystals (0.35 g, 1.25 mmol, 3%, Rf = 0.26 (EtOAc)). 1H-NMR (400 MHz, CDCl3): δ 4.10 (dd, J = 9.4, 4.9 Hz, 2H), 3.53 (dd, J = 9.4, 4.9 Hz, 2H), 3.24 (s, 6H), 3.08 ppm (s, 6H). 13C {1H}-NMR (101 MHz, CDCl3): δ 167.1 (2C), 151.6 (2C), 53.7 (2C), 44.5 (2C), 33.8 (2C), 28.1 ppm (2C). HRMS (ESI) m/z calcd. for [C12H17N4O4]+: 281.1250 [M + H]+, found 281.1245.
- (b)
- t,s-DMU<>DMU: White crystals (1.12 g, 4.00 mmol, 8%, Rf = 0.18 (EtOAc)).1H-NMR (400 MHz, CDCl3): δ 3.87 (d, J = 8.0 Hz, 2H), 3.62 (d, J = 8.0 Hz, 2H), 3.25 (s, 6H), 3.06 ppm (s, 6H). 13C {1H}-NMR (101 MHz, CDCl3): δ 168.1 (2C), 151.8 (2C), 59.3 (2C), 39.1 (2C), 35.1 (2C), 28.3 ppm (2C). HRMS (ESI) m/z calcd. for [C12H17N4O4]+: 281.1250 [M + H]+, found 281.1245.
- (c)
- c,a-DMU<>DMU: White crystals (0.56 g, 2.00 mmol, 4%, Rf = 0.08 (EtOAc)). 1H-NMR (400 MHz, CDCl3): δ 4.09 (t, J = 8.5 Hz, 2H), 3.77 (t, J = 8.6 Hz, 2H), 3.12 (s, 6H), 3.11 ppm (s, 6H). 13C {1H}-NMR (101 MHz, CDCl3): δ 166.1 (2C), 152.2 (2C), 49.3 (2C), 45.4 (2C), 35.7 (2C), 27.8 ppm (2C). HRMS (ESI) m/z calcd. for [C12H17N4O4]+: 281.1250 [M + H]+, found 281.1245.
- (d)
- c,s-DMU<>DMU: White crystals (1.54 g, 5.49 mmol, 11%, Rf = 0.04 (EtOAc)). 1H-NMR (400 MHz, CDCl3): δ 4.06 (dd, J = 6.1, 3.9 Hz, 2H), 3.78 (dd, J = 6.1, 3.9 Hz, 2H), 3.16 (s, 6H), 3.00 ppm (s, 6H). 13C {1H}-NMR (101 MHz, CDCl3): δ 165.8 (2C), 152.8 (2C), 55.5 (2C), 39.6 (2C), 35.7 (2C), 28.0 ppm (2C). HRMS (ESI) m/z calcd. for [C12H17N4O4]+: 281.1250 [M + H]+, found 281.1245.
Synthesis of DMT<>DMT Isomers
DMT (9.30 g, 60.30 mmol) was dimerized with an overall yield of 29% (2.73 g, 8.85 mmol).
- (a)
- c,a-DMT<>DMT: White crystals (0.70 g, 2.27 mmol, 7%, Rf = 0.04 (1:1 n-pentane/EtOAc)). 1H-NMR (400 MHz, CDCl3): δ 3.27 (s, 2H), 3.16 (s, 6H), 3.08 (s, 6H), 1.58 ppm (s, 6H). 13C {1H}-NMR (101 MHz, CDCl3): δ 169.6 (2C), 151.9 (2C), 64.4 (2C), 48.8 (2C), 36.3 (2C), 27.9 (2C), 25.0 ppm (2C). HRMS (ESI) m/z calcd. for [C14H21N4O4]+: 309.1563 [M + H]+, found 309.1557.
- (b)
- c,s-DMT<>DMT: White crystals (2.03 g, 6.58 mmol, 22%, Rf = 0.10 (EtOAc)). 1H-NMR (500 MHz, CDCl3): δ 3.72 (s, 2H), 3.14 (s, 6H), 3.00 (s, 6H), 1.50 ppm (s, 6H). 13C {1H}-NMR (126 MHz, CDCl3): δ 169.5 (2C), 152.5 (2C), 60.6 (2C), 47.6 (2C), 35.9 (2C), 28.3 (2C), 19.4 ppm (2C). HRMS (ESI) m/z calcd. for [C14H21N4O4]+: 309.1563 [M + H]+, found 309.1582.
Synthesis of DMU6-Me<>DMU6-Me Isomers
DMU6-Me (7.71 g, 50.00 mmol) was dimerized with an overall yield of 67% (5.16 g, 16.70 mmol). The crystal structure of t,s-DMU6-Me<>DMU6-Me could not be solved, and this compound was, therefore, characterized by comparing the NMR data with ref. [22].
- (a)
- t,s-DMU6-Me<>DMU6-Me: White crystals (0.21 g, 0.67 mmol, 3%, Rf = 0.05 (1:1 n-pentane/EtOAc)). 1H-NMR (400 MHz, CDCl3): δ 3.27 (s, 6H), 3.23 (s, 2H), 3.00 (s, 6H), 1.42 ppm (s, 6H). 13C {1H}-NMR (101 MHz, CDCl3): δ 168.1 (2C), 152.4 (2C), 62.2 (2C), 45.1 (2C), 31.2 (2C), 28.5 (2C), 22.3 ppm (2C). HRMS (ESI) m/z calcd. for [C14H21N4O4]+: 309.1563 [M + H]+, found 309.1556.
- (b)
- c,a-DMU6-Me<>DMU6-Me: White crystals (4.11 g, 13.30 mmol, 53%, Rf = 0.19 (EtOAc)). 1H-NMR (400 MHz, CDCl3): δ 3.21 (s, 6H), 3.16 (s, 2H), 2.82 (s, 6H), 1.65 ppm (s, 6H). 13C {1H}-NMR (101 MHz, CDCl3): δ 165.3 (2C), 152.0 (2C), 58.0 (2C), 53.6 (2C), 31.2 (2C), 28.2 (2C), 27.8 ppm (2C). HRMS (ESI) m/z calcd. for [C14H21N4O4]+: 309.1563 [M + H]+, found 309.1557.
- (c)
- c,s-DMU6-Me<>DMU6-Me: White crystals (0.82 g, 2.67 mmol, 11%, Rf = 0.03 (2:1 n-pentane/EtOAc)). 1H-NMR (400 MHz, CDCl3): δ 3.41 (s, 2H), 3.16 (s, 6H), 2.92 (s, 6H), 1.52 ppm (s, 6H). 13C {1H}-NMR (101 MHz, CDCl3): δ 165.6 (2C), 152.8 (2C), 62.5 (2C), 44.9 (2C), 32.2 (2C), 28.0 (2C), 21.6 ppm (2C). HRMS (ESI) m/z calcd. for [C14H21N4O4]+: 309.1563 [M + H]+, found 309.1557.
2.2. X-ray Data
Intensity data were collected on a Rigaku XtalLAB Synergy at 100.0(1) K diffractometer using either Cu-Kα or Mo-Kα radiation at 100.0(1) K. The temperature was maintained using an Oxford Cryostream cooling device. The structures were solved by direct methods and difference Fourier synthesis [23]. The thermal ellipsoid plot was generated using the program Mercury-3 [24] integrated within the WINGX [25] suite of programs. Detailed information on the crystal structures is provided in the Supplementary Material. X-ray crystal structures have been deposited at the Cambridge Structural Database and assigned the CCDC codes 1995805–1995812.
2.3. Laser Flash Photolysis Studies
Kinetic measurements were performed using an Edinburgh Instruments LP920 spectrometer using the third harmonic of a Quantel Brilliant B Nd:YAG laser (6 ns pulse; 10–20 mJ/pulse, λ = 355 nm) to generate the radical transient. A Hamamatsu R2856 photomultiplier tube (PMT) interfaced with a Tektronix TDS 3012C Digital Phosphor Oscilloscope was used for the detection system. Samples were exposed to laser light in Starna Spectrosil Quartz fluorometer cells (10 × 10 × 48 mm).
All kinetic experiments were carried out under pseudo-first-order conditions following the established procedure described in refs. [26,27,28], with the pyrimidine monomer and cyclobutane dimer as the excess component ([substrate] = 0.2–11 mM, [CAN] = 0.33 mM; using the molar extinction coefficient of ε = 1350 M−1 cm−1 at λ = 630 nm in acetonitrile [29], [NO3•] in the range of 108–135 µM was generated). Each data point was determined from the average of three measurements. Experimental details of the kinetic measurements and data evaluation are given in the Supplementary Material (Figures S1–S4).
2.4. DFT Calculations
Density functional theory (DFT) calculations were carried out with the Gaussian suite of programs [30] using the M062X method [31,32,33] in combination with the 6-31+G* basis set, which has been employed previously to investigate reactions of NO3• with biomolecules [26,28]. Calculations in acetonitrile were performed using the conductor-like polarizable continuum model (CPCM) [34]. All equilibrium geometries and transition structures were verified by vibrational frequency analysis at the same level of theory, and all identified transition structures showed only one imaginary frequency. The spin expectation value, <s2>, was very close to 0.75 after spin annihilation. The Gaussian archive entries for all optimized geometries, including free energy data and imaginary frequencies of the transition structures, are given in the Supplementary Material.
3. Results
The pyrimidine cyclobutane dimers studied in this work are shown in Figure 1. These compounds were prepared by photochemical dimerization of the respective N,N’-dimethylated pyrimidine using acetone as triplet sensitizer, as described previously (see Materials and Methods section) [17]. Under such conditions, dimerization was not stereospecific, and formation of syn- and anti-isomers with both cis and trans stereochemistry at the cyclobutane ring could principally occur. However, only in the case of DMU, all four possible stereoisomers were formed in synthetically useful yield, whereas, for DMT, only the cis-configured isomers, and, in the case of DMU6-Me, all isomers, except for the trans,anti-isomer, were obtained.
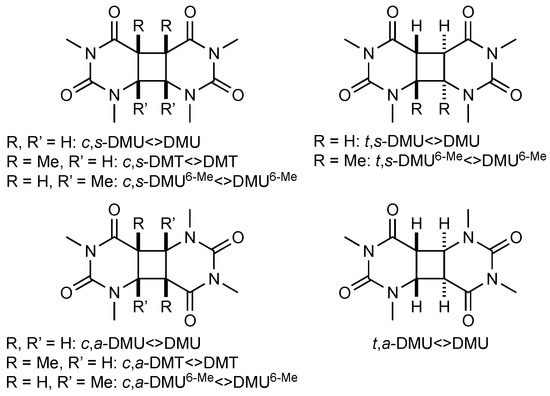
Figure 1.
Pyrimidine cyclobutane dimers studied in this work. DMU = 1,3-dimethyluracil, DMT = 1,3-dimethylthymine, DMU6-Me = 1,3,6-trimethyluracil; c = cis, t = trans, s = syn, a = anti.
The laser flash photolysis experiments were performed in acetonitrile under pseudo-first-order conditions with the dimer as an excess component by monitoring the decay of the NO3• signal at λ = 630 nm. In previous work, we found that purging the solutions to remove oxygen was detrimental to the NO3• signal for reasons not yet understood [26]. However, since the lifetime of NO3• is independent of the presence of oxygen [29], the rate data were measured without degassing the reaction mixtures.
The decay profiles depicted in Figure 2a for the exemplary reaction of NO3• with different excess concentrations of c,s-DMT<>DMT clearly showed that the rate of NO3• consumption increased with increasing [c,s-DMT<>DMT]. Determination of the second-order rate coefficient of the reaction, k, was obtained from the slope of the plot of the pseudo-first-order rate coefficient kobs vs. [c,s-DMT<>DMT] (Figure 2b).
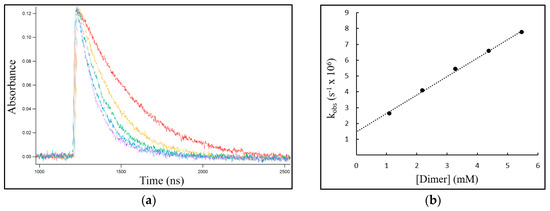
Figure 2.
The reaction of NO3• with c,s-DMT<>DMT. (a) Concentration-time profiles for NO3• recorded at λ = 630 nm with different excess concentrations of c,s-DMT<>DMT: red 1.1 mM, yellow 2.2 mM, green 3.3 mM, blue 4.4 mM, purple 5.5 mM. (b) The plot of the pseudo-first-order rate coefficient kobs vs. [dimer] to determine the second-order rate coefficient k from the slope.
The intercept in Figure 2b could be attributed to the background reaction of NO3• with the solvent acetonitrile, which proceeded via hydrogen abstraction (HAT) with a rate coefficient of 9 × 102 M−1 s−1 and led to the depletion of the NO3• signal after about 300 μs [26]. However, this decay was much slower than the consumption of NO3• through reaction with the cyclobutane dimers, which occurred on the nanosecond timescale and could, therefore, be neglected.
The second-order rate coefficients k for the reactions of NO3• with the pyrimidine cyclobutane dimers and with the respective monomers are compiled in Table 2.
Table 2.
Absolute second-order rate coefficients k for the reaction of NO3• with pyrimidine cyclobutane dimers and the corresponding monomers using nanosecond laser flash photolysis 1,2.
The rate data ranged from 8–280 × 107 M−1 s−1, revealing a very high susceptibility of all pyrimidine cyclobutane dimers towards reaction with NO3•. This high reactivity was further illustrated by comparison with the rate coefficients for the reaction of NO3• with amino acids and short peptides, which were in the range of 106–108 M−1 s−1, with the fastest reactions occurring with aromatic amino acids and proline [26,27,28,35]. In general, for a particular pyrimidine, the syn isomers reacted faster than the anti-configured isomers by a factor of about 2–4 (entries 1 vs. 3, 2 vs. 4, 5 vs. 6, and 8 vs. 9), and the trans isomers reacted faster than the cis isomers by a factor of about 1.5–3 (entries 1 vs. 2, 3 vs. 4, and 7 vs. 8). This finding largely supported the observations from the previous competition studies (see Table 1; for example, entries 8 vs. 9 and entry 10). Overall, the reactivity increased in the order DMU6-Me<>DMU6-Me → DMU<>DMU → DMT<>DMT for dimers with similar geometry and configuration at the cyclobutane ring. For example, the ratio of the rate coefficients for c,s-DMU6-Me<>DMU6-Me and c,s-DMT<>DMT was about 0.3 (entry 8 vs. 5) and only about 0.15 for the pair c,a-DMU6-Me<>DMU6-Me and c,a-DMT<>DMT (entry 9 vs. 6). This finding was remarkable since the only difference between the latter two dimers was the site of the methyl groups at the cyclobutane ring.
Rate coefficients were also determined for the reaction of NO3• with the monomers formed upon dimer cleavage (see Scheme 2). The data, which are included in Table 2, revealed also a very high reactivity with these pyrimidines. According to previous work by us, the initial step in the reaction of NO3• with pyrimidines likely proceeded through oxidative ET, leading to different products depending on the nature of the pyrimidine [36,37]. However, since the kinetic measurements were performed with a large excess of the dimers to ensure pseudo-first-order conditions, errors arising from the second-order consumption of NO3• through reaction with the respective monomers should be negligible.
4. Discussion
The overall trend that for the same stereochemistry and configuration at the cyclobutane ring (where available), DMT<>DMT is the most reactive and DMU6-Me<>DMU6-Me the least reactive dimer system is remarkable: it indicates that not only the absence or presence of a methyl group at the cyclobutane ring but also the site of methyl substitution significantly impacts the reaction rate. For example, the rate coefficients for the cis,syn-configured dimers, which would be expected to react with NO3• through ET with similar rates on the basis of the available E0 data (see Table 1), varied by a factor of 3 (entries 2, 5, vs. 8). This finding raised the question of whether these reactions proceeded indeed via initial ET, in particular since reactions of NO3• with biomolecules, such as peptides, commonly also occur through HAT [26,28]. Therefore, we explored possible alternative mechanisms in the reaction of NO3• with selected pyrimidine cyclobutane dimers using density functional theory (DFT) calculations (see Materials and Methods section).
Using c,a-DMU<>DMU as a model system, potential HAT pathways were examined by calculating the energies associated for hydrogen abstraction from the four different positions in the dimer, i.e., from C(6), C(5), and from the methyl groups at N(1) and N(3), respectively (Scheme 3a).
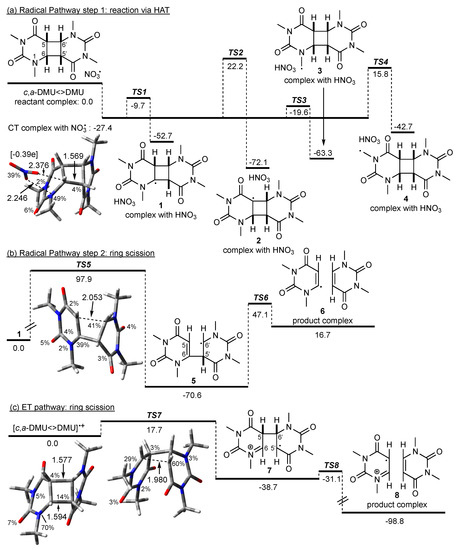
Scheme 3.
Possible pathways in the reaction of NO3• with c,a-DMU<>DMU. M062X/6-31+G* geometries and free energies in kJ mol−1 (in acetonitrile). Spin density in percentage, distances in Å, Mulliken charge on the NO3 fragment in square brackets. For the HAT pathway (a), the energy of the reactant complex without CT interactions was used as a reference point; the energy for the radical ring scission in (b) was given relative to the dimer radical 1; for the ET pathway (c), the energy of the dimer radical cation was used as a reference point (see text).
The computations predicted the formation of a charge-transfer (CT) complex between the dimer and NO3• prior to any actual reaction. The optimized geometry of the CT complex, which is included in Scheme 3, showed that NO3• coordinated via two of its oxygen atoms to N(1) and the hydrogen at C(6) of the dimer, but the structure of c,a-DMU<>DMU itself was very similar to that of the ‘intact’ dimer (not shown). However, analysis of the spin density in this reactant complex showed a partition of the spin between NO3• and N(1). Furthermore, the Mulliken charges showed a value of −0.39e on NO3•, indicating an accumulation of negative charge on this moiety. The calculated energy of a reactant complex without such CT interactions was higher by 27.4 kJ mol−1 (not shown), and this complex was used as a reference point in Scheme 3a with its energy set to 0 kJ mol−1.
NO3•-induced HAT from all four positions in c,a-DMU<>DMU was thermodynamically highly favorable with reaction energies exceeding −40 kJ mol−1. Of the four possible HAT pathways, abstraction from the methyl group at N(1) via TS3 is predicted to be the kinetically most preferred pathway, which is only about 8 kJ mol−1 above the energy of the CT complex. Hydrogen abstraction from C(6) is slightly less favorable, with TS1 being about 10 kJ mol−1 higher in energy than TS3. In contrast to this, the barriers for HAT from C(5) via TS2 and from the methyl group at N(3) via TS4 are considerably higher, and both pathways are, therefore, not kinetically competitive compared with the reaction channels via TS1 and TS3 [38].
It is highly unlikely that the dimer radical 3 resulting from the kinetically most favorable HAT from the N(1)-Me group could lead to cleavage of the cyclobutane ring in a straightforward fashion. On the other hand, to explore whether the formation of a radical site at the cyclobutane ring itself could principally lead to ring scission, which has been suggested for the reaction of pyrimidine cyclobutane dimers with other O-centered radicals [12], homolytic fragmentation in the C(6) radical 1 was exemplary calculated. The energy profile for this reaction is given in Scheme 3b, which showed that the stepwise bond scissions were associated with high barriers TS5 and TS6. The geometry of TS5 was characterized by a similar spin density at C(6) and C(6′) on both pyrimidine moieties and a considerably large distance between C(5) and C(6′) of 2.053 Å, suggesting a relatively late transition state. Furthermore, while the formation of 5 through scission of the C(5)–C(6′) bond was exothermic, the subsequent cleavage of the C(6)–C(5′) bond and formation of the product complex consisting of the repaired monomer and the monomer vinyl radical was endothermic, rendering repair through a radical-only mechanism as highly unlikely. Based on these data, NO3•-induced HAT in c,a-DMU<>DMU could, therefore, not be considered as a ‘productive’ pathway with regards to cleavage of the cyclobutane ring under these conditions.
In contrast to this, oxidation of c,a-DMU<>DMU by NO3• led to the dimer radical cation [c,a-DMU<>DMU]•+, which still had an intact cyclobutane ring where most of the spin was localized on N(1). The energy diagram for the cleavage of c,a-DMU<>DMU]•+ is shown in Scheme 3c. Thus, fragmentation of the C(6)–C(5′) bond was associated with a barrier TS7 of only about 18 kJ mol−1 and gave the singly bonded dimer 7 in an exothermic process. The distance between C(6) and C(5′) in TS7 was considerably shorter than that of the breaking bond in TS5, which was in line with an earlier, energetically more favorable process. Subsequent homolytic scission of the C(5)–C(6′) bond via TS8 was only about 8 kJ mol−1 higher in energy than 7 and should occur rapidly to give the product complex 8 in a highly exothermic reaction.
Scheme 4a shows the calculated energy profile for the reaction of NO3• with the isomeric dimer c,s-DMU<>DMU. Interestingly, the formation of a complex with a broken C(6)–C(6′) bond occurred, as revealed by the distance of 1.955 Å (Scheme 4a). A complex with an intact cyclobutane ring could not be located, which suggested that c,s-DMU<>DMU underwent barrierless ring scission upon encountering NO3•. Analysis of the spin density and Mulliken charges in this complex showed complete loss of radical character and accumulation of negative charge on the NO3 fragment. The spin was distributed over the dimer with 27% located on each of C(6) and C(6′) and about 20% on each of N(1) and N(1′). These data suggested that the complex should, in fact, be regarded as a nitrate-dimer radical cation pair resulting from ET, i.e., [NO3−] [c,s-DMU<>DMU]•+. The energy of [NO3−] [c,s-DMU<>DMU]•+ was about 43 kJ mol−1 lower than the sum of the energies of the free reactants, which were used as a reference point for the energy profile. A reactant complex without any CT contribution (similar to c,a-DMU<>DMU) could not be located computationally for c,s-DMU<>DMU.
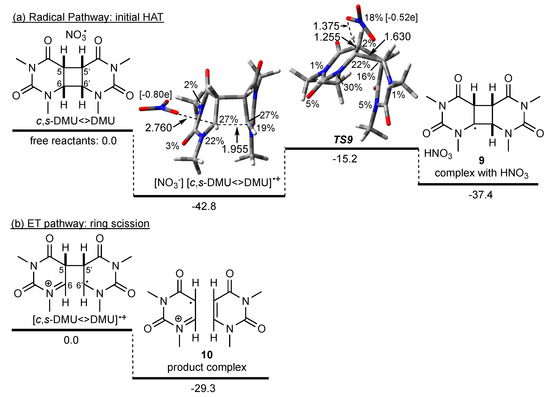
Scheme 4.
Possible pathways in the reaction of NO3• with c,s-DMU<>DMU. M062X/6-31+G* geometry and free energies in kJ mol−1 (in acetonitrile). Spin density in percentage, distances in Å, Mulliken charge on NO3 fragment in square brackets. For the HAT pathway (a), the sum of the energies of the free reactants was used as a reference point; for the ET pathway (b), the energy of the dimer radical cation was used as a reference point (see text).
To explore a potential homolytic fragmentation of the cyclobutane ring in c,s-DMU<>DMU, the first step through NO3•-induced HAT from C(6) was calculated. This process was associated with a barrier (TS9) of about 28 kJ mol−1 relative to [NO3−] [c,s-DMU<>DMU]•+ and led to the product complex of the C(6) dimer radical 9 and HNO3 in an overall exothermic reaction with respect to the free reactants. Inspection of the spin density in TS9 revealed that only 18% of the spin was remaining on the NO3 fragment, indicating a relatively late transition state that was also supported by the comparably long C(6)–H distance of 1.255 Å compared with 1.087 Å in the dimer (not shown). Interestingly, in TS9, the C(6)–C(6′) distance was only 1.630 Å, indicating a nearly restored cyclobutane ring (1.568 Å in the intact dimer, not shown). Intrinsic reaction coordinate (IRC) calculations confirmed that the complex and TS9 were connected and that with increasing C(6)–H distance, the C(6)–C(6′) bond snapped close. Together with the reduced negative charge on the NO3 moiety in TS9 (Mulliken charge −0.52e), these findings suggested that if a radical HAT from C(6) starting from [NO3−] [c,s-DMU<>DMU]•+ were to occur, a reverse charge transfer from the dimer to the NO3 moiety would be required to traverse TS9.
On the other hand, scission of the remaining C(5)–C(5′) bond in the dimer radical cation [c,s-DMU<>DMU]•+, which according to the calculations should be immediately formed when NO3• encounters c,s-DMU<>DMU, was both barrier-less (i.e., a TS could not be located) and led to the product complex 10 in an exothermic reaction, relative to [c,s-DMU<>DMU]•+ (Scheme 4b). From these data, it could, therefore, be concluded that repair of c,s-DMU<>DMU was initiated by NO3•-induced ET, whereas a HAT pathway was not competitive.
Calculations of the reactant complexes with other pyrimidine cyclobutane dimers used in the kinetic studies clearly revealed that all syn-configured dimers formed similar nitrate-dimer radical cation pairs, where the dimer had an already cleaved C(6)–C(6′) bond and should undergo barrierless scission of the remaining C(5)–C(5′) bond. This behavior was not limited to acetonitrile as a solvent but was similarly also found in water (data not shown). It should be noted that it was not possible to locate a geometry for any syn-configured dimer radical cation in which the cyclobutane ring was not broken.
The reactant complexes between NO3• and anti-configured dimers generally constituted CT complexes with a largely intact cyclobutane ring, in which the spin density was distributed over both NO3• and dimer and negative charge accumulating on the NO3 moiety. These data suggested a less favorable ET process, which was in line with the higher E0 value determined for anti-configured dimers (see Table 1). The scission of the cyclobutane ring was associated with a barrier, which slowed the overall repair process down. Because ET was less favorable, it should be noted that it could not be excluded that a competing HAT reaction at the methyl group at N(1) or at C(6) could occur in anti-configured dimers to some extent. However, the rate coefficient determined experimentally for HAT from Namide-alkyl groups of about 6–10 × 107 M−1 s−1 [35] suggested that such a reaction should only be a minor contributor to the overall reaction of NO3• with the dimer.
The finding that the trans-configured dimers reacted faster with NO3• than the isomeric cis-configured dimers of the same pyrimidine could be rationalized by steric effects. Analysis of the spin densities in the NO3• complexes with the dimers (see Scheme 3 and Scheme 4) suggested that oxidation occurred at the amide nitrogen N(1) in both pyrimidine moieties. Therefore, accessibility of this site for NO3• should be a factor that modulates the rate of oxidation—a proposition that is supported by recent findings that steric hindrance at tertiary amides slows down the rate of oxidation by NO3• [35]. Figure 3 shows in the top row the X-ray structures of t,s-DMU<>DMU and c,s-DMU<>DMU. t,s-DMU<>DMU had a largely linear shape, where N(1) in both pyrimidine rings could be accessed by NO3• from both above and below the molecular plane, as indicated by the green arrows. In contrast to this, c,s-DMU<>DMU clearly had a freely accessible convex side, but the approach of NO3• from the concave side was sterically hindered, which should lower the rate of oxidation.
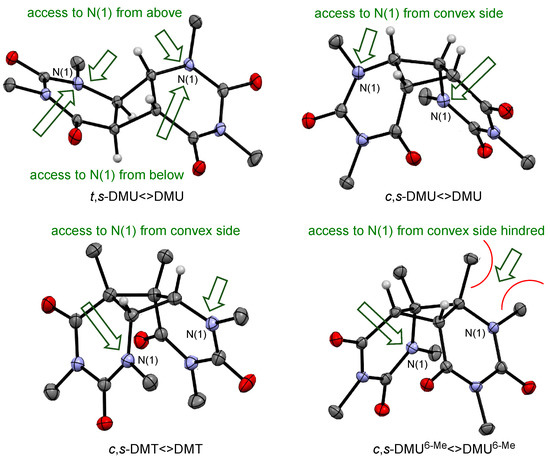
Figure 3.
X-ray structures for selected dimers and likely approach by NO3• (green arrow).
Similarly, steric hindrance could also explain the slower reaction of the dimers of DMU6-Me, compared with those of DMU and DMT with similar configurations. Figure 3 presents in the bottom row the X-ray structures of c,s-DMT<>DMT and c,s-DMU6-Me<>DMU6-Me. It would be expected that the higher degree of alkyl substitution at the cyclobutane ring in both dimers, compared with the isomeric c,s-DMU<>DMU, should facilitate oxidation. Indeed, the rate coefficient, shown in Table 2, for the reaction with c,s-DMT<>DMT was about 30% higher than with c,s-DMU<>DMU. On the other hand, the considerably lower reactivity of c,s-DMU6-Me<>DMU6-Me suggested that steric hindrance caused by the methyl substituents at C(6) was impeding NO3• access to the adjacent amide N(1) in both pyrimidine moieties, which slowed down the rate of oxidation.
5. Conclusions
NO3• reacted with pyrimidine cyclobutane dimers via a rapid ET at N(1), which triggered cleavage of the cyclobutane ring and regeneration of a pyrimidine monomer and a monomer radical cation.
The four possible geometric isomers of the pyrimidine cyclobutane dimers differed considerably in their reactivity towards NO3•, with the syn-configured dimers being more reactive than their anti-configured counterparts. The higher reactivity of the former was manifested by the formation of a nitrate/dimer radical cation pair, where the negative charge had accumulated on the nitrate moiety, and the C(6)–C(6′) bond in the dimer was already broken. The scission of the remaining C(5)–C(5)’ bond was barrierless and exothermic. In the case of the anti-configured dimers, ET was less progressed, which was in line with the higher E0 value, and a CT complex with NO3• was formed. The cyclobutane ring in this complex, as well as in the corresponding dimer radical cation, was still intact. Therefore, ring-opening in the dimer radical cation required sequential cleavage of two bonds, of which the first step was associated with a moderate barrier. However, because of the slower ET in the anti-configured dimers, it could not be excluded that NO3•-induced HAT at the methyl group of N(1) and from C(6) at the cyclobutane ring might also occur to some extent. However, subsequent fragmentation of the cyclobutane ring through radical-only pathways was not competitive with the cleavage process in a dimer radical cation.
Steric effects were likely responsible for the higher reactivity of trans-configured dimers compared with their cis isomers. Thus, the geometry of the trans dimers allowed the approach of NO3• to the N(1) atoms in the pyrimidines from both sides of the molecular plane, whereas, in the cis dimers, only the convex side was readily accessible for NO3•. Likewise, the order of reactivity of DMU6-Me<>DMU6-Me → DMU<>DMU → DMT<>DMT towards NO3• was a result of steric and electronic effects. The dimers of DMT generally showed the highest reactivity due to the electron-donating methyl groups at C(5,5′) on the cyclobutane ring that increased their susceptibility to oxidation by NO3•. Remarkably, methyl substituents at C(6,6′) in the DMU6-Me dimers lowered their reactivity, even below that of the DMU dimers. This finding could be rationalized by the difficulty for NO3• to approach N(1) in DMU6-Me<>DMU6-Me due to steric hindrance caused by the adjacent methyl groups.
Overall, this study clearly revealed that the rate of the NO3• reaction was not only determined by the redox potential of the dimers but also to a considerable extent by the accessibility of the reaction site for NO3•. In future work, we will further explore the role of steric effects on the radical-induced oxidative damage of DNA constituents, such as pyrimidine deoxy nucleosides and their cyclobutane dimers.
Supplementary Materials
The following are available online at https://www.mdpi.com/2624-8549/2/2/27/s1, 1H and 13C-NMR spectra of substrates used in the laser flash photolysis study, crystallographic data for pyrimidine cyclobutane dimers, details of laser flash photolysis experiments and kinetic plots, Gaussian archive entries.
Author Contributions
Conceptualization, U.W.; methodology, T.H., J.G.N., and U.W.; validation, T.H. and J.G.N.; formal analysis, T.H., J.M.W., and J.G.N.; investigation, T.H., J.M.W, J.G.N., and U.W.; resources, T.H., J.G.N., and U.W.; data curation, T.H. and J.G.N.; writing—original draft preparation, J.G.N. and U.W.; writing—review and editing, T.H., J.M.W., J.G.N., and U.W.; visualization, J.G.N. and U.W.; supervision, J.G.N. and U.W.; project administration, U.W.; funding acquisition, U.W. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by The University of Melbourne and the Australian Research Council (LE0989197 and LE170100065).
Acknowledgments
We acknowledge the support of this work by Oliver Krüger.
Conflicts of Interest
The authors declare no conflict of interest.
References and Notes
- D’Orazio, J.; Jarrett, S.; Amaro-Ortiz, A.; Scott, T. UV Radiation and the Skin. Int. J. Mol. Sci. 2013, 14, 12222–12248. [Google Scholar] [CrossRef] [PubMed]
- Douki, T.; Sauvaigo, S.; Odin, F.; Cadet, J. Formation of the Main UV-induced Thymine Dimeric Lesions within Isolated and Cellular DNA as Measured by High Performance Liquid Chromatography-Tandem Mass Spectrometry. J. Biol. Chem. 2000, 275, 11678–11685. [Google Scholar] [CrossRef] [PubMed]
- Barak, Y.; Cohen-Fix, O.; Livneh, Z. Deamination of Cytosine-containing Pyrimidine Photodimers in UV-irradiated DNA. Significance for UV Light Mutagenesis. J. Biol. Chem. 1995, 270, 24174–24179. [Google Scholar] [CrossRef] [PubMed]
- Ravanat, J.-L.; Douki, T.; Cadet, J. Direct and indirect effects of UV radiation on DNA and its components. J. Photochem. Photobiol. B 2001, 63, 88–102. [Google Scholar] [CrossRef]
- Thomas, D.C.; Kunkel, T.A. Replication of UV-irradiated DNA in human cell extracts: Evidence for mutagenic bypass of pyrimidine dimers. Proc. Natl. Acad. Sci. USA 1993, 90, 7744–7748. [Google Scholar] [CrossRef] [PubMed]
- Burroughs, A.M.; Aravind, L. RNA damage in biological conflicts and the diversity of responding RNA repair systems. Nucleic Acids Res. 2016, 44, 8525–8555. [Google Scholar] [CrossRef]
- Krokan, H.E.; Standal, R.; Slupphaug, G. DNA glycosylases in the base excision repair of DNA. Biochem. J. 1997, 325, 1–16. [Google Scholar] [CrossRef]
- Brettel, K.; Byrdin, M. Reaction mechanisms of DNA photolyase. Curr. Opin. Struct. Biol. 2010, 20, 693–701. [Google Scholar] [CrossRef]
- Friedel, M.G.; Gierlich, J.; Carell, T. Cyclobutane Pyrimidine Dimers as UV-Induced DNA Lesions. In PATAI’S Chemistry of Functional Groups; Rappoport, Z., Ed.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2009. [Google Scholar] [CrossRef]
- Huntley, J.J.; Nieman, R.A.; Rose, S.D. Development and Investigation of a Novel Oxidative Pyrimidine Dimer Splitting Model. Photochem. Photobiol. 1999, 69, 1–7. [Google Scholar] [CrossRef]
- Treml, J.; Smejkal, K. Flavonoids as Potent Scavengers of Hydroxyl Radicals. Compr. Rev. Food Sci. F. 2016, 15, 720–738. [Google Scholar] [CrossRef]
- Heelis, P.; Deeble, D.; Kim, S.-T.; Sancar, A. Splitting of Cis-syn Cyclobutane Thymine-thymine Dimers by Radiolysis and Its Relevance to Enzymatic Photoreactivation. Int. J. Radiat. Biol. 1992, 62, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.-Q.; Song, Q.-H.; Hei, X.-M.; Guo, Q.-X.; Lin, W.-Z. Oxidative Splitting of a Pyrimidine Cyclobutane Dimer: A Pulse Radiolysis Study. Chin. J. Chem. 2003, 21, 16–19. [Google Scholar] [CrossRef]
- Alfassi, Z.B.; Harriman, A.; Huie, R.E.; Mosseri, S.; Neta, P. The redox potential of the azide/azidyl couple. J. Phys. Chem. 1987, 91, 2120–2122. [Google Scholar] [CrossRef]
- Ito, T.; Shinohara, H.; Hatta, H.; Nishimoto, S.-I.; Fujita, S.-I. Radiation-Induced and Photosensitized Splitting of C5−C5’-Linked Dihydrothymine Dimers: Product and Laser Flash Photolysis Studies on the Oxidative Splitting Mechanism. J. Phys. Chem. A 1999, 103, 8413–8420. [Google Scholar] [CrossRef]
- Baciocchi, E.; Del Giacco, T.; Murgia, S.M.; Sebastiani, G.V. Rate and mechanism for the reaction of the nitrate radical with aromatic and alkylaromatic compounds in acetonitrile. J. Chem. Soc. Chem. Commun. 1987, 1246–1248. [Google Scholar] [CrossRef]
- Krüger, O.; Wille, U. Oxidative Cleavage of a Cyclobutane Pyrimidine Dimer by Photochemically Generated Nitrate Radicals (NO3•). Org. Lett. 2001, 3, 1455–1458. [Google Scholar] [CrossRef]
- Boussicault, F.; Krüger, O.; Robert, M.; Wille, U. Dissociative electron transfer to and from pyrimidine cyclobutane dimers: An electrochemical study. Org. Biomol. Chem. 2004, 2, 2742–2750. [Google Scholar] [CrossRef]
- Neta, P.; Huie, R.E.; Ross, A.B. Rate Constants for Reactions of Inorganic Radicals in Aqueous Solution. J. Phys. Chem. Ref. Data 1988, 17, 1027–1284. [Google Scholar] [CrossRef]
- Wenska, G.; Paszyc, S. Electron-acceptor-sensitized splitting of cyclobutane-type thymine dimers. J. Photochem. Photobiol. B 1990, 8, 27–37. [Google Scholar] [CrossRef]
- Pac, C.; Kubo, J.; Majima, T.; Sakurai, H. Structure-reactivity relationships in redox-photosensitized splitting of pyrimidine dimers and unusual enhancing effect of molecular oxygen. Photochem. Photobiol. 1982, 36, 273–282. [Google Scholar] [CrossRef]
- Krüger, O. Oxidative Schädigung und Reparatur von Nucleobasen und Nucleosiden durch NO3 Radikale. Ph.D. Thesis, Christian-Albrechts University, Kiel, Germany, 2002. [Google Scholar]
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edlington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Cryst. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Nathanael, J.G.; Hancock, A.N.; Wille, U. Reaction of Amino Acids, Di- and Tripeptides with the Environmental Oxidant NO3•: A Laser Flash Photolysis and Computational Study. Chem. Asian J. 2016, 11, 3188–3195. [Google Scholar] [CrossRef]
- Nathanael, J.G.; Gamon, L.F.; Cordes, M.; Rablen, P.R.; Bally, T.; Fromm, K.F.; Giese, B.; Wille, U. Amide Neighbouring-Group Effects in Peptides: Phenylalanine as Relay Amino Acid in Long-Distance Electron Transfer. ChemBioChem 2018, 19, 922–926. [Google Scholar] [CrossRef]
- Nathanael, J.G.; Wille, U. Oxidative Damage in Aliphatic Amino Acids and Di- and Tripeptides by the Environmental Free Radical Oxidant NO3•: The Role of the Amide Bond Revealed by Kinetic and Computational Studies. J. Org. Chem. 2019, 84, 3405–3418. [Google Scholar] [CrossRef]
- Del Giacco, T.; Baciocchi, E.; Steenken, S. One-Electron Oxidation of Alkylbenzenes in Acetonitrile by Photochemically Produced Nitrate Radical: Evidence for an Inner-Sphere Mechanism. J. Phys. Chem. 1993, 97, 5451–5456. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Goerigk, L.; Hansen, A.; Bauer, C.; Ehrlich, S.; Najibi, A.; Grimme, S. A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 32184–32215. [Google Scholar] [CrossRef]
- Goerigk, L.; Grimme, S. A thorough benchmark of density functional methods for general main group thermochemistry, kinetics, and noncovalent interactions. Phys. Chem. Chem. Phys. 2011, 13, 6670–6688. [Google Scholar] [CrossRef]
- Takano, Y.; Houk, K.N. Benchmarking the Conductor-like Polarizable Continuum Model (CPCM) for Aqueous Solvation Free Energies of Neutral and Ionic Organic Molecules. J. Chem. Theory Comput. 2005, 1, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Nathanael, J.G.; White, J.M.; Richter, A.; Nuske, M.; Wille, U. Radical-induced oxidative damage of proline is influenced by its position in a peptide: A kinetic and product study. manuscript in preparation.
- Goeschen, C.; White, J.M.; Gable, R.W.; Wille, U. Oxidative Damage of Pyrimidine Nucleosides by the Environmental Free Radical Oxidant NO3• in the Absence and Presence of NO2• and Other Radical and Non-Radical Oxidants. Aust. J. Chem. 2012, 65, 427–437. [Google Scholar] [CrossRef]
- The higher reactivity of DMT and DMU6-Me could be due to the additional methyl group at C(5) and C(6), respectively, which should lower the redox potential compared with that of DMU.
- Quantum mechanical tunneling was not considered in the HAT calculations, as these were aimed to qualitatively assess the most reactive sites and not to predict rate coefficients for these processes. Experimental rate data have been determined previously (see ref. [35]).








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).