Reactive Sterol Electrophiles: Mechanisms of Formation and Reactions with Proteins and Amino Acid Nucleophiles †


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
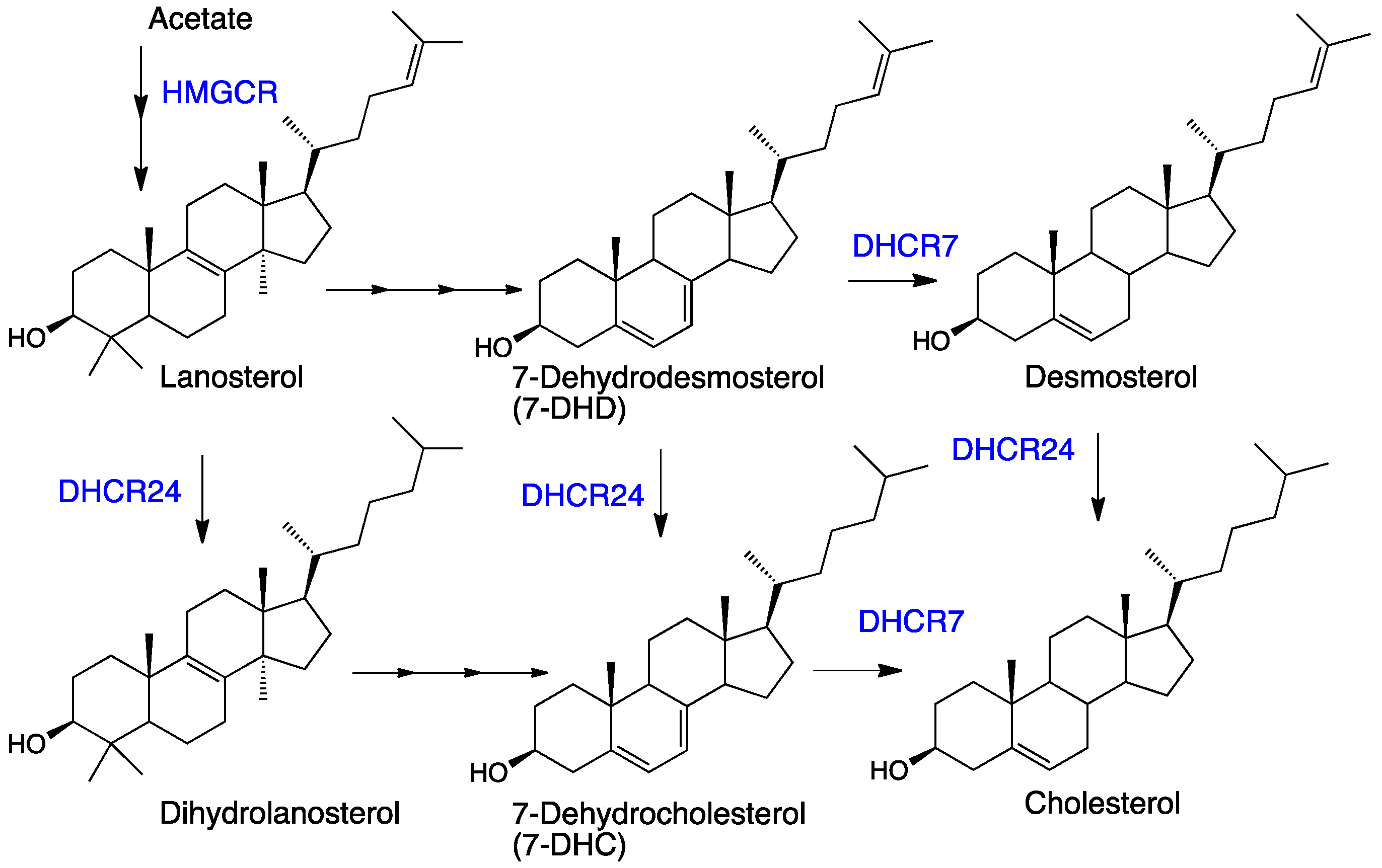
:1. Introduction
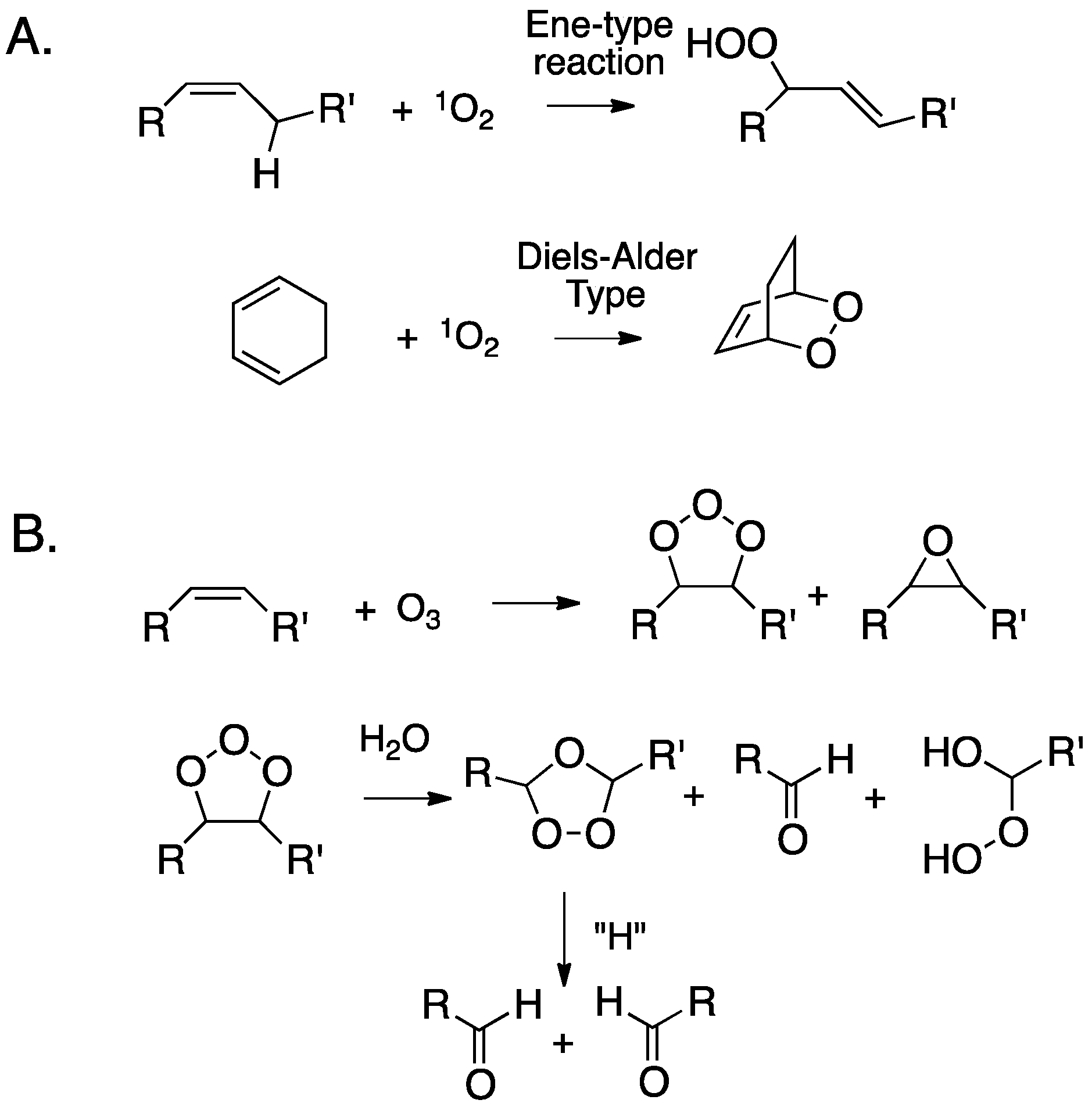
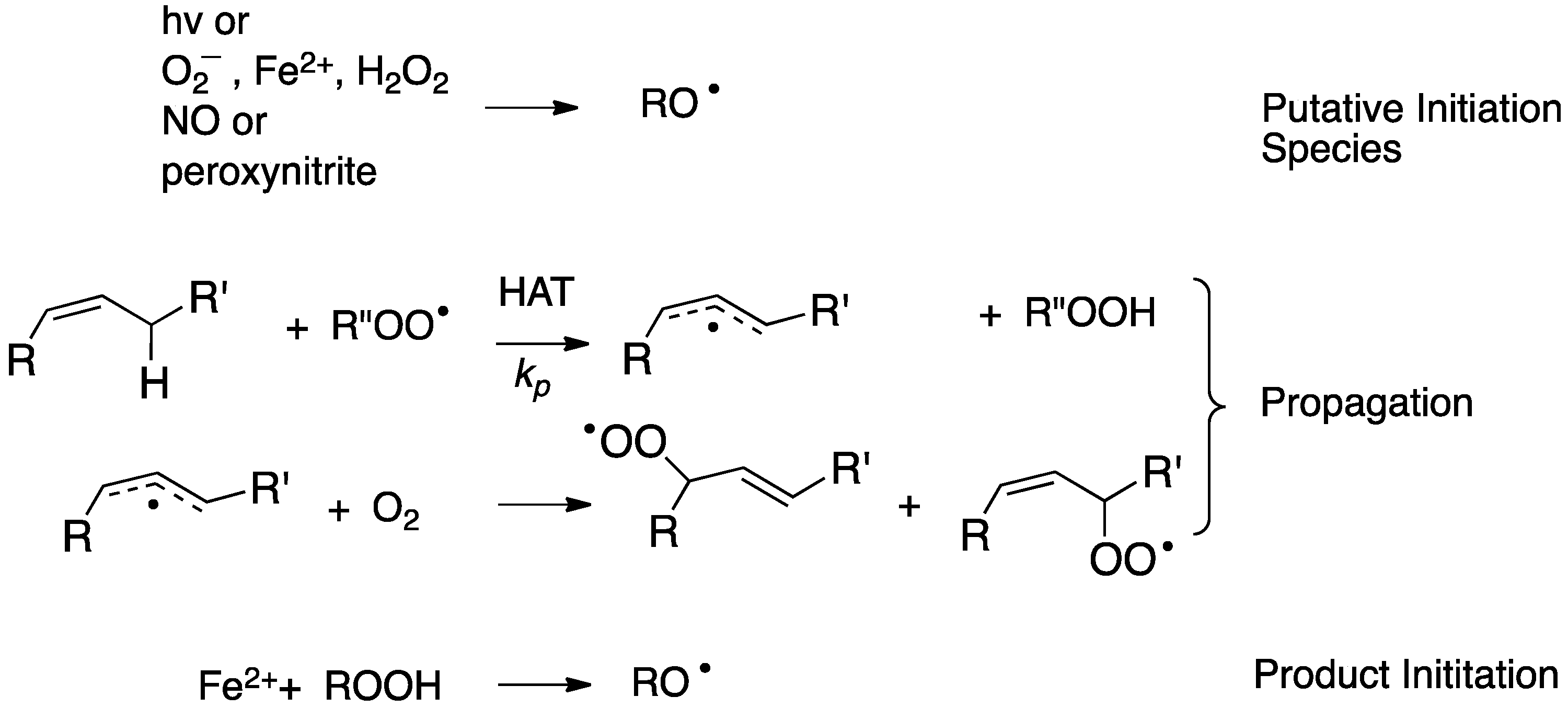
2. Primary Reactions
3. Cholesterol Autoxidation
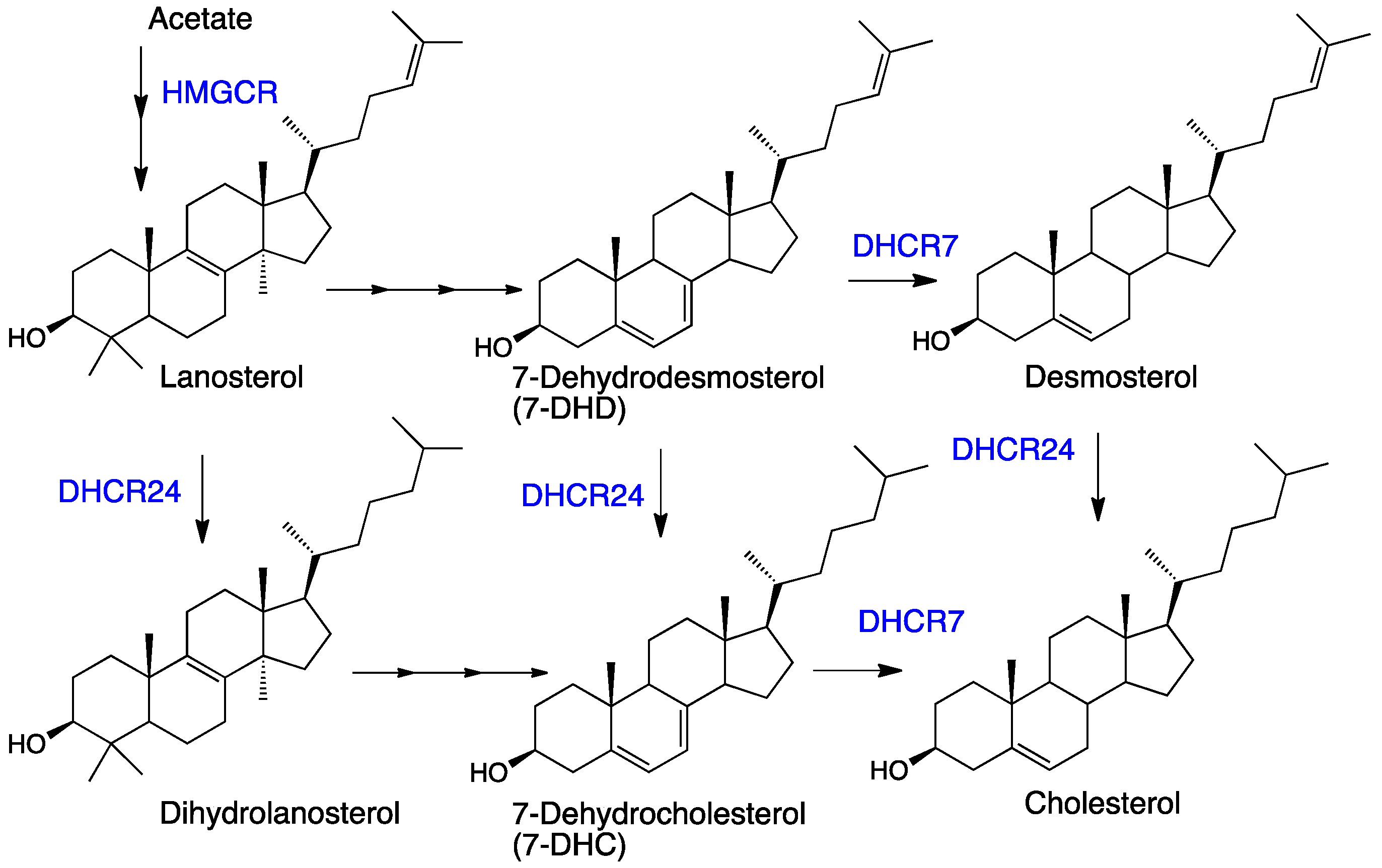
4. Cholesterol-Derived Electrophiles
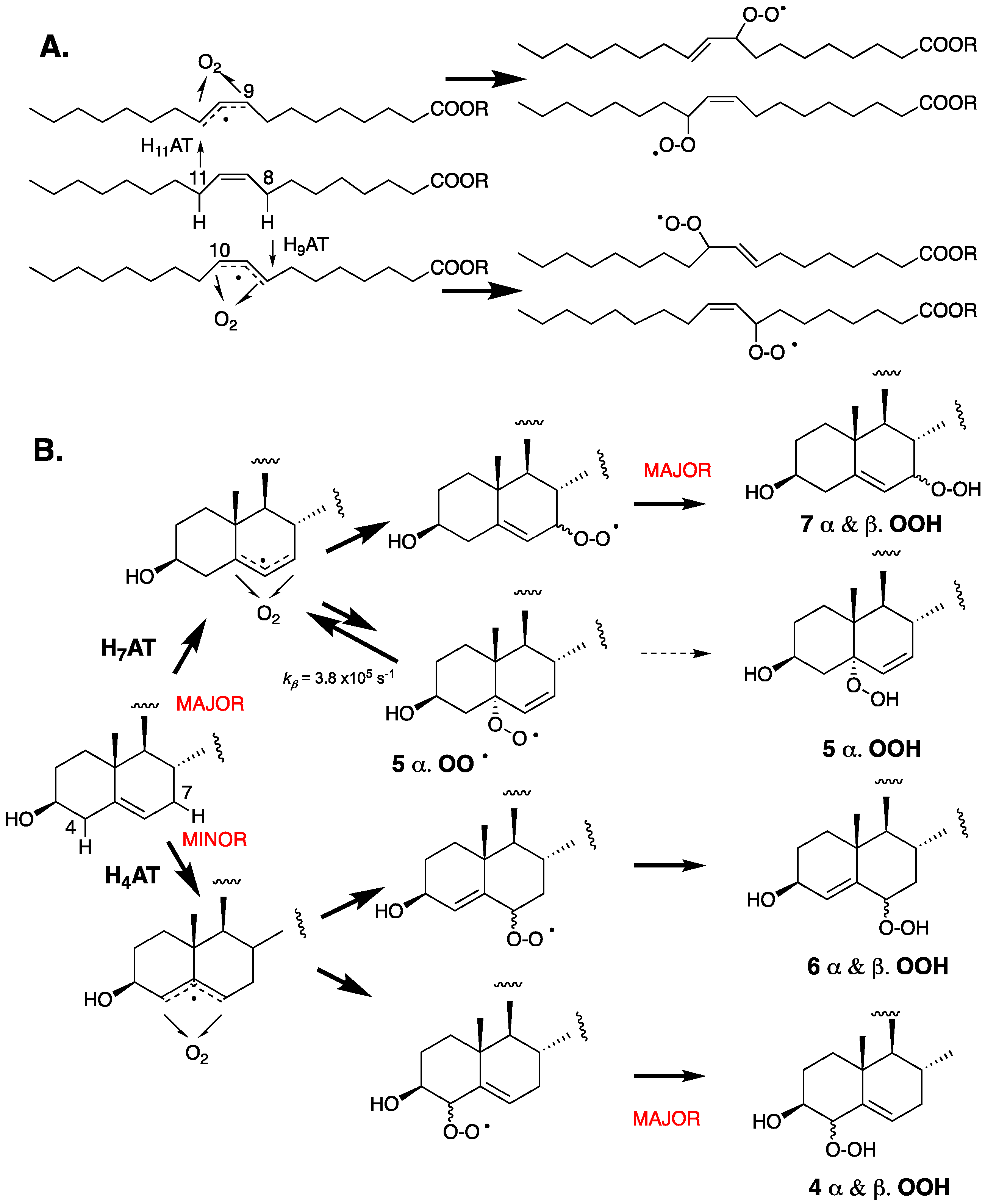
5. Autoxidation of 7- and 8-Dehydrocholesterols
6. 7- and 8-Dehydrocholesterol-Derived Electrophiles
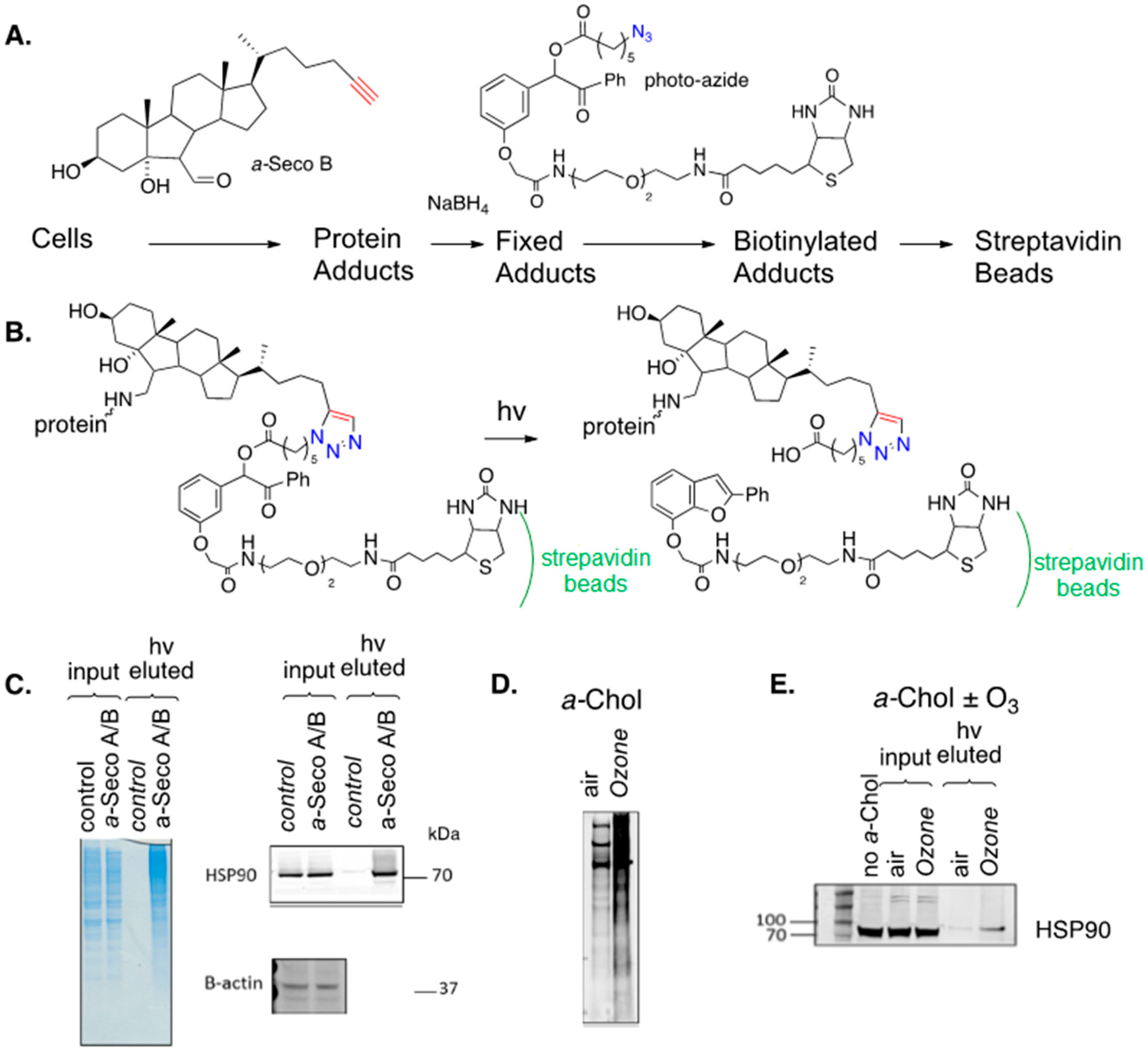
7. Protein Adduction of Lipid-Derived Electrophiles
7.1. PUFA-Derived Protein Adducts
7.2. Cholesterol-Derived Protein Adducts
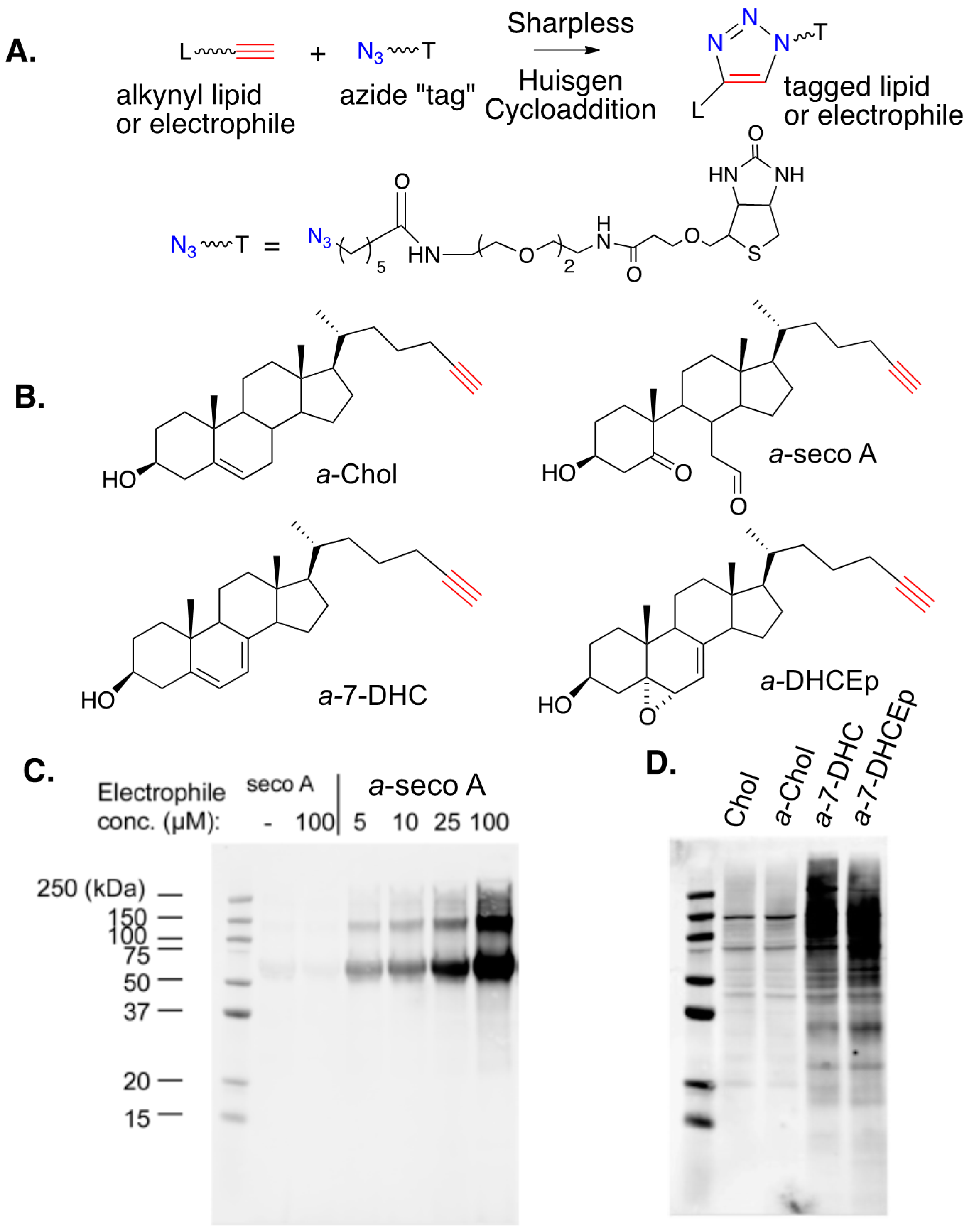
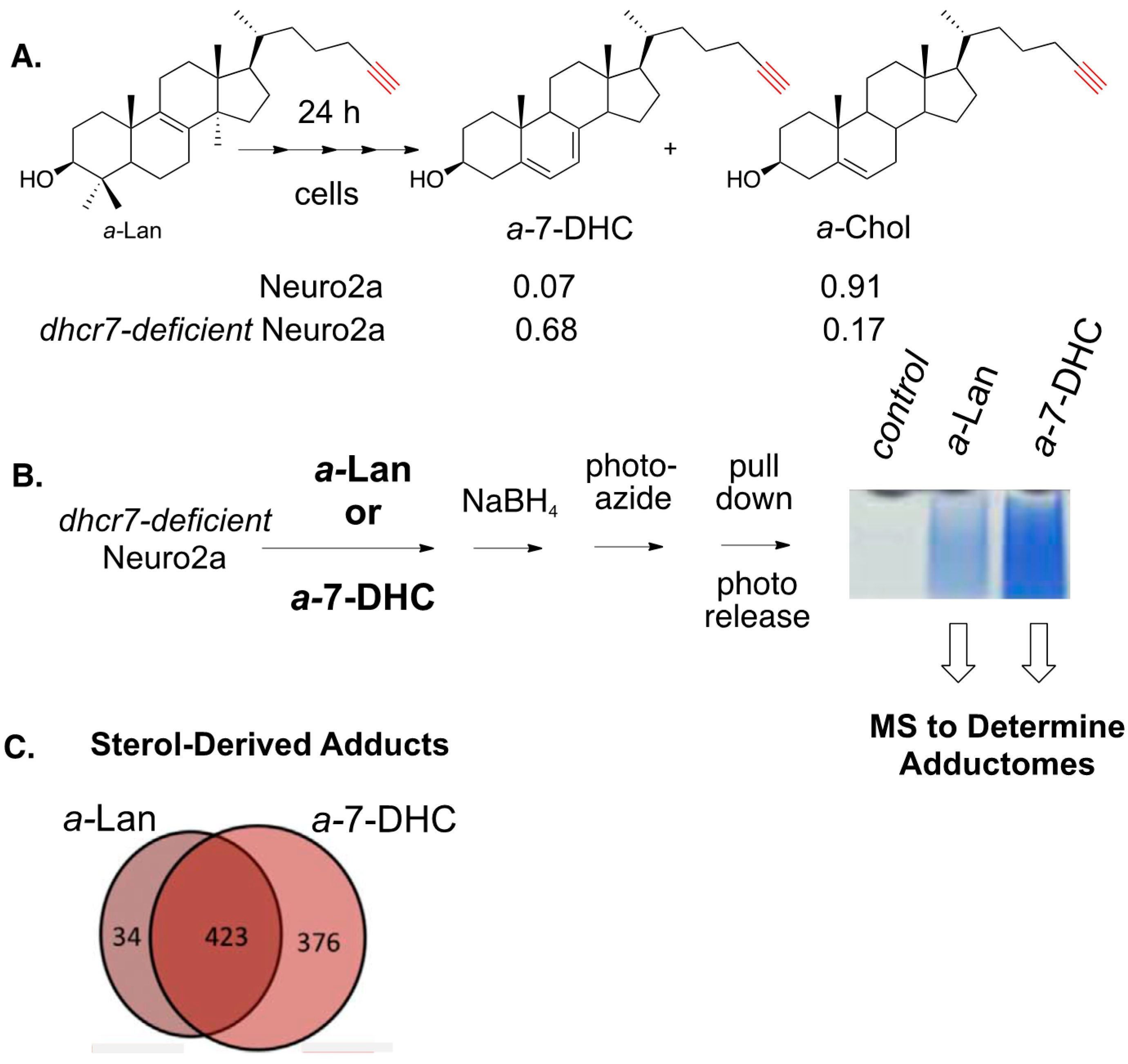
8. Alkynyl-Sterol Probes
9. Questions and Prospects
Funding
Acknowledgments
Conflicts of Interest
References
- EPA. National Ambient Air Quality Standards (NAAQS)—Ozone (O3); EPA: Washington, DC, USA, 2014.
- Hollingsworth, J.W.; Kleeberger, S.R.; Foster, W.M. Ozone and Pulmonary Innate Immunity. Proc. Am. Thorac. Soc. 2007, 4, 240–246. [Google Scholar] [CrossRef] [Green Version]
- Antczak, A.; Nowak, D.; Shariati, B.; Król, M.; Piasecka, G.; Kurmanowska, Z. Increased hydrogen peroxide and thiobarbituric acid-reactive products in expired breath condensate of asthmatic patients. Eur. Respir. J. 1997, 10, 1235–1241. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B. Lipid peroxidation, antioxidants and cardiovascular disease: How should we move forward? Cardiovasc. Res. 2000, 47, 410–418. [Google Scholar] [CrossRef] [Green Version]
- Bouhajja, H.; Kacem, F.H.; Abdelhedi, R.; Ncir, M.; Dimitrov, J.D.; Marrakchi, R.; Jamoussi, K.; Rebaï, A.; El Feki, A.; Abid, M.; et al. Potential Predictive Role of Lipid Peroxidation Markers for Type 2 Diabetes in the Adult Tunisian Population. Can. J. Diabetes 2018, 42, 263–271. [Google Scholar] [CrossRef]
- Polidori, M.C.; Praticó, D.; Savino, K.; Rokach, J.; Stahl, W.; Mecocci, P. Increased F2 isoprostane plasma levels in patients with congestive heart failure are correlated with antioxidant status and disease severity. J. Card. Fail. 2004, 10, 334–338. [Google Scholar] [CrossRef]
- Bastos, A.D.S.; Graves, D.T.; Loureiro, A.P.D.M.; Júnior, C.R.; Corbi, S.; Frizzera, F.; Scarel-Caminaga, R.; Câmara, N.O.S.; Andriankaja, O.M.; Hiyane, M.I.; et al. Diabetes and increased lipid peroxidation are associated with systemic inflammation even in well-controlled patients. J. Diabetes Complicat. 2016, 30, 1593–1599. [Google Scholar] [CrossRef] [Green Version]
- Mishra, S.; Mishra, B.B. Study of Lipid Peroxidation, Nitric Oxide End Product, and Trace Element Status in Type 2 Diabetes Mellitus with and without Complications. Int. J. Appl. Basic Med Res. 2017, 7, 88–93. [Google Scholar] [CrossRef] [Green Version]
- Di Domenico, F.; Tramutola, A.; Butterfield, D.A. Role of 4-hydroxy-2-nonenal (HNE) in the pathogenesis of alzheimer disease and other selected age-related neurodegenerative disorders. Free. Radic. Boil. Med. 2017, 111, 253–261. [Google Scholar] [CrossRef]
- Usui, K.; Hulleman, J.D.; Paulsson, J.F.; Siegel, S.J.; Powers, E.; Kelly, J.W. Site-specific modification of Alzheimer’s peptides by cholesterol oxidation products enhances aggregation energetics and neurotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 18563–18568. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Powers, E.; Nieva, J.; Huff, M.E.; Dendle, M.A.; Bieschke, J.; Glabe, C.G.; Eschenmoser, A.; Wentworth, P.; Lerner, R.A.; et al. Metabolite-initiated protein misfolding may trigger Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 4752–4757. [Google Scholar] [CrossRef] [Green Version]
- Simonian, N.A.; Coyle, J.T. Oxidative stress in neurodegenerative diseases. Annu. Rev. Pharmacol. Toxicol. 1996, 36, 83–106. [Google Scholar] [CrossRef]
- Björkhem, I.; Cedazo-Mínguez, A.; Leoni, V.; Meaney, S. Oxysterols and neurodegenerative diseases. Mol. Asp. Med. 2009, 30, 171–179. [Google Scholar] [CrossRef]
- Marnett, L.J. Lipid peroxidation—DNA damage by malondialdehyde. Mutat. Res. Mol. Mech. Mutagen. 1999, 424, 83–95. [Google Scholar] [CrossRef]
- Marnett, L.J. Oxyradicals and DNA damage. Carcinogenesis 2000, 21, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, I.; Larrayoz, I. Cholesterol oxidation in the retina: Implications of 7KCh formation in chronic inflammation and age-related macular degeneration. J. Lipid Res. 2010, 51, 2847–2862. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Ryurin, V.A.; Hammond, V.J.; Herback, N.; Aichler, M.; Walch, A.; Eggenfhofer, E.; et al. Inactivation of the ferrotopsis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Conrad, M.; Pratt, D.A. The chemical basis of ferroptosis. Nat. Methods 2019, 15, 1137–1147. [Google Scholar] [CrossRef]
- Kohn, H.I.; Liversedge, N. On a New Aerobic Metabolite Whose Production by Brain is Inhibited by Apomorphine, Emetine, Ergotamine, Epinephrine and Menadione. J. Pharmacol. Exper. Therap. 1944, 82, 292–300. [Google Scholar]
- Nair, V.; Turner, G.A. The thiobarbituric acid test for lipid peroxidation: Structure of the adduct with malondialdehyde. Lipids 1984, 19, 804–805. [Google Scholar] [CrossRef]
- Bernheim, F.; Bernheim, M.L.C.; Wilbur, K.M. The reaction between thiobarbituric acid and the oxidation products of certain lipides. J. Boil. Chem. 1948, 174, 257–264. [Google Scholar]
- Ingold, K.U. Peroxy radicals. Accounts Chem. Res. 1969, 2, 1–9. [Google Scholar] [CrossRef]
- Ingold, K.U. 60 Years of Research on Free Radical Physical Organic Chemistry; American Chemical Society: Washington, DC, USA, 2015; pp. 223–250. [Google Scholar]
- Porter, N.A. Mechanisms for the autoxidation of polyunsaturated lipids. Accounts Chem. Res. 1986, 19, 262–268. [Google Scholar] [CrossRef]
- Niki, E. Biomarkers of lipid peroxidation in clinical material. Biochim. Biophys. Acta 2014, 1840, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Zielinski, Z.; Pratt, D.A. Lipid Peroxidation: Kinetics, Mechanisms, and Products. J. Org. Chem. 2017, 82, 2817–2825. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Xu, L.; Porter, N.A.; Yin, H. Free Radical Lipid Peroxidation: Mechanisms and Analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef]
- Xu, L.; Porter, N.A. Free radical oxidation of cholesterol and its precursors: Implications in cholesterol biosynthesis disorders. Free. Radic. Res. 2015, 49, 835–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, N.A. A Perspective on Free Radical Autoxidation: The Physical Organic Chemistry of Polyunsaturated Fatty Acid and Sterol Peroxidation. J. Org. Chem. 2013, 78, 3511–3524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, L.L. Cholesterol Autoxidation; Plenum Press: New York, NY, USA, 1981. [Google Scholar]
- Smith, L.L. Cholesterol autoxidation 1981–1986. Chem. Phys. Lipids 1987, 44, 87–125. [Google Scholar] [CrossRef]
- Smith, L.L. Oxygen, oxysterols, ouabain, and ozone: A cautionary tale. Free Radic. Biol. Med. 2004, 37, 318–324. [Google Scholar] [CrossRef]
- Girotti, A.W. Mechanisms of lipid peroxidation. J. Free. Radicals Boil. Med. 1985, 1, 87–95. [Google Scholar] [CrossRef]
- Girotti, A.W. Lipid hydroperoxide generation, turnover, and effector action in biological systems. J. Lipid Res. 1998, 39, 1529–1542. [Google Scholar]
- Girotti, A.W.; Korytowski, W. Cholesterol as a natural probe for free radical-mediated lipid peroxidation in biological membranes and lipoproteins. J. Chromatogr. B 2016, 1019, 202–209. [Google Scholar] [CrossRef] [Green Version]
- Girotti, A.W.; Korytowski, W. Cholesterol Hydroperoxide Generation, Translocation, and Reductive Turnover in Biological Systems. Cell Biophys. 2017, 75, 413–419. [Google Scholar] [CrossRef]
- Girotti, A.W.; Korytowski, W. Cholesterol Peroxidation as a Special Type of Lipid Oxidation in Photodynamic Systems. Photochem. Photobiol. 2018, 95, 73–82. [Google Scholar] [CrossRef]
- Zielinski, Z.; Pratt, D.A. Cholesterol Autoxidation Revisited: Debunking the Dogma Associated with the Most Vilified of Lipids. J. Am. Chem. Soc. 2016, 138, 6932–6935. [Google Scholar] [CrossRef]
- Schaefer, E.L.; Zopyrus, N.; Zielinski, Z.A.M.; Facey, G.A.; Pratt, D.A. On the Products of Cholesterol Autoxidation in Phospholipid Bilayers and the Formation of Secosterols Derived Therefrom. Angew. Chem. Int. Ed. 2020, 59, 2089–2094. [Google Scholar] [CrossRef]
- Zielinski, Z.A.M.; Pratt, D.A. H-Atom Abstraction vs Addition: Accounting for the Diverse Product Distribution in the Autoxidation of Cholesterol and Its Esters. J. Am. Chem. Soc. 2019, 141, 3037–3051. [Google Scholar] [CrossRef] [PubMed]
- Iuliano, L. Pathways of cholesterol oxidation via non-enzymatic mechanisms. Chem. Phys. Lipids 2011, 164, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Pryor, W.A.; Stanley, J.P. Letter: A suggested mechanism for the production of malonaldehyde during the autoxidation of polyunsaturated fatty acids. Nonenzymatic production of prostaglandin endoperoxides during autoxidation. J. Org. Chem. 1975, 40, 3615–3617. [Google Scholar] [CrossRef] [PubMed]
- Porter, N.A.; Funk, M.O. Peroxy radical cyclization as a model for prostaglandin biosynthesis. J. Org. Chem. 1975, 40, 3614–3615. [Google Scholar] [CrossRef] [PubMed]
- Pryor, W.A.; Porter, N.A. Suggested mechanisms for the production of 4-hydroxy-2-nonenal from the autoxidation of polyunsaturated fatty acids. Free. Radic. Boil. Med. 1990, 8, 541–543. [Google Scholar] [CrossRef]
- Schneider, C.; Porter, N.A.; Brash, A.R. Routes to 4-hydroxynonenal: Fundamental issues in the mechanisms of lipid peroxidation. J. Boil. Chem. 2008, 283, 15539–15543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benedetti, A.; Comporti, M.; Esterbauer, H. Identification of 4-hydroxynonenal as a cytotoxic product originating from the peroxidation of liver microsomal lipids. Biochim. Biophys. Acta 1980, 620, 281–296. [Google Scholar] [CrossRef]
- Esterbauer, H.; Zollern, H. Methods for determination of aldehydic lipid peroxidation products. Free. Radic. Boil. Med. 1989, 7, 197–203. [Google Scholar] [CrossRef]
- Zhong, H.; Yin, H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: Focusing on mitochondria. Redox Boil. 2014, 4, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Salomon, R.G.; Bi, W. Isolevuglandin Adducts in Disease. Antioxid. Redox Signal. 2015, 22, 1703–1718. [Google Scholar] [CrossRef] [Green Version]
- Brame, C.J.; Salomon, R.G.; Morrow, J.D.; Roberts, L.J. Identification of extremely reactive gamma-ketoaldehydes (isolevuglandins) as products of the isoprostane pathway and characterization of their lysyl protein adducts. J. Boil. Chem. 1999, 274, 13139–13146. [Google Scholar] [CrossRef] [Green Version]
- Davies, S.S.; Zhang, L. Isolevuglandins and cardiovascular disease. Prostaglandins Lipid Mediat. 2018, 139, 29–35. [Google Scholar] [CrossRef]
- Zhang, L.; Yermalitsky, V.; Huang, J.; Pleasent, T.; Borja, M.S.; Oda, M.; Jerome, W.G.; Yancey, P.G.; Linton, E.F.; Davies, S.S. Modification by isolevuglandins, highly reactive γ-ketoaldehydes, deleteriously alters high-density lipoprotein structure and function. J. Boil. Chem. 2018, 293, 9176–9187. [Google Scholar] [CrossRef] [Green Version]
- Uchida, K. Lipofuscin-like fluorophores originated from malondialdehyde. Free. Radic. Res. 2006, 40, 1335–1338. [Google Scholar] [CrossRef] [PubMed]
- Liebler, D.; Zimmerman, L.J. Targeted Quantitation of Proteins by Mass Spectrometry. Biochemistry 2013, 52, 3797–3806. [Google Scholar] [CrossRef] [PubMed]
- Connor, R.E.; Codreanu, S.G.; Marnett, L.J.; Liebler, D. Targeted protein capture for analysis of electrophile-protein adducts. Breast Cancer 2013, 987, 163–176. [Google Scholar]
- Codreanu, S.G.; Liebler, D. Novel approaches to identify protein adducts produced by lipid peroxidation. Free. Radic. Res. 2015, 49, 881–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobley, J.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Boil. 2018, 15, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Carrié, I.; Clément, M.; De Javel, D.; Francès, H.; Bourre, J.M. Specific phospholipid fatty acid composition of brain regions in mice. Effects of n-3 polyunsaturated fatty acid deficiency and phospholipid supplementation. J. Lipid Res. 2000, 41, 465–472. [Google Scholar] [PubMed]
- Bosco, D.A.; Fowler, D.M.; Zhang, Q.; Nieva, J.; Powers, E.; Wentworth, A.D.; Lerner, R.A.; Kelly, J.W. Elevated levels of oxidized cholesterol metabolites in Lewy body disease brains accelerate α-synuclein fibrilization. Nat. Methods 2006, 2, 249–253. [Google Scholar] [CrossRef]
- Xu, L.; Davis, T.A.; Porter, N.A. Rate Constants for Peroxidation of Polyunsaturated Fatty Acids and Sterols in Solution and in Liposomes. J. Am. Chem. Soc. 2009, 131, 13037–13044. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Korade, Z.; Porter, N.A. Oxysterols from Free Radical Chain Oxidation of 7-Dehydrocholesterol: Product and Mechanistic Studies. J. Am. Chem. Soc. 2010, 132, 2222–2232. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Korade, Z.; Rosado, D.A.; Liu, W.; Lamberson, C.R.; Porter, N.A. An oxysterol biomarker for 7-dehydrocholesterol oxidation in cell/mouse models for Smith-Lemli-Opitz syndrome[S]. J. Lipid Res. 2011, 52, 1222–1233. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.W.; Lemli, L.; Opitz, J.M. A newly recognized syndrome of multiple congenital anomalies. J. Pediatr. 1964, 64, 210–217. [Google Scholar] [CrossRef]
- Porter, F.D. Smith–Lemli–Opitz syndrome: Pathogenesis, diagnosis and management. Eur. J. Hum. Genet. 2008, 16, 535–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Korade, Z.; Rosado, D.A.; Mirnics, K.; Porter, N.A. Metabolism of oxysterols derived from nonenzymatic oxidation of 7-dehydrocholesterol in cells. J. Lipid Res. 2013, 54, 1135–1143. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Liu, W.; Sheflin, L.G.; Fliesler, S.J.; Porter, N.A. Novel oxysterols observed in tissues and fluids of AY9944-treated rats: A model for Smith-Lemli-Opitz syndrome. J. Lipid Res. 2011, 52, 1810–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Mirnics, K.; Bowman, A.B.; Liu, W.; Da, J.; Porter, N.A.; Korade, Z. DHCEO accumulation is a critical mediator of pathophysiology in a Smith–Lemli–Opitz syndrome model. Neurobiol. Dis. 2012, 45, 923–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Windsor, K.; Genaro-Mattos, T.; Kim, H.-Y.H.; Liu, W.; Tallman, K.A.; Miyamoto, S.; Korade, Z.; Porter, N.A. Probing lipid-protein adduction with alkynyl surrogates: Application to Smith-Lemli-Opitz syndrome. J. Lipid Res. 2013, 54, 2842–2850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pryor, W.A.; Squadrito, G.L.; Friedman, M. The cascade mechanism to explain ozone toxicity: The role of lipid ozonation products. Free. Radic. Boil. Med. 1995, 19, 935–941. [Google Scholar] [CrossRef]
- Uppu, R.; Cueto, R.; Squadrito, G.; Pryor, W. What Does Ozone React with at the Air Lung Interface? Model Studies Using Human Red Blood Cell Membranes. Arch. Biochem. Biophys. 1995, 319, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Pulfer, M.K.; Harrison, K.; Murphy, R.C. Direct electrospray tandem mass spectrometry of the unstable hydroperoxy bishemiacetal product derived from cholesterol ozonolysis. J. Am. Soc. Mass Spectrom. 2004, 15, 194–202. [Google Scholar] [CrossRef] [Green Version]
- Pulfer, M.K.; Murphy, R.C.; Ishida, J.; Nishiwaki, S.; Iguchi, T.; Matsuzaki, H.; Shiota, N.; Okunishi, H.; Sugiyama, F.; Kasuya, Y.; et al. Formation of Biologically Active Oxysterols during Ozonolysis of Cholesterol Present in Lung Surfactant. J. Boil. Chem. 2004, 279, 26331–26338. [Google Scholar] [CrossRef] [Green Version]
- Pulfer, M.K.; Taube, C.; Gelfand, E.; Murphy, R.C. Ozone Exposure in Vivo and Formation of Biologically Active Oxysterols in the Lung. J. Pharmacol. Exp. Ther. 2005, 312, 256–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maillard, B.; Ingold, K.U.; Scaiano, J.T. Rate constants for the reactions of free radicals with oxygen in solution. J. Am. Chem. Soc. 1983, 105, 5095–5099. [Google Scholar] [CrossRef]
- Mccord, J.M.; Day, E.D. Superoxide-dependent production of hydroxyl radical catalyzed by iron-EDTA complex. FEBS Lett. 1978, 86, 139–142. [Google Scholar] [CrossRef] [Green Version]
- Gardner, H.W.; Kleiman, R.; Weisleder, D. Homolytic decomposition of linoleic acid hydroperoxide: Identification of fatty acid products. Lipids 1974, 9, 696–706. [Google Scholar] [CrossRef]
- Gardner, H.W.; Weisleder, D.; Kleiman, R. Addition ofN-acetylcysteine to linoleic acid hydroperoxide. Lipids 1976, 11, 127–134. [Google Scholar] [CrossRef]
- Gardner, H.W.; Kleiman, R. Degradation of linoleic acid hydroperoxides by a cysteine. FeCl3 catalyst as a model for similar biochemical reactions. II. Specificity in formation of fatty acid epoxides. Biochim. Biophys. Acta 1981, 665, 113–125. [Google Scholar] [CrossRef]
- Dix, T.A.; Marnett, L.J. Conversion of linoleic acid hydroperoxide to hydroxy, keto, epoxyhydroxy, and trihydroxy fatty acids by hematin. J. Boil. Chem. 1985, 260, 5351–5357. [Google Scholar]
- Schaich, K.M. Metals and lipid oxidation. Contemporary issues. Lipids 1992, 27, 209–218. [Google Scholar] [CrossRef]
- Porter, N.A.; Mills, K.A.; Carter, R.L. A Mechanistic Study of Oleate Autoxidation: Competing Peroxyl H-Atom Abstraction and Rearrangement. J. Am. Chem. Soc. 1994, 116, 6690–6696. [Google Scholar] [CrossRef]
- Pratt, D.A.; Tallman, K.A.; Porter, N.A. Free Radical Oxidation of Polyunsaturated Lipids: New Mechanistic Insights and the Development of Peroxyl Radical Clocks. Accounts Chem. Res. 2011, 44, 458–467. [Google Scholar] [CrossRef] [Green Version]
- Tallman, K.A.; Pratt, D.A.; Porter, N.A. Kinetic products of linoleate peroxidation: Rapid beta-fragmentation of nonconjugated peroxyls. J. Am. Chem. Soc. 2001, 123, 11827–11828. [Google Scholar] [CrossRef] [PubMed]
- Tallman, K.A.; Rector, C.L.; Porter, N.A. Substituent Effects on Regioselectivity in the Autoxidation of Nonconjugated Dienes. J. Am. Chem. Soc. 2009, 131, 5635–5641. [Google Scholar] [CrossRef] [Green Version]
- Tallman, K.A.; Roschek, B.; Porter, N.A. Factors Influencing the Autoxidation of Fatty Acids: Effect of Olefin Geometry of the Nonconjugated Diene. J. Am. Chem. Soc. 2004, 126, 9240–9247. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Farmer, L.A.; Zilka, O.; Van Kessel, A.T.; Pratt, D.A. Beyond DPPH: Use of Fluorescence-Enabled Inhibited Autoxidation to Predict Oxidative Cell Death Rescue. Cell Chem. Boil. 2019, 26, 1594–1607. [Google Scholar] [CrossRef] [PubMed]
- Paillasse, M.R.; Saffon, N.; Gornitzka, H.; Silvente-Poirot, S.; Poirot, M.; De Medina, P. Surprising unreactivity of cholesterol-5,6-epoxides towards nucleophiles. J. Lipid Res. 2012, 53, 718–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Medina, P.; Paillasse, M.R.; Segala, G.; Poirot, M.; Silvente-Poirot, S. Identification and pharmacological characterization of cholesterol-5,6-epoxide hydrolase as a target for tamoxifen and AEBS ligands. Proc. Natl. Acad. Sci. USA 2010, 107, 13520–13525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meijer, J.; DePierre, J.W.; Jörnvall, H. Cytosolic epoxide hydrolase from liver of control and clofibrate-treated mice. Structural comparison by HPLC peptide mapping. Biosci. Rep. 1987, 7, 891–896. [Google Scholar] [CrossRef]
- Nashed, N.T.; Michaud, D.P.; Levin, W.; Jerina, N.M. Properties of liver microsomal cholesterol 5,6-oxide hydrolase. Arch. Biochem. Biophys. 1985, 241, 149–162. [Google Scholar] [CrossRef]
- Silvente-Poirot, S.; Poirot, M. Cholesterol epoxide hydrolase and cancer. Curr. Opin. Pharmacol. 2012, 12, 696–703. [Google Scholar] [CrossRef]
- Watabe, T.; Ozawa, N.; Ishii, H.; Chiba, K.; Hiratsuka, A. Hepatic microsomal cholesterol epoxide hydrolase: Selective inhibition by detergents and separation from xenobiotic epoxide hydrolase. Biochem. Biophys. Res. Commun. 1986, 140, 632–637. [Google Scholar] [CrossRef]
- Dalenc, F.; Poirot, M.; Silvente-Poirot, S. Dendrogenin A: A Mammalian Metabolite of Cholesterol with Tumor Suppressor and Neurostimulating Properties. Curr. Med. Chem. 2015, 22, 3533–3549. [Google Scholar] [CrossRef]
- De Medina, P.; Paillasse, M.R.; Segala, G.; Voisin, M.; Mhamdi, L.; Dalenc, F.; Lacroix-Triki, M.; Filleron, T.; Pont, F.; Al Saati, T.; et al. Dendrogenin A arises from cholesterol and histamine metabolism and shows cell differentiation and anti-tumour properties. Nat. Commun. 2013, 4, 1840. [Google Scholar] [CrossRef] [Green Version]
- Fransson, A.; De Medina, P.; Paillasse, M.R.; Silvente-Poirot, S.; Poirot, M.; Ulfendahl, M. Dendrogenin A and B two new steroidal alkaloids increasing neural responsiveness in the deafened guinea pig. Front. Aging Neurosci. 2015, 7, 145. [Google Scholar] [CrossRef] [PubMed]
- Poirot, M.; Silvente-Poirot, S. Oxysterols and related sterols: Implications in pharmacology and pathophysiology. Biochem. Pharmacol. 2013, 86, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Poirot, M.; Silvente-Poirot, S. Cholesterol-5,6-epoxides: Chemistry, biochemistry, metabolic fate and cancer. Biochimie 2013, 95, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Poirot, M.; Silvente-Poirot, S. When cholesterol meets histamine, it gives rise to dendrogenin A: A tumour suppressor metabolite1. Biochem. Soc. Trans. 2016, 44, 631–637. [Google Scholar] [CrossRef] [Green Version]
- Segala, G.; David, M.; De Medina, P.; Poirot, M.C.; Serhan, N.; Vergez, F.; Mougel, A.; Saland, E.; Carayon, K.; Leignadier, J.; et al. Dendrogenin A drives LXR to trigger lethal autophagy in cancers. Nat. Commun. 2017, 8, 1903. [Google Scholar] [CrossRef]
- Silvente-Poirot, S.; De Medina, P.; Record, M.; Poirot, M. From tamoxifen to dendrogenin A: The discovery of a mammalian tumor suppressor and cholesterol metabolite. Biochimie 2016, 130, 109–114. [Google Scholar] [CrossRef]
- Windsor, K.; Genaro-Mattos, T.; Miyamoto, S.; Stec, N.F.; Kim, H.-Y.H.; Tallman, K.A.; Porter, N.A. Assay of Protein and Peptide Adducts of Cholesterol Ozonolysis Products by Hydrophobic and Click Enrichment Methods. Chem. Res. Toxicol. 2014, 27, 1757–1768. [Google Scholar] [CrossRef]
- Hock, H.; Lang, S. Autoxydation von Koblen-wasserstoffen, IX. Mitteil: Uber Peroxyde von Benzol-Derivaten. Chem. Ber. 1944, 77, 257–264. [Google Scholar] [CrossRef]
- Lee, J.B.; Uff, B.C. Organic reactions involving electrophilic oxygen. Q. Rev. Chem. Soc. 1967, 21, 429. [Google Scholar] [CrossRef]
- Frimer, A.A. The reaction of singlet oxygen with olefins: The question of mechanism. Chem. Rev. 1979, 79, 359–387. [Google Scholar] [CrossRef]
- Brinkhorst, J.; Nara, S.J.; Pratt, D.A. Hock Cleavage of Cholesterol 5α-Hydroperoxide: An Ozone-Free Pathway to the Cholesterol Ozonolysis Products Identified in Arterial Plaque and Brain Tissue. J. Am. Chem. Soc. 2008, 130, 12224–12225. [Google Scholar] [CrossRef] [PubMed]
- Pryor, W.A. How far does ozone penetrate into the pulmonary air/tissue boundary before it reacts? Free Radic. Boil. Med. 1992, 12, 83–88. [Google Scholar] [CrossRef]
- Pryor, W.A. Mechanisms of radical formation from reactions of ozone with target molecules in the lung. Free Radic. Boil. Med. 1994, 17, 451–465. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M.; Valente, M.; Ferri, L.; Gregolin, C. Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim. Biophys. Acta 1982, 710, 197–211. [Google Scholar] [CrossRef]
- Korytowski, W.; Geiger, P.G.; Girotti, A.W. Enzymatic Reducibility in Relation to Cytotoxicity for Various Cholesterol Hydroperoxides. Biochemistry 1996, 35, 8670–8679. [Google Scholar] [CrossRef]
- Ruan, B.; Wilson, W.K.; Pang, J.; Schroepfer, G.J. Synthesis of [3alpha-3H]cholesta-5,8-dien-3beta-ol and tritium-labeled forms of other sterols of potential importance in the Smith-Lemli-Optiz syndrome. Steroids 2000, 65, 29–39. [Google Scholar] [CrossRef]
- Porter, F.D.; Herman, G.E. Malformation syndromes caused by disorders of cholesterol synthesis. J. Lipid Res. 2010, 52, 6–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, W.L. Genetic disorders of Vitamin D biosynthesis and degradation. J. Steroid Biochem. Mol. Boil. 2017, 165, 101–108. [Google Scholar] [CrossRef]
- Prabhu, A.V.; Luu, W.; Li, D.; Sharpe, L.J.; Brown, A.J. DHCR7: A vital enzyme switch between cholesterol and vitamin D production. Prog. Lipid Res. 2016, 64, 138–151. [Google Scholar] [CrossRef] [PubMed]
- Tanret, C. Sur un Nouveau Principe Immediat de l”Ergot de Seigle. l’Ergosterine. Ann. Chim. Phys. 1890, 6, 289–293. [Google Scholar]
- Tanret, C. Sur l’Ergosterine et la Fongisterine. Ann. Chim. Phys. 1908, 8, 313–317. [Google Scholar]
- Meyer, K. On Catalytic Oxidations V. The Oxidation of Ergosterol. J. Biol. Chem. 1933, 103, 607–616. [Google Scholar]
- Weenen, H.; Porter, N.A. Autoxidation of model membrane systems: Cooxidation of polyunsaturated lecithins with steroids, fatty acids, and alpha.-tocopherol. J. Am. Chem. Soc. 1982, 104, 5216–5221. [Google Scholar] [CrossRef]
- Porter, N.A.; Weber, B.A.; Weenen, H. Autoxidation of polyunsaturated lipids. Factors controlling the stereochemistry of product hydroperoxides. J. Am. Chem. Soc. 1980, 102, 5597–5601. [Google Scholar] [CrossRef]
- Roschek, B.; Tallman, K.A.; Rector, C.L.; Gillmore, J.; Pratt, D.A.; Punta, C.; Porter, N.A. Peroxyl Radical Clocks. J. Org. Chem. 2006, 71, 3527–3532. [Google Scholar] [CrossRef]
- Xu, L.; Porter, N.A. Reactivities and Products of Free Radical Oxidation of Cholestadienols. J. Am. Chem. Soc. 2014, 136, 5443–5450. [Google Scholar] [CrossRef]
- Rajeev, R.; Sunoj, R.B. Mechanism and Stereoselectivity of Biologically Important Oxygenation Reactions of the 7-Dehydrocholesterol Radical. J. Org. Chem. 2013, 78, 7023–7029. [Google Scholar] [CrossRef]
- Korade, Z.; Xu, L.; Mirnics, K.; Porter, N.A. Lipid biomarkers of oxidative stress in a genetic mouse model of Smith-Lemli-Opitz syndrome. J. Inherit. Metab. Dis. 2012, 36, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Korade, Z.; Xu, L.; Shelton, R.; Porter, N.A. Biological activities of 7-dehydrocholesterol-derived oxysterols: Implications for Smith-Lemli-Opitz syndrome. J. Lipid Res. 2010, 51, 3259–3269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, B.A.; Xu, L.; Porter, N.A.; Rao, S.R.; Fliesler, S.J. Differential cytotoxic effects of 7-dehydrocholesterol-derived oxysterols on cultured retina-derived cells: Dependence on sterol structure, cell type, and density. Exp. Eye Res. 2016, 145, 297–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watabe, T.; Kanai, M.; Isobe, M.; Ozawa, N. The hepatic microsomal biotransformation of delta 5-steroids to 5 alpha, 6 beta-glycols via alpha- and beta-epoxides. J. Boil. Chem. 1981, 256, 2900–2907. [Google Scholar]
- Mohana, K.; Achary, A. Human cytosolic glutathione-S-transferases: Quantitative analysis of expression, comparative analysis of structures and inhibition strategies of isozymes involved in drug resistance. Drug Metab. Rev. 2017, 49, 318–337. [Google Scholar] [CrossRef]
- Meyer, D.J.; Ketterer, B. 5 alpha,6 alpha-Epoxy-cholestan-3 beta-ol (cholesterol alpha-oxide): A specific substrate for rat liver glutathione transferase B. FEBS Lett. 1982, 150, 499–502. [Google Scholar] [CrossRef] [Green Version]
- Fliesler, S.J.; Peachey, N.S.; Herron, J.; Hines, K.; Weinstock, N.I.; Rao, S.R.; Xu, L. Prevention of Retinal Degeneration in a Rat Model of Smith-Lemli-Opitz Syndrome. Sci. Rep. 2018, 8, 1286. [Google Scholar] [CrossRef] [Green Version]
- Kapphahn, R.J.; Richards, M.; Ferrington, D.A.; Fliesler, S.J. Lipid-derived and other oxidative modifications of retinal proteins in a rat model of Smith-Lemli-Opitz syndrome. Exp. Eye Res. 2019, 178, 247–254. [Google Scholar] [CrossRef]
- Esterbauer, H. Estimation of peroxidative damage. A critical review. Pathol. Boil. 1996, 44, 25–28. [Google Scholar]
- Doorn, J.A.; Petersen, D.R. Covalent Modification of Amino Acid Nucleophiles by the Lipid Peroxidation Products 4-Hydroxy-2-nonenal and 4-Oxo-2-nonenal. Chem. Res. Toxicol. 2002, 15, 1445–1450. [Google Scholar] [CrossRef]
- Nadkarni, D.V.; Sayre, L.M. Structural Definition of Early Lysine and Histidine Adduction Chemistry of 4-Hydroxynonenal. Chem. Res. Toxicol. 1995, 8, 284–291. [Google Scholar] [CrossRef]
- Uchida, K. 4-Hydroxy-2-nonenal: A product and mediator of oxidative stress. Prog. Lipid Res. 2003, 42, 318–343. [Google Scholar] [CrossRef]
- Uchida, K.; Stadtman, E.R. Modification of histidine residues in proteins by reaction with 4-hydroxynonenal. Proc. Natl. Acad. Sci. USA 1992, 89, 4544–4548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, J.D.; Marnett, L.J. Alterations in Gene Expression Induced by the Lipid Peroxidation Product, 4-Hydroxy-2-nonenal. Chem. Res. Toxicol. 2005, 18, 1642–1653. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Reed, T.; Perluigi, M.; De Marco, C.; Coccia, R.; Cini, C.; Sultana, R. Elevated protein-bound levels of the lipid peroxidation product, 4-hydroxy-2-nonenal, in brain from persons with mild cognitive impairment. Neurosci. Lett. 2006, 397, 170–173. [Google Scholar] [CrossRef]
- Long, E.K.; Picklo, M.J., Sr. Trans-4-hydroxy-2-hexenal, a product of n-3 fatty acid peroxidation: Make some room HNE. Free. Radic. Boil. Med. 2010, 49, 1–8. [Google Scholar] [CrossRef]
- Codreanu, S.G.; Kim, H.-Y.H.; Porter, N.A.; Liebler, D. Biotinylated probes for the analysis of protein modification by electrophiles. Breast Cancer 2012, 803, 77–95. [Google Scholar]
- Codreanu, S.G.; Ullery, J.C.; Zhu, J.; Tallman, K.A.; Beavers, W.N.; Porter, N.A.; Marnett, L.J.; Zhang, B.; Liebler, D. Alkylation damage by lipid electrophiles targets functional protein systems. Mol. Cell. Proteom. 2014, 13, 849–859. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-Y.H.; Tallman, K.A.; Liebler, D.; Porter, N.A. An azido-biotin reagent for use in the isolation of protein adducts of lipid-derived electrophiles by streptavidin catch and photorelease. Mol. Cell. Proteom. 2009, 8, 2080–2089. [Google Scholar] [CrossRef] [Green Version]
- Sun, R.; Fu, L.; Liu, K.; Tian, C.; Yang, Y.; Tallman, K.A.; Porter, N.A.; Liebler, D.C.; Yang, J. Chemoproteomics Reveals Chemical Diversity and Dynamics of 4-Oxo-2-nonenal Modifications in Cells. Mol. Cell. Proteom. 2017, 16, 1789–1800. [Google Scholar] [CrossRef] [Green Version]
- Tallman, K.A.; Kim, H.-Y.H.; Ji, J.-X.; Szapacs, M.E.; Yin, H.; McIntosh, T.J.; Liebler, D.C.; Porter, N.A.; Yin, H. Phospholipid−Protein Adducts of Lipid Peroxidation: Synthesis and Study of New Biotinylated Phosphatidylcholines. Chem. Res. Toxicol. 2007, 20, 227–234. [Google Scholar] [CrossRef]
- Vila, A.; Tallman, K.A.; Jacobs, A.T.; Liebler, D.; Porter, N.A.; Marnett, L.J. Identification of Protein Targets of 4-Hydroxynonenal Using Click Chemistry for ex Vivo Biotinylation of Azido and Alkynyl Derivatives. Chem. Res. Toxicol. 2008, 21, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Vila, A.; Tallman, K.A.; Porter, N.; Liebler, D.C.; Marnett, L.J. Proteomic analysis of azido-HNE adducted proteins in RKO cells. Chem. Res. Tox. 2007, 20, 2010. [Google Scholar]
- Higdon, A.N.; Dranka, B.; Hill, B.; Oh, J.-Y.; Johnson, M.S.; Landar, A.; Darley-Usmar, V. Methods for imaging and detecting modification of proteins by reactive lipid species. Free. Radic. Boil. Med. 2009, 47, 201–212. [Google Scholar] [CrossRef] [Green Version]
- Sayre, L.M.; Lin, D.; Yuan, Q.; Zhu, X.; Tang, X. Protein Adducts Generated from Products of Lipid Oxidation: Focus on HNE and ONE. Drug Metab. Rev. 2006, 38, 651–675. [Google Scholar] [CrossRef]
- Wentworth, A.D.; Nieva, J.; Takeuchi, C.; Galve, R.; Wentworth, A.D.; Dilley, R.B.; Delaria, G.A.; Saven, A.; Babior, B.M.; Janda, K.D.; et al. Evidence for Ozone Formation in Human Atherosclerotic Arteries. Science 2003, 302, 1053–1056. [Google Scholar] [CrossRef] [Green Version]
- Wentworth, A.D.; Song, B.-D.; Nieva, J.; Shafton, A.; Tripurenani, S.; Wentworth, P. The ratio of cholesterol 5,6-secosterols formed from ozone and singlet oxygen offers insight into the oxidation of cholesterol in vivo. Chem. Commun. 2009, 3098–3100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheinost, J.C.; Witter, D.P.; Boldt, G.E.; Offer, J.; Wentworth, P., Jr. Cholesterol secosterol adduction inhibits the misfolding of a mutant prion protein fragment that induces neurodegeneration. Angew Chem. Int. Ed. Engl. 2009, 48, 9469–9472. [Google Scholar] [CrossRef] [PubMed]
- Nieva, J.; Song, B.-D.; Rogel, J.K.; Kujawara, D.; Altobel, L.; Izharrudin, A.; Boldt, G.E.; Grover, R.K.; Wentworth, A.D.; Wentworth, P. Cholesterol Secosterol Aldehydes Induce Amyloidogenesis and Dysfunction of Wild-Type Tumor Protein p53. Chem. Boil. 2011, 18, 920–927. [Google Scholar] [CrossRef] [Green Version]
- Genaro-Mattos, T.; Appolinário, P.P.; Mugnol, K.C.U.; Bloch, C., Jr.; Nantes-Cardos, I.L.; Di Mascio, P.; Miyamoto, S. Covalent Binding and Anchoring of Cytochrome c to Mitochondrial Mimetic Membranes Promoted by Cholesterol Carboxyaldehyde. Chem. Res. Toxicol. 2013, 26, 1536–1544. [Google Scholar] [CrossRef]
- Rostovtsev, V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Best, M.D. Click Chemistry and Bioorthogonal Reactions: Unprecedented Selectivity in the Labeling of Biological Molecules. Biochemistry 2009, 48, 6571–6584. [Google Scholar] [CrossRef] [PubMed]
- Dantas, L.S.; Filho, A.B.C.; Coelho, F.R.; Genaro-Mattos, T.C.; Tallman, K.A.; Porter, N.A.; Augusto, O.; Miyamoto, S. Cholesterol secosterol aldehyde adduction and aggregation of Cu, Zn-superoxide dismutase: Potential implications in ALS. Redox Boil. 2018, 19, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Pratt, A.J.; Shin, D.S.; Merz, G.E.; Rambo, R.P.; Lancaster, W.A.; Dyer, K.N.; Borbat, P.P.; Poole, F.L.; Adams, M.W.W.; Freed, J.H.; et al. Aggregation propensities of superoxide dismutase G93 hotspot mutants mirror ALS clinical phenotypes. Proc. Natl. Acad. Sci. USA 2014, 111, E4568–E4576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speen, A.M.; Kim, H.-Y.H.; Bauer, R.N.; Meyer, M.; Gowdy, K.M.; Fessler, M.B.; Duncan, K.E.; Liu, W.; Porter, N.A.; Jaspers, I. Ozone-derived Oxysterols Affect Liver X Receptor (LXR) Signaling. J. Boil. Chem. 2016, 291, 25192–25206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearl, L.; Prodromou, C.; Workman, P. The Hsp90 molecular chaperone: An open and shut case for treatment. Biochem. J. 2008, 410, 439–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connor, R.E.; Marnett, L.J.; Liebler, D. Protein-Selective Capture to Analyze Electrophile Adduction of Hsp90 by 4-Hydroxynonenal. Chem. Res. Toxicol. 2011, 24, 1275–1282. [Google Scholar] [CrossRef]
- Tallman, K.A.; Kim, H.-Y.H.; Korade, Z.; Genaro-Mattos, T.; Wages, P.; Liu, W.; Porter, N.A. Probes for protein adduction in cholesterol biosynthesis disorders: Alkynyl lanosterol as a viable sterol precursor. Redox Boil. 2017, 12, 182–190. [Google Scholar] [CrossRef]
- Schopfer, F.J.; Cipollina, C.; Freeman, B.A. Formation and Signaling Actions of Electrophilic Lipids. Chem. Rev. 2011, 111, 5997–6021. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Bermudez, J.; Baudrier, L.; Bayraktar, E.C.; Shen, Y.; La, K.; Guarecuco, R.; Yucel, B.; Fiore, D.; Tavora, B.; Freinkman, E.; et al. Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature 2019, 567, 118–122. [Google Scholar] [CrossRef]
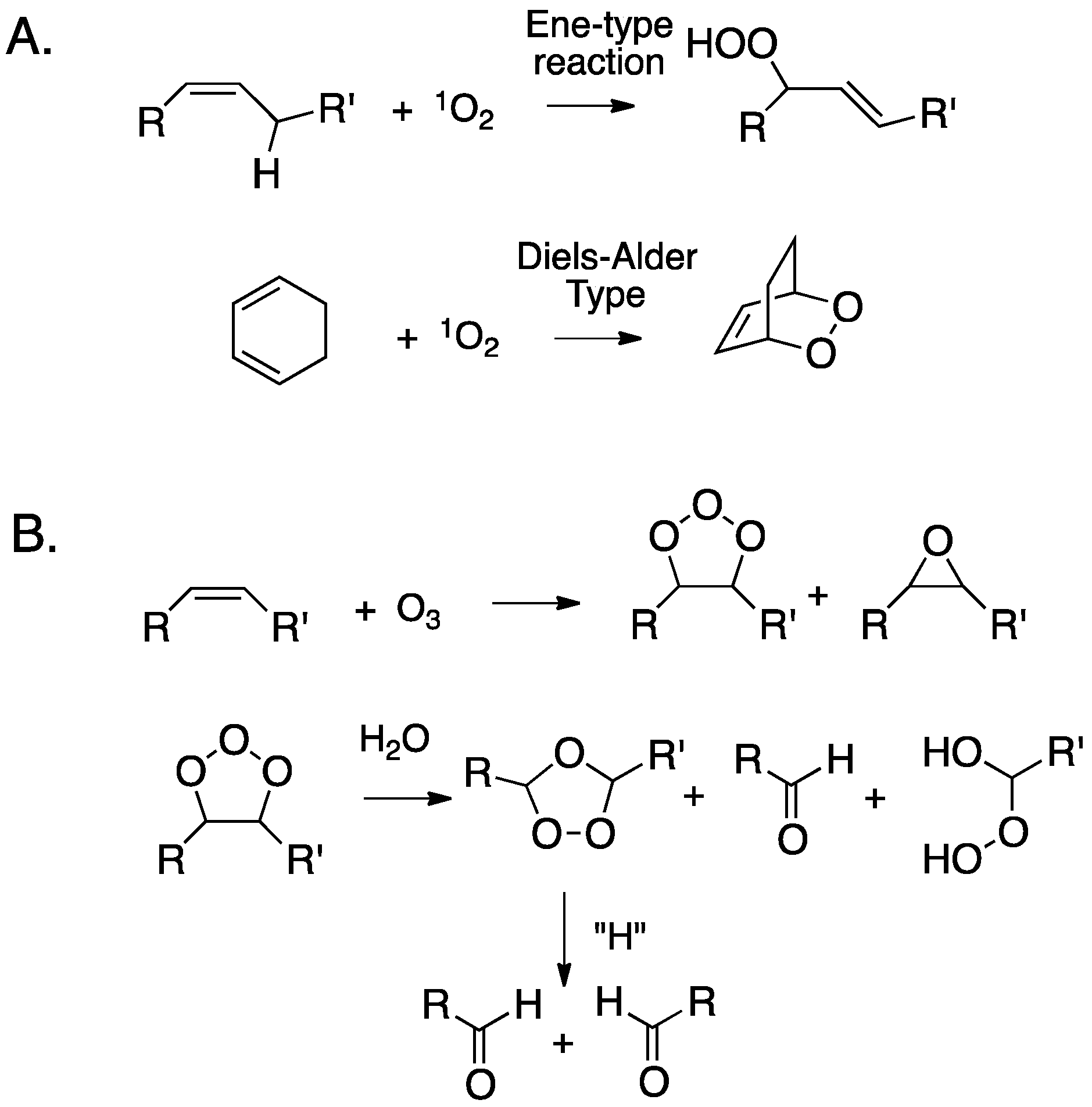
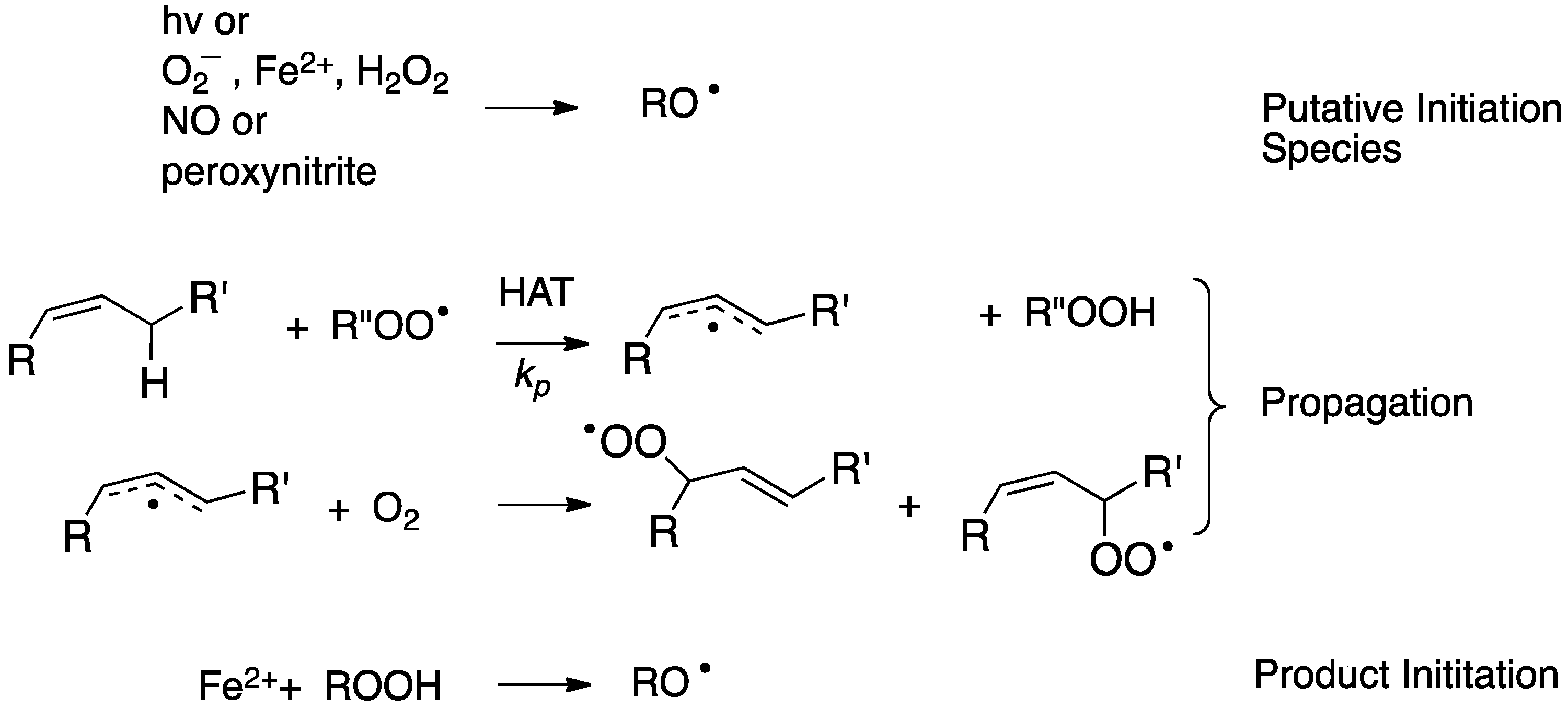

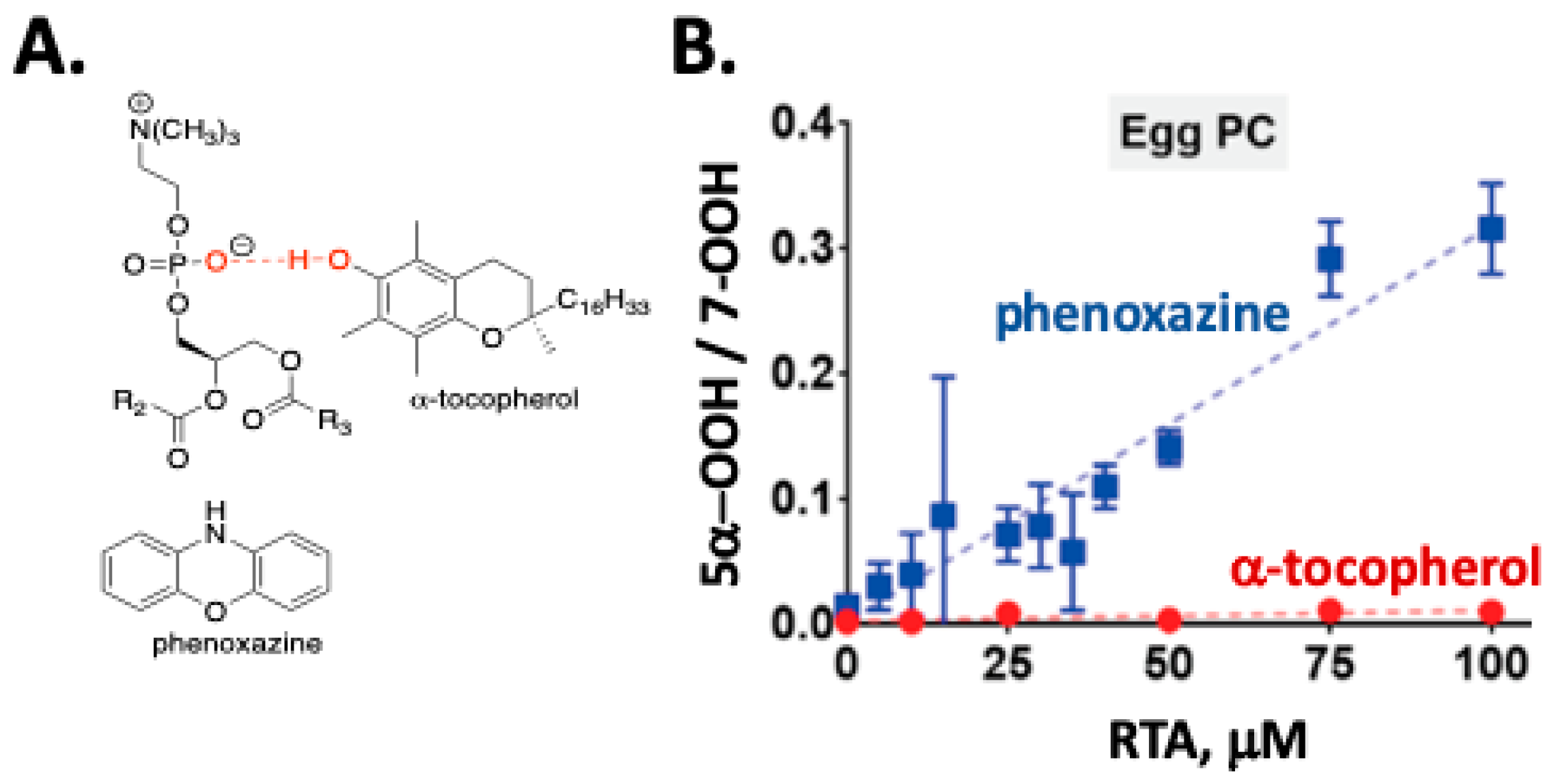
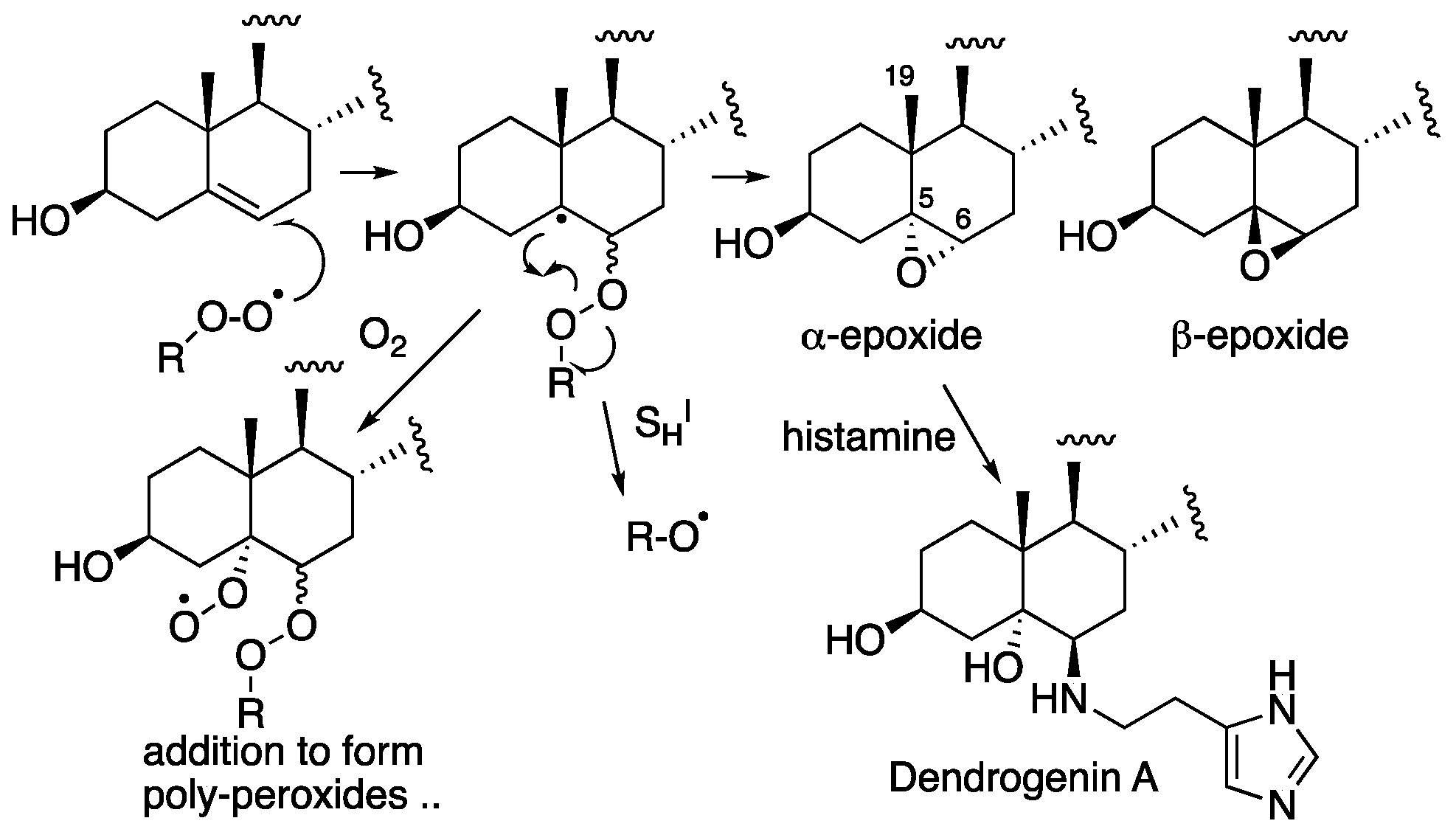
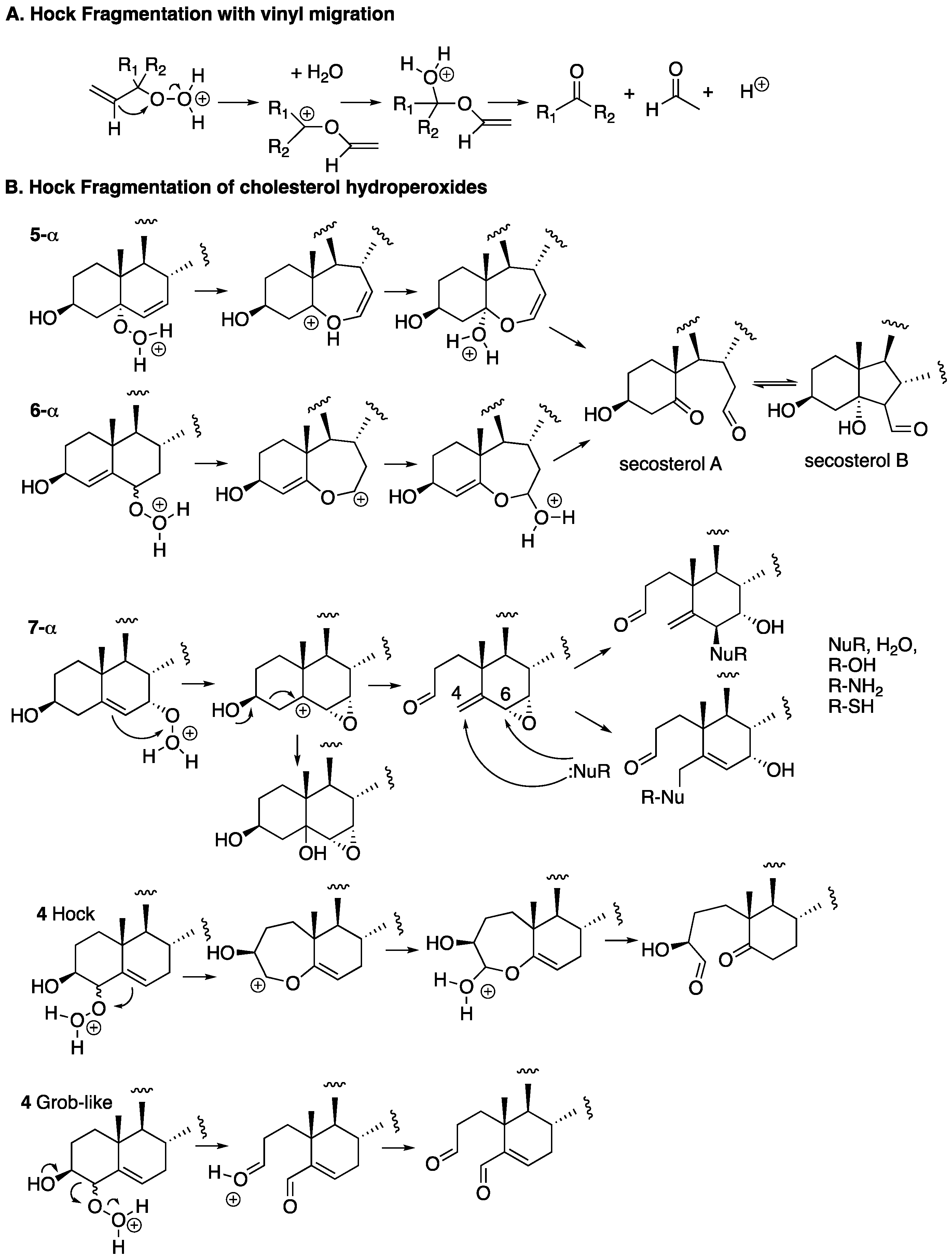
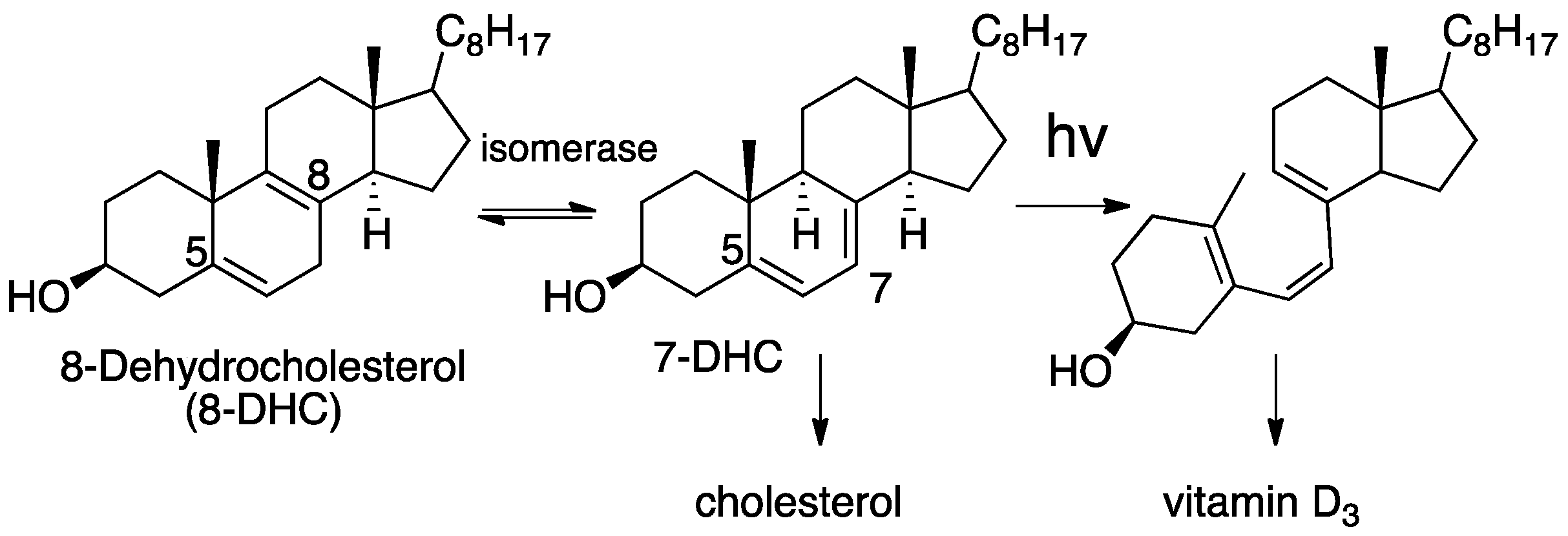
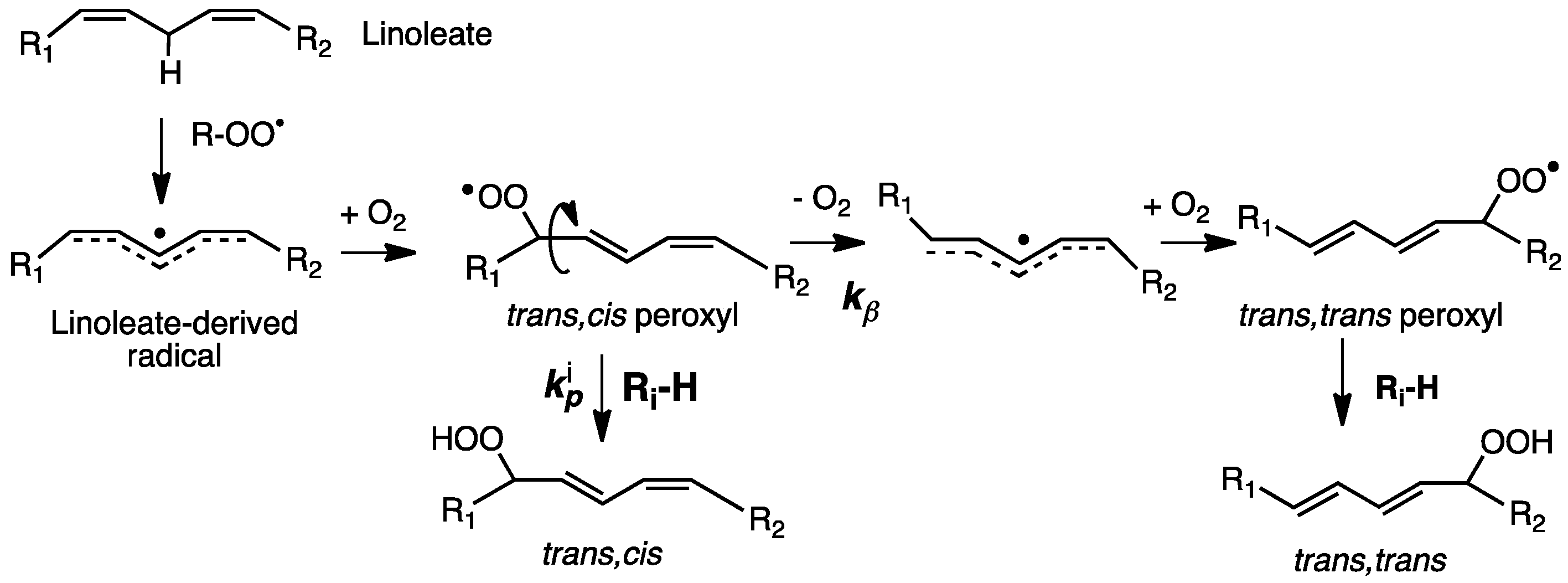
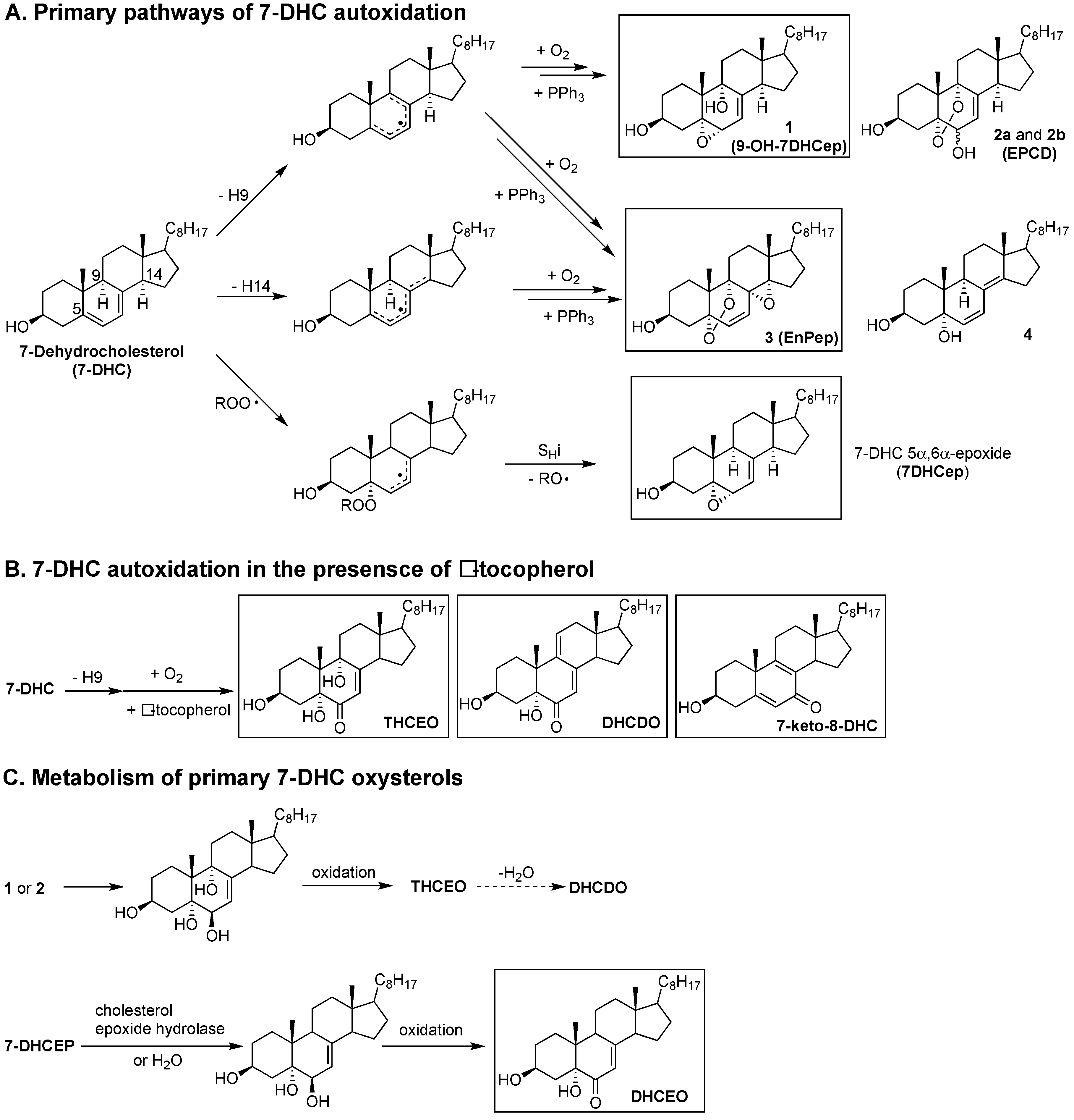
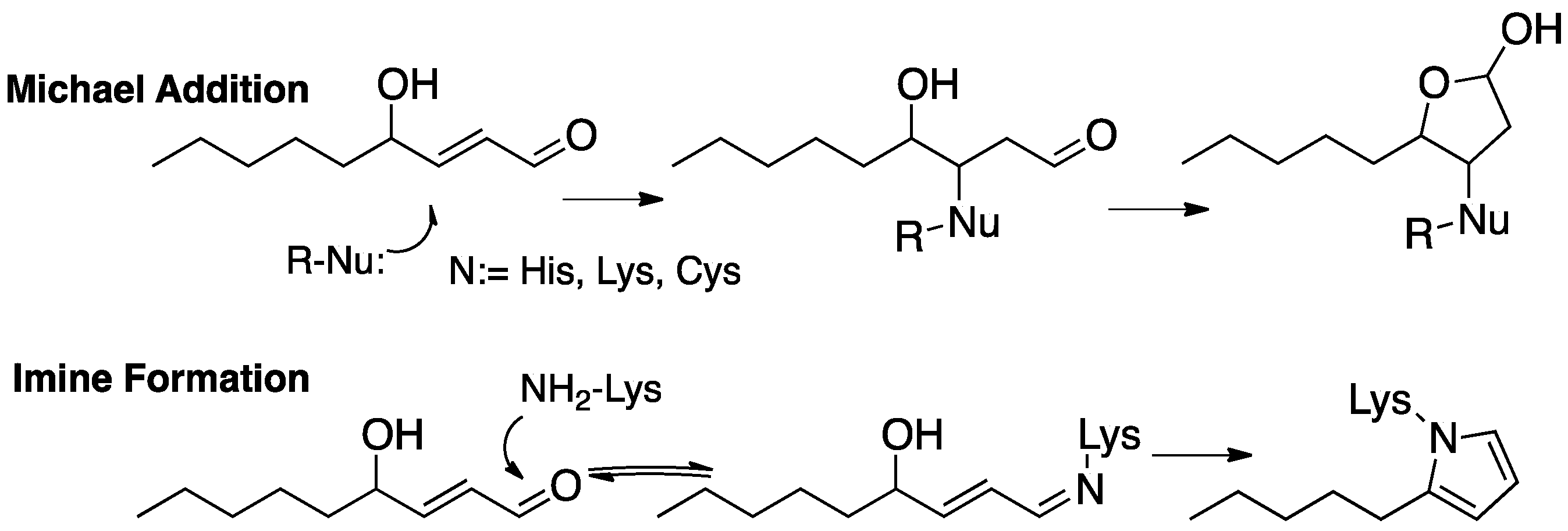
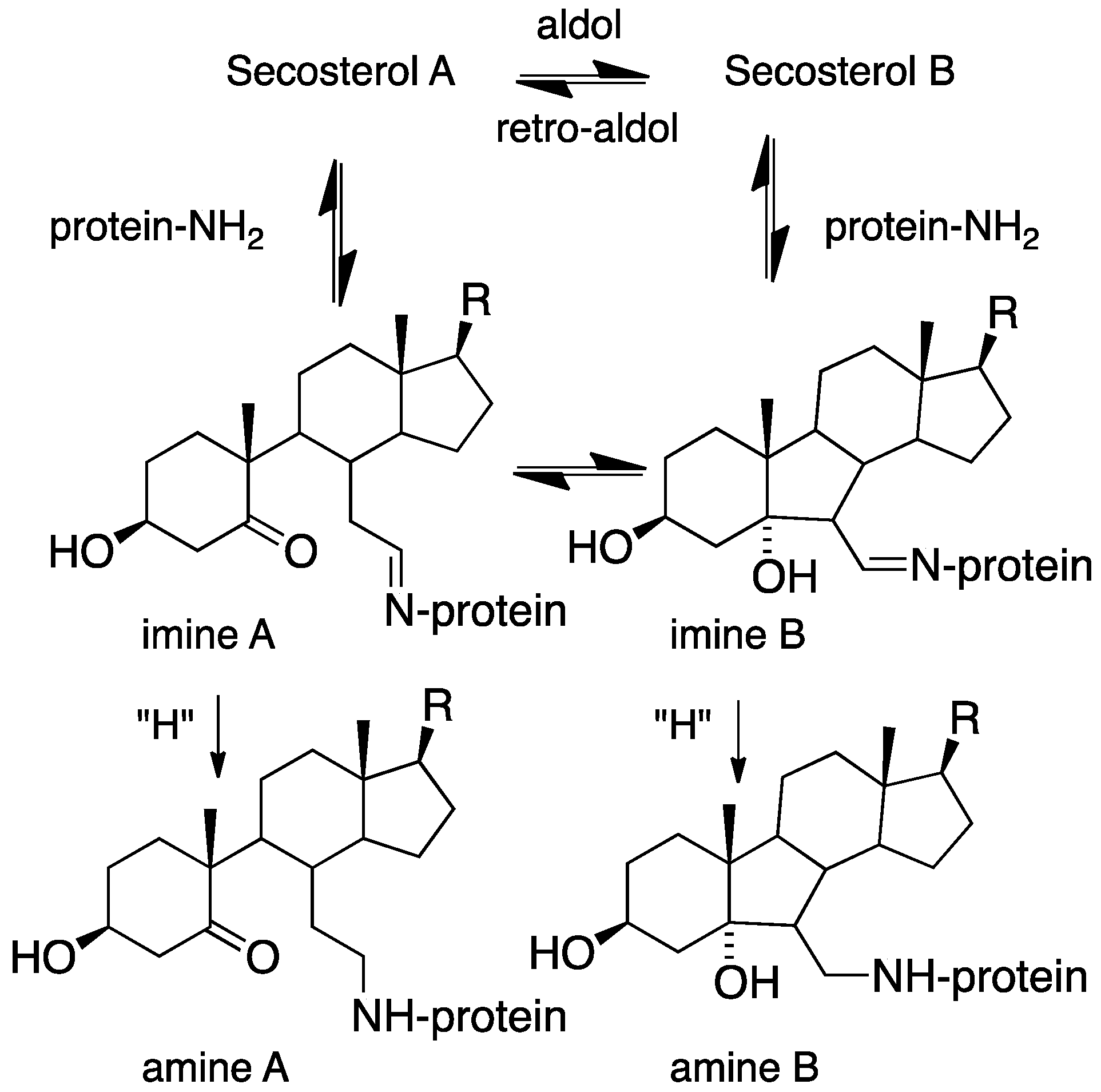
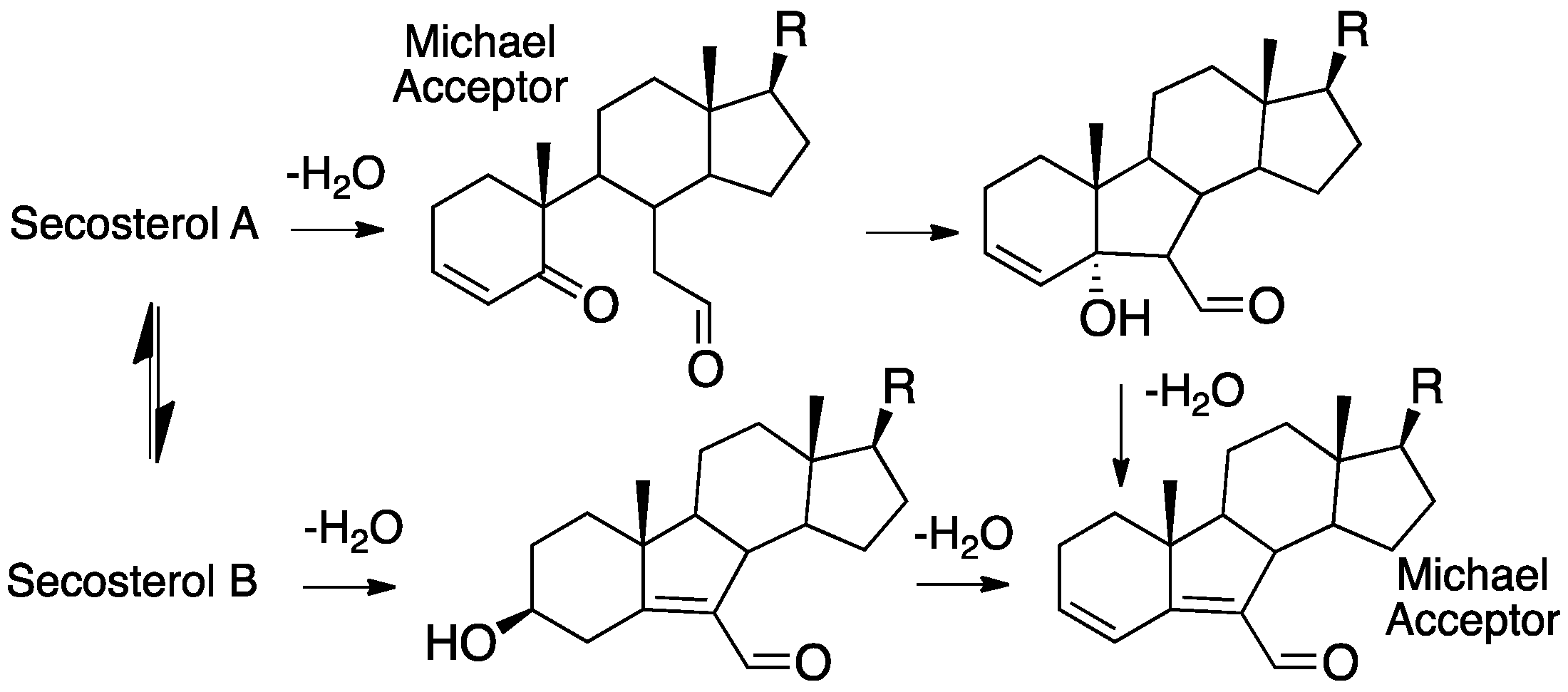
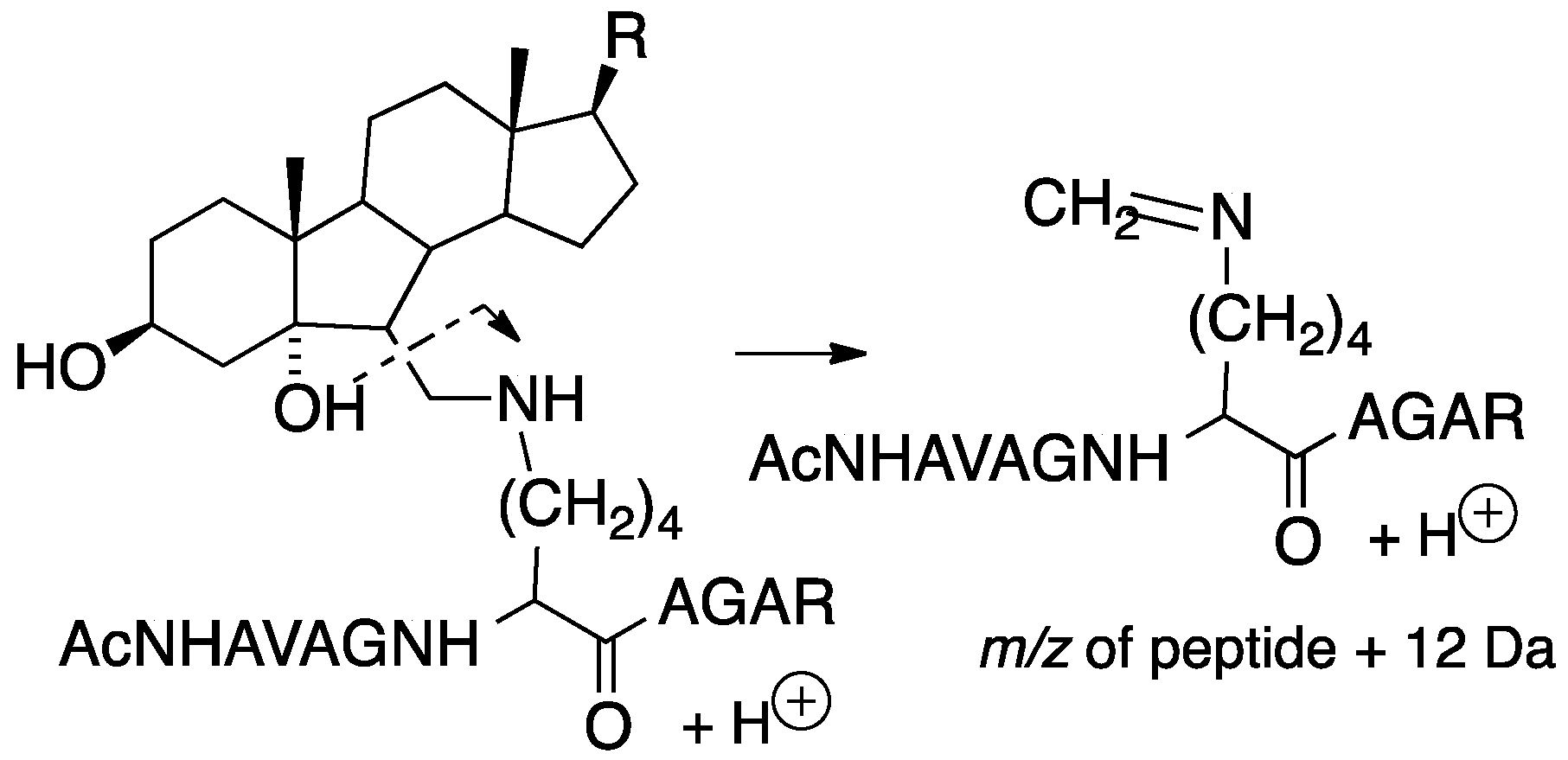

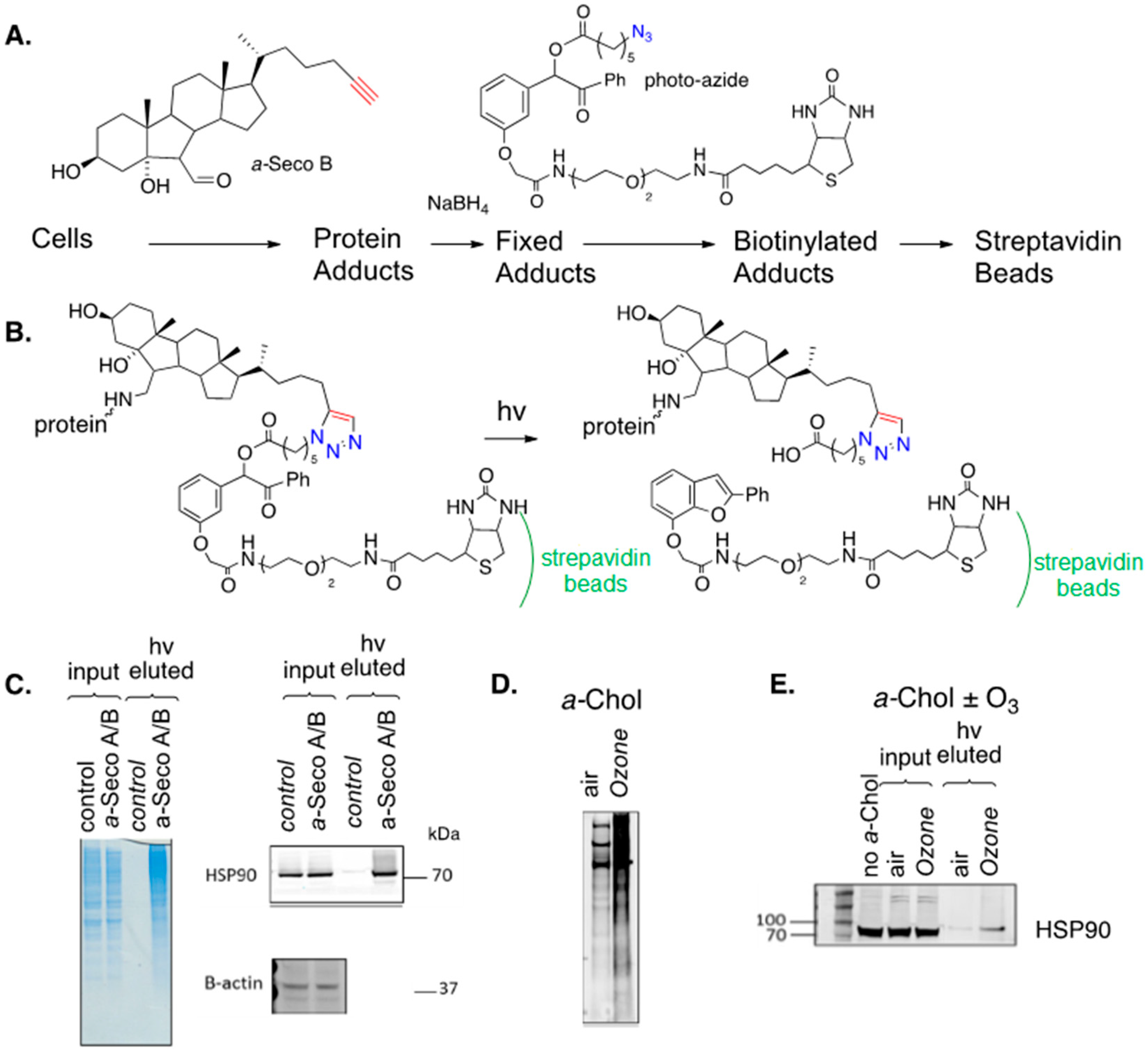
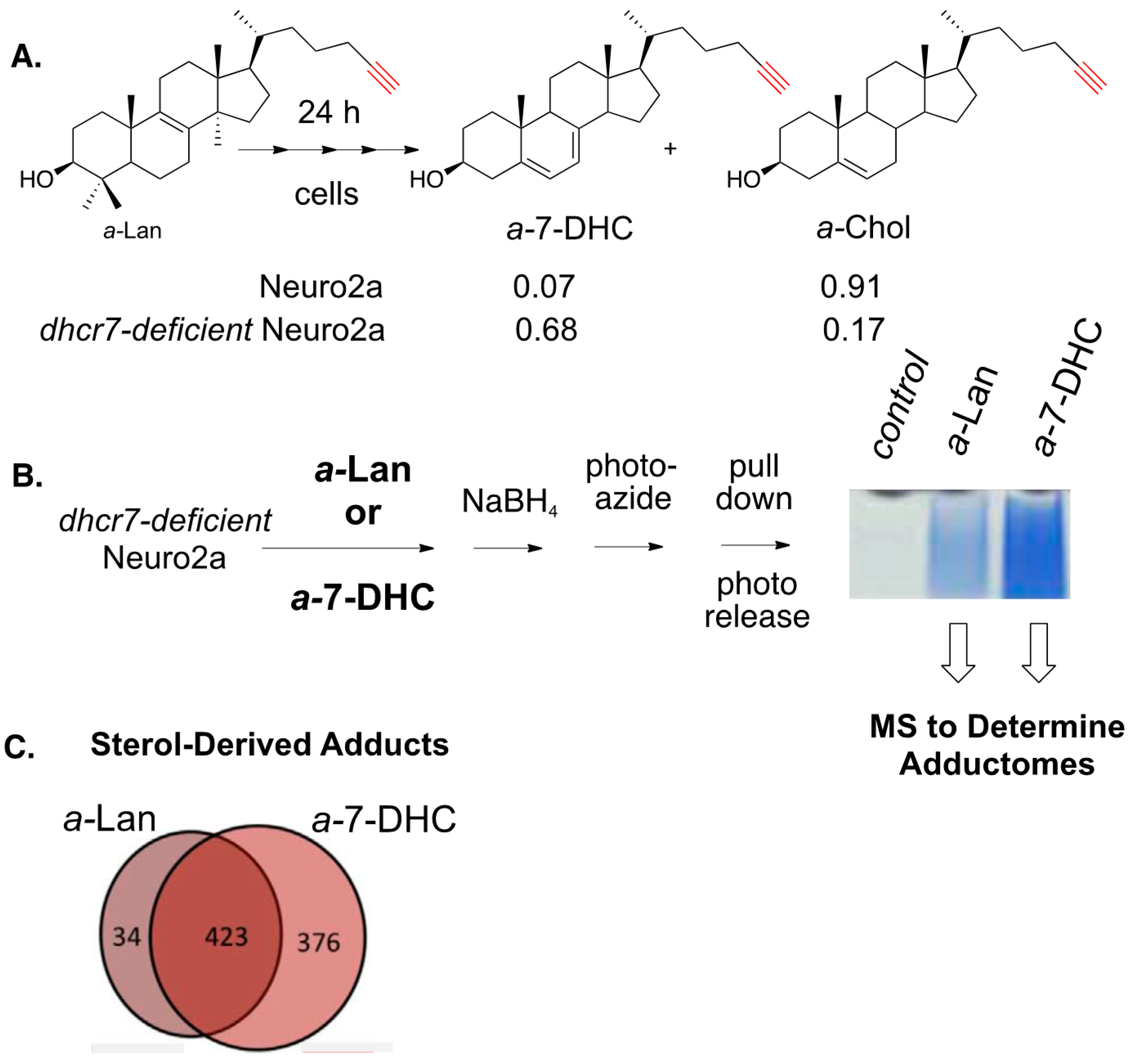
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porter, N.A.; Xu, L.; Pratt, D.A. Reactive Sterol Electrophiles: Mechanisms of Formation and Reactions with Proteins and Amino Acid Nucleophiles. Chemistry 2020, 2, 390-417. https://doi.org/10.3390/chemistry2020025
Porter NA, Xu L, Pratt DA. Reactive Sterol Electrophiles: Mechanisms of Formation and Reactions with Proteins and Amino Acid Nucleophiles. Chemistry. 2020; 2(2):390-417. https://doi.org/10.3390/chemistry2020025
Chicago/Turabian StylePorter, Ned A., Libin Xu, and Derek A. Pratt. 2020. "Reactive Sterol Electrophiles: Mechanisms of Formation and Reactions with Proteins and Amino Acid Nucleophiles" Chemistry 2, no. 2: 390-417. https://doi.org/10.3390/chemistry2020025
APA StylePorter, N. A., Xu, L., & Pratt, D. A. (2020). Reactive Sterol Electrophiles: Mechanisms of Formation and Reactions with Proteins and Amino Acid Nucleophiles. Chemistry, 2(2), 390-417. https://doi.org/10.3390/chemistry2020025