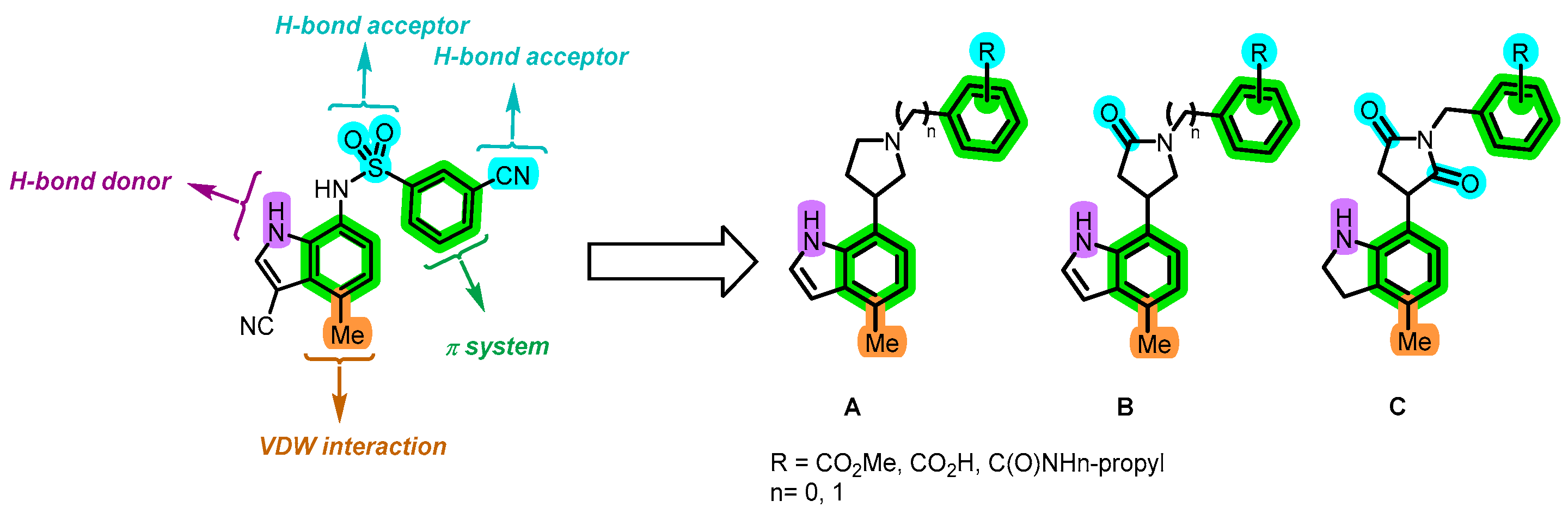
5.3. Experiments
Methyl ester derivative (1.0 equiv.) was dissolved in MeOH (40 µM), and a solution of 2 N NaOH (5.0 equiv.) was added. The resulting solution was stirred for 24 h at room temperature. After completion, the reaction mixture was then evaporated to dryness. The residue was dissolved in CH2Cl2 and washed with 2 N HCl and brine. Drying over Na2SO4, filtration, and concentration yielded the crude carboxylic acid.
HATU (1.3 equiv.), 4-Dimethylaminopyridine (0.2 equiv.), and N,N-Diisopropylethylamine (2.2 equiv.) were added to a stirred solution of carboxylic acid (1.0 eq.) in dry DMF (30 µM). The reaction mixture turned yellow. After 1 h, Propylamine (2.0 equiv.) was added. The resulting mixture was stirred at room temperature overnight. The reaction was quenched with 3 volumes of 1 M HCl, diluted with 3 volumes of EtOAc, and extracted with EtOAc (3 × 2 volumes). The organic layer was collected and washed sequentially with 1 M HCl (4 × 4 volumes), NaHCO3 (2 × 2 volumes), and brine (2 × 2 volumes). It was then dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was quickly purified by chromatography to yield the corresponding amide.
To a solution of protected indole (1.0 equiv.) in THF (59 µM), tetrabutylammonium fluoride (10 equiv.) was added, and the mixture was heated at 80 °C overnight. The reaction mixture was cooled to rt, diluted with EtOAc (2 volumes), and washed sequentially with water (2 volumes × 3) and brine (2 volumes × 1). The organic layer was dried with Na2SO4, and the solvent was removed under reduced pressure and purified by chromatography to afford unprotected indole.
To a solution of protected indole (1.0 equiv.) in THF (59 µM), ethylenediamine (6.0 equiv.) and tetrabutylammonium fluoride (10 equiv.) were added, and the mixture was heated at 80 °C overnight. The reaction mixture was cooled to rt, diluted with EtOAc (2 volumes), and washed with water (3 × 2 volumes) and brine (1 × 2 volumes). The organic layer was dried over Na2SO4, and the solvent was removed under reduced pressure. The crude product was purified by chromatography to afford the unprotected indole.
To a pre-dried seal tube, indoline derivatives (0.2 mmol, 1.0 equiv.), [RhCp*Cl2]2 (5 mol%), AgSbF6 (20 mol%), Ag2O (2.0 equiv.), maleimide derivatives (1.5 equiv.), and AcOH (3.0 equiv.) were added. To this mixture, DCE (2 mL) was then added. The vial was flushed with argon gas while tightly capped and placed in a pre-heated (100 °C) oil bath. After 24 h, the reaction mixture was cooled to room temperature, diluted with ethyl acetate, and passed through a short pad of Celite. The organic layer was concentrated under reduced pressure, and the crude product was purified on a silica gel column using an ethyl acetate/hexane mixture to afford 65–95% yield (a trace amount of Heck-type product was also observed).
To a pre-dried seal tube, C7 alkylated indoline derivatives (0.2 mmol, 1.0 equiv.) was added, and to this, 4M HCl in dioxane was added (1 mL). The vial was capped and placed in a pre-heated (100 °C) oil bath. After 30 min, the reaction mixture was cooled to room temperature and diluted with ethyl acetate; the reaction mixture was neutralized with sat, aq, and sodium bicarbonate solution; and the aqueous layer was extracted twice with EtOAc. The combined organic layer was dried over sodium sulfate. The organic layer was concentrated under reduced pressure (25 °C), and the crude product was purified on a silica gel column using ethyl acetate/hexane (if the reaction proceeded for more than 30 min, the formation of pyroquilon product was observed).
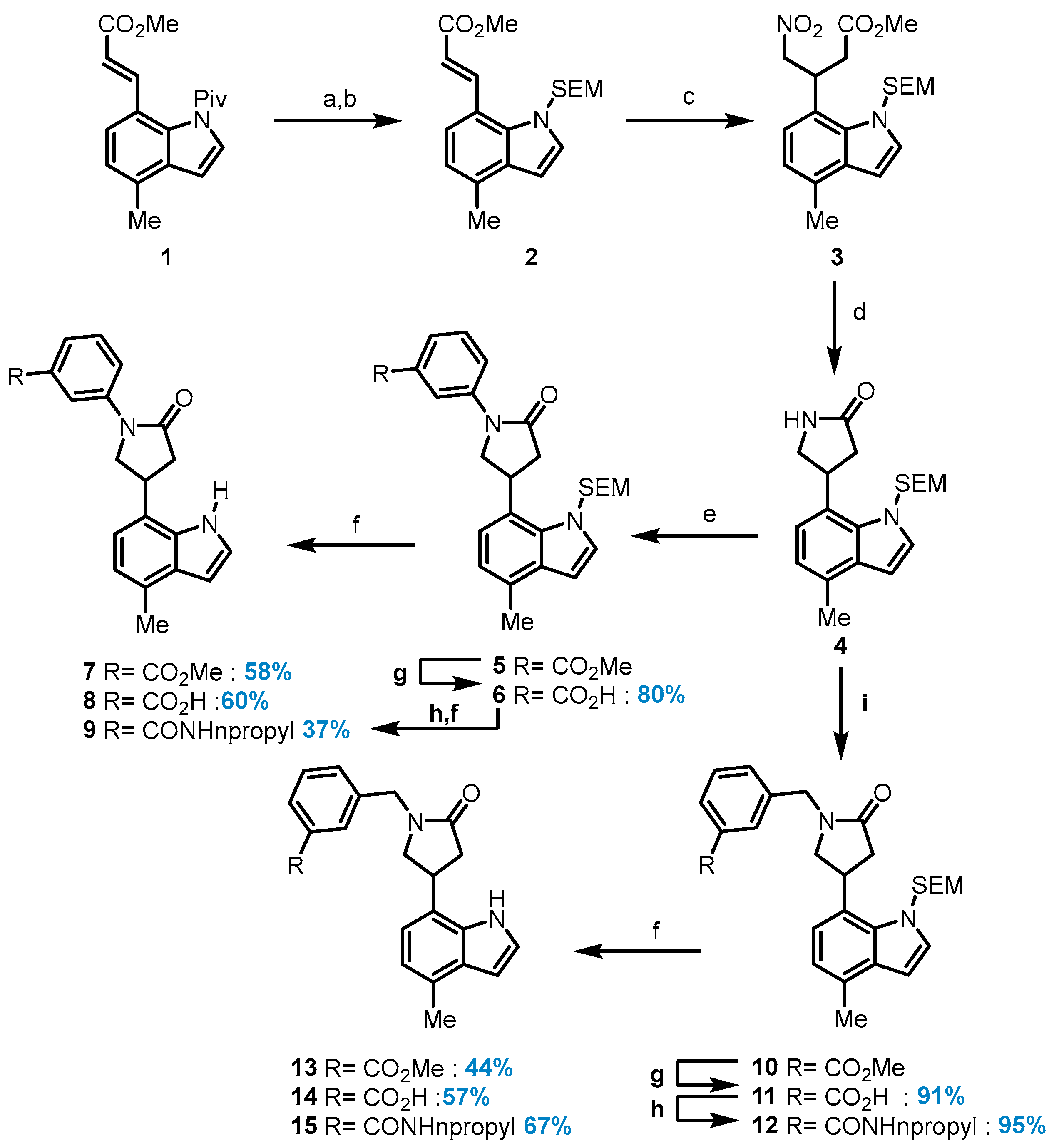
Methyl (E)-3-(4-methyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indol-7-yl)acrylate (2): To a solution of indole 1 (2.58 g, 8.62 mmol) in MeOH (17.2 mL) at room temperature, triethylamine (18.5 mL, 132 mmol) was added. The reaction mixture was stirred for 24 h under reflux and then concentrated under reduced pressure. The residue was washed with hexane to afford the free indole as a yellow solid. (1.86 g, 90%). 1H NMR (500 MHz, Chloroform-d) δ 8.55 (s, 1H), 8.00 (d, J = 16.0 Hz, 1H), 7.34 (d, J = 7.5 Hz, 1H), 7.28 (dd, J = 3.3, 2.5 Hz, 1H), 6.96 (d, J = 7.5 Hz, 1H), 6.63 (dd, J = 3.3, 2.0 Hz, 1H), 6.45 (d, J = 16.0 Hz, 1H), 3.84 (s, 3H), 2.59 (s, 3H). 13C NMR (101 MHz, Chloroform-d) δ 167.9, 141.6, 133.9, 128.7, 124.2, 122.9, 120.7, 116.3, 116.0, 101.9, 51.8, 19.1. A stirring, ice-cooled solution of the free indole (250 mg, 1.16 mmol) in DMF (13.0 mL) was treated with sodium hydride (74.3 mg, 1.86 mmol), and the reaction mixture was stirred at room temperature for 45 min. After cooling to 0 °C, 2-(trimethylsilyl)ethoxymethyl chloride (308 µL, 1.74 mmol) was added dropwise. The reaction mixture was stirred at room temperature overnight and then partitioned between H2O (50 mL) and Et2O (50 mL). The aqueous layer was extracted with Et2O (2 × 50 mL), and the combined organic phases were washed with water and brine and then dried over MgSO4. The product was purified by chromatography to afford 2 (390 mg, 96%) as a white solid. 1H NMR (500 MHz, Chloroform-d) δ 8.61 (d, J = 15.6 Hz, 1H), 7.33 (d, J = 7.5 Hz, 1H), 7.13 (d, J = 3.3 Hz, 1H), 7.00–6.94 (m, 1H), 6.54 (d, J = 3.3 Hz, 1H), 6.38 (d, J = 15.6 Hz, 1H), 5.47 (s, 2H), 3.81 (s, 3H), 3.63–3.56 (m, 2H), 2.56 (s, 3H), 1.00–0.93 (m, 2H), 0.02 (s, 9H). 13C NMR (101 MHz, Chloroform-d) δ 167.5, 143.1, 133.0, 130.5, 129.8, 122.1, 121.0, 118.8, 118.7, 100.9, 65.4, 51.6, 34.7, 31.6, 25.3, 22.7, 18.8, 17.7, 1.4; HRMS (M+H)+ calcd. for C19H28NO3Si 346.18330, found 346.18270.
Methyl 3-(4-methyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indol-7-yl)-4-nitrobutanoate (3): Indole 2 (3.17 g, 9.17 mmol) was dissolved in nitromethane, >96% (13.8 mL, 241 mmol), after which 1,1,3,3-tetramethylguanidine (576 µL, 4.59 mmol) was added with stirring, and the mixture was heated overnight at 50 °C. An additional amount of base (1 equiv.) was added, and the reaction was stirred at 50 °C for an additional period (1 h). The reaction mixture was concentrated at reduced pressure to afford an orange-colored oil which was purified by chromatography (10% EtOAc in Hexane) to give 3 as an oil (2.85 g, 76%). 1H NMR (400 MHz, Chloroform-d) δ 7.14 (d, J = 3.3 Hz, 1H), 7.00–6.93 (m, 2H), 6.54 (d, J = 3.3 Hz, 1H), 5.91 (d, J = 11.4 Hz, 1H), 5.63 (d, J = 11.4 Hz, 1H), 5.02–4.91 (m, 1H), 4.85–4.76 (m, 2H), 3.64 (s, 3H), 3.58–3.50 (m, 2H), 3.05–2.85 (m, 2H), 2.54 (d, J = 0.7 Hz, 3H), 0.93 (ddd, J = 9.5, 8.2, 7.1 Hz, 2H), 0.02 (s, 9H). 13C NMR (101 MHz, Chloroform-d) δ 171.7, 130.6, 129.9, 120.9, 120.6, 120.3, 100.6, 79.2, 65.4, 51.8, 38.1, 33.4, 18.5, 17.6; HRMS (M+H)+ calcd. for C20H31N2O5Si 407.19968, found 407.19890.
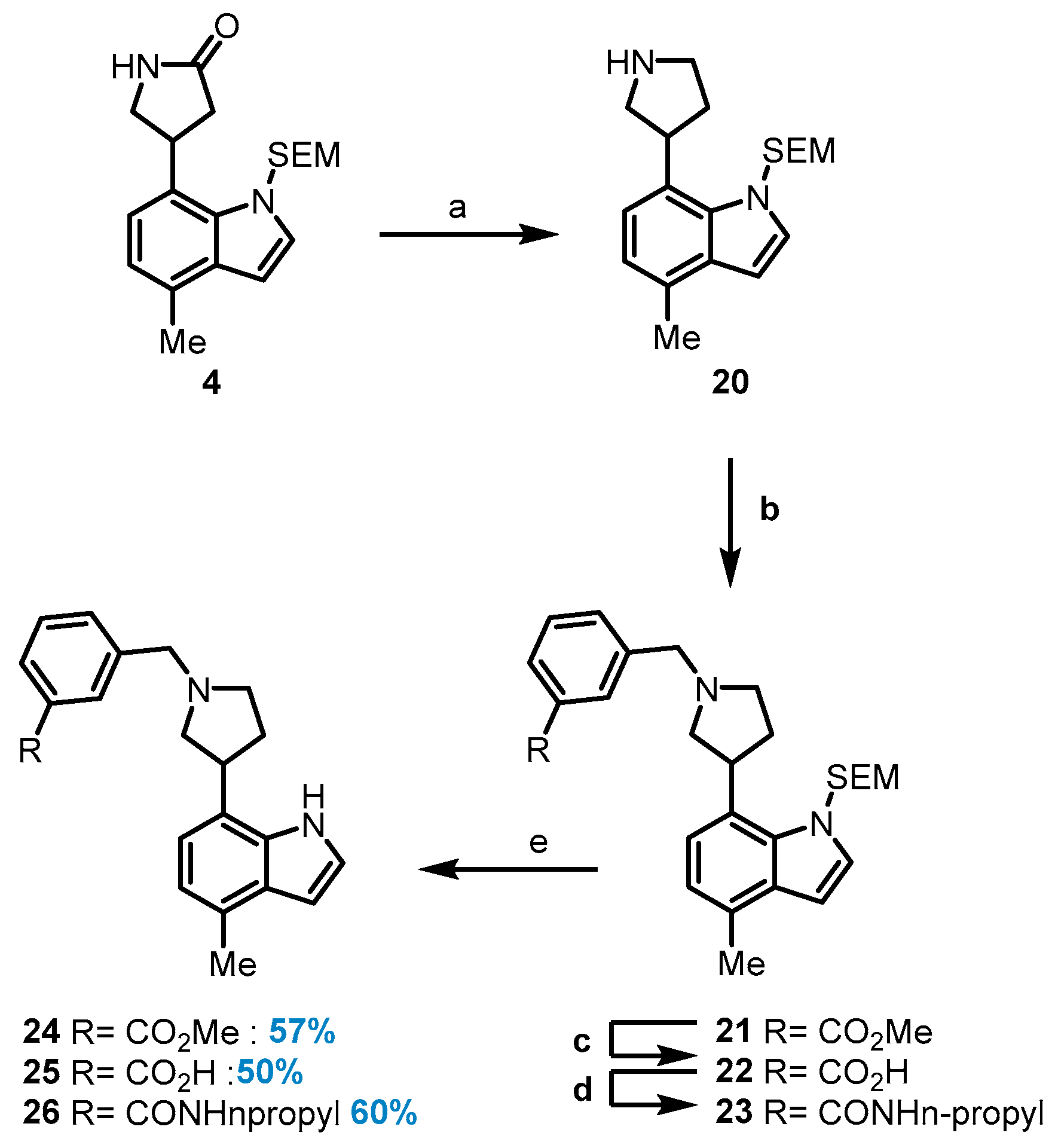
4-(4-Methyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indol-7-yl) pyrrolidine-2-one (4): To a solution of compound 3 (1.54 g, 3.79 mmol) and nickel(ii) chloride hexahydrate (908 mg, 3.79 mmol) in EtOH (20.2 mL), sodium borohydride (1.52 g, 41.7 mmol) was added at 4 °C. The reaction mixture was stirred at 4 °C for 1 h before it was diluted with EtOH. A 6 M NaOH solution was added, and the mixture was stirred for 30 min at room temperature before being treated slowly with saturated NH4Cl solution. The solution was extracted with CH2Cl2, dried over Na2SO4, filtered through Celite, and concentrated under reduced pressure, and it was then purified by chromatography (10% EtOAc in Hexane) to give compound 4 (1.29 g, 99%) as a white foam. 1H NMR (500 MHz, Chloroform-d) δ 7.18 (d, J = 7.5 Hz, 1H), 7.12 (d, J = 3.3 Hz, 1H), 7.01 (dd, J = 7.5, 1.0 Hz, 1H), 6.57 (d, J = 3.3 Hz, 1H), 5.80 (s, 1H), 5.58 (d, J = 11.8 Hz, 1H), 5.51 (d, J = 11.8 Hz, 1H), 4.67 (p, J = 7.4 Hz, 1H), 3.88 (dd, J = 9.4, 8.1 Hz, 1H), 3.57 (dd, J = 9.5, 5.6 Hz, 1H), 3.53–3.48 (m, 2H), 2.85 (dd, J = 17.0, 9.1 Hz, 1H), 2.64 (dd, J = 17.0, 6.7 Hz, 1H), 2.57 (d, J = 0.8 Hz, 3H), 0.95–0.86 (m, 2H), 0.01 (s, 9H). 13C NMR (126 MHz, Chloroform-d) δ 177.6, 133.0, 131.0, 130.4, 129.2, 124.9, 121.1, 120.1, 100.8, 65.4, 50.3, 38.9, 33.9, 18.5, 17.8; HRMS (M+K)+ calcd. for C19H28N2O2SiK 383.15516, found 383.15554.
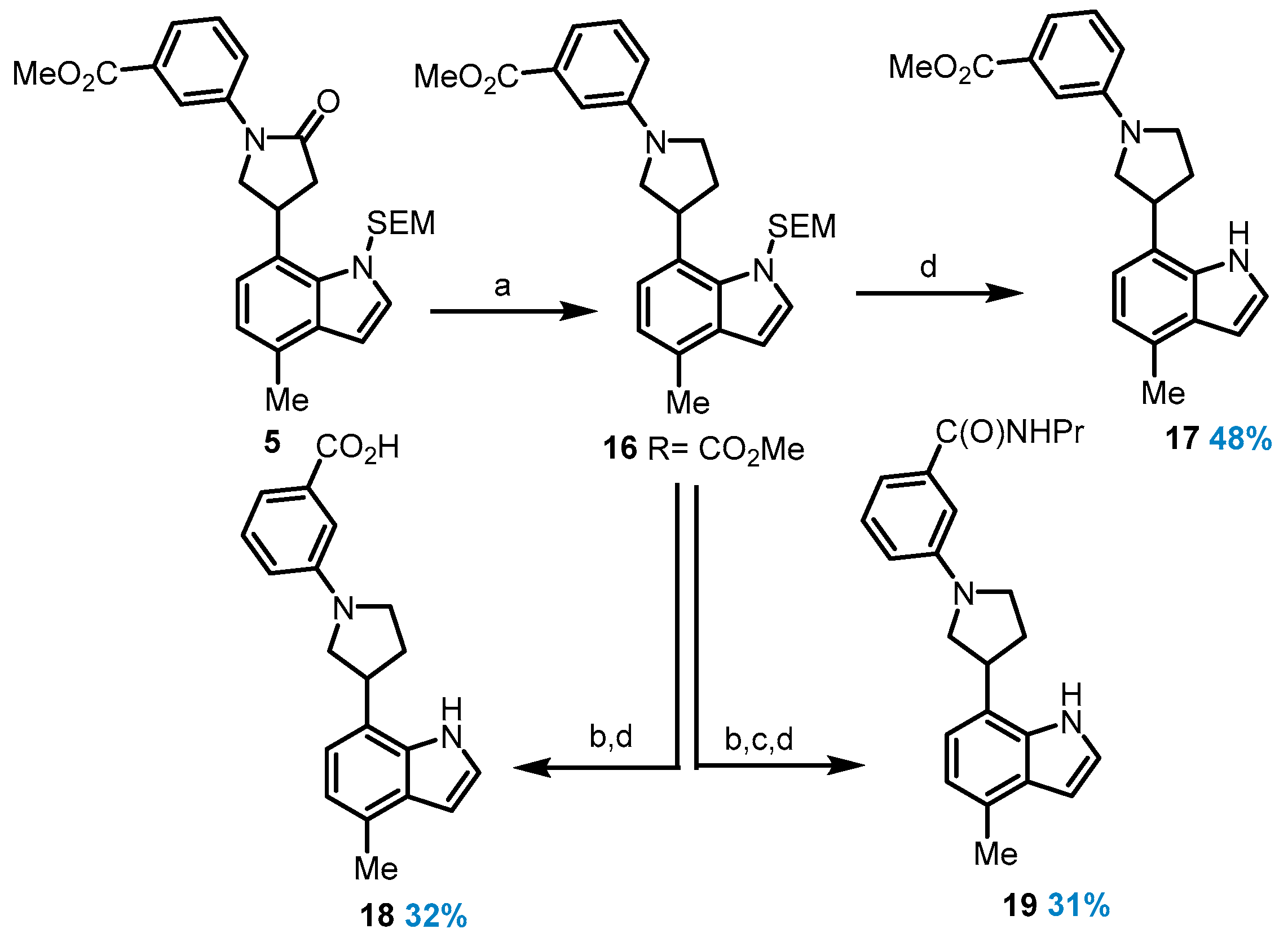
Methyl 3-(4-(4-methyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indol-7-yl)-2-oxopyrrolidin-1-yl)benzoate (5): A flame-dried pressure vial was charged with 4,5-Bis(diphenylphosphino)-9,9-dimethylxanthene (36.2 mg, 62.6 mmol), compound 4 (89.7 mg, 417 mmol), tris(dibenzylideneacetone)-dipalladium (0) (19.5 mg, 20.9 mmol), and cesium carbonate (192 mg, 584 mmol). The vial was capped with a septum, evacuated, and then filled with argon twice. 1,4-Dioxane (1 mL/mmol aryl halide) was added using a syringe, and then the septum was replaced with a Teflon screwcap, and the mixture was stirred at 100 °C for 16 h until the starting aryl halide had been completely consumed. The reaction mixture was then allowed to cool to room temperature, diluted with dichloromethane (10 mL), filtered over Celite, and concentrated under reduced pressure. The crude material was purified by chromatography on silica gel to afford compound 5 as a white solid (182 mg, 91%). 1H NMR (500 MHz, Chloroform-d) δ 8.16 (ddd, J = 8.2, 2.4, 1.1 Hz, 1H), 8.10 (t, J = 1.9 Hz, 1H), 7.87 (dt, J = 7.8, 1.3 Hz, 1H), 7.50 (t, J = 8.0 Hz, 1H), 7.17 (d, J = 7.5 Hz, 1H), 7.14 (d, J = 3.2 Hz, 1H), 7.00 (d, J = 7.6 Hz, 1H), 6.58 (d, J = 3.3 Hz, 1H), 5.64–5.54 (m, 2H), 4.68 (q, J = 7.2 Hz, 1H), 4.36 (dd, J = 9.6, 7.8 Hz, 1H), 4.08 (dd, J = 9.6, 5.5 Hz, 1H), 3.95 (s, 3H), 3.53 (ddd, J = 9.3, 8.1, 3.3 Hz, 2H), 3.16 (dd, J = 17.2, 8.8 Hz, 1H), 2.96 (dd, J = 17.1, 6.5 Hz, 1H), 2.57 (s, 3H), 0.93 (d, J = 8.3 Hz, 2H), 0.01 (s, 9H). 13C NMR (101 MHz, Chloroform-d) δ 173.5, 166.8, 139.5, 133.1, 131.2, 130.8, 130.5, 129.4, 129.0, 125.6, 124.8, 124.2, 121.2, 120.2, 120.1, 119.9, 100.9, 65.5, 56.5, 52.3, 41.4, 30.7, 18.5, 17.8; HRMS (M+H)+ calcd. for C27H35N2O4Si 479.23606, found 479.23650.
3-(4-(4-Methyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indol-7-yl)-2-oxopyrrolidin-1-yl)benzoic acid (6): Compound 6 was prepared following General Procedure A from methyl ester 5 (59.0 mg, 113 mmol). Compound 6 was obtained as a white solid (46.0 mg, 80%). 1H NMR (400 MHz, Chloroform-d) δ 8.26 (ddd, J = 8.3, 2.4, 1.0 Hz, 1H), 8.12 (t, J = 2.0 Hz, 1H), 7.93 (dt, J = 7.9, 1.3 Hz, 1H), 7.53 (t, J = 8.0 Hz, 1H), 7.23–7.13 (m, 2H), 7.01 (dd, J = 7.3, 1.0 Hz, 1H), 6.59 (d, J = 3.3 Hz, 1H), 4.77–4.61 (m, 1H), 4.39 (dd, J = 9.6, 7.8 Hz, 1H), 4.13–4.06 (m, 1H), 3.59–3.48 (m, 2H), 3.23–3.12 (m, 1H), 3.01 (d, J = 6.5 Hz, 1H), 2.57 (d, J = 0.8 Hz, 3H), 0.98–0.89 (m, 2H), 0.01 (s, 9H). 13C NMR (101 MHz, Chloroform-d) δ 173.68, 170.9, 133.1, 131.2, 130.5, 130.0, 129.4, 129.1, 128.9, 126.2, 125.6, 124.2, 121.2, 120.6, 120.2, 119.9, 100.9, 65.6, 56.5, 41.3, 30.7, 18.5, 17.8; HRMS (M+Na)+ calcd. for C26H32N2O4SiNa 487.20236, found 487.20329.
Methyl 3-(4-(4-methyl-1H-indol-7-yl)-2-oxopyrrolidin-1-yl)benzoate (7): Compound 7 was prepared following General Procedure C from the protected indole 5 (40.0 mg, 83.6 µmol) and obtained as a white solid (17.0 mg, 58%). 1H NMR (500 MHz, Chloroform-d) δ 8.66 (s, 1H), 8.09–8.04 (m, 2H), 7.83 (dt, J = 7.8, 1.3 Hz, 1H), 7.47–7.41 (m, 1H), 7.05 (d, J = 7.4 Hz, 1H), 6.95 (dd, J = 7.4, 1.0 Hz, 1H), 6.65 (dd, J = 3.3, 2.0 Hz, 1H), 4.28 (dd, J = 9.6, 8.1 Hz, 1H), 4.17–4.11 (m, 1H), 4.02 (t, J = 8.3 Hz, 1H), 3.91 (s, 3H), 3.15–2.98 (m, 2H), 2.58 (d, J = 0.8 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 173.3, 166.7, 139.2, 133.6, 130.8, 129.8, 129.1, 128.4, 125.8, 124.7, 123.9, 121.0, 120.3, 120.2, 119.3, 102.1, 54.3, 52.3, 39.1, 33.2, 18.6; HRMS (M+H)+ calcd. for C21H20N2O3 349.1554, found 349.1546.
3-(4-(4-Methyl-1H-indol-7-yl)-2-oxopyrrolidin-1-yl)benzoic acid (8): Compound 8 was prepared following General Procedure C from the protected indole 6 (36.0 mg, 77.5 µmol) and obtained as a colorless oil (12.5 mg, 48%). 1H NMR (400 MHz, Methanol-d4) δ 8.02 (s, 1H), 7.92 (d, J = 8.1 Hz, 1H), 7.80 (d, J = 7.6 Hz, 1H), 7.40 (t, J = 7.9 Hz, 1H), 7.27 (d, J = 3.2 Hz, 1H), 7.01 (d, J = 7.3 Hz, 1H), 6.83 (d, J = 7.3 Hz, 1H), 6.53 (d, J = 3.2 Hz, 1H), 4.45–4.36 (m, 1H), 4.13 (dd, J = 15.4, 8.1 Hz, 2H), 3.07 (dd, J = 16.8, 8.2 Hz, 1H), 2.93 (dd, J = 16.7, 8.0 Hz, 1H), 2.51 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 134.5, 128.3, 128.0, 125.1, 119.6, 118.3, 100.7, 54.2, 39.0, 23.5, 18.9, 13.9. HRMS (M+H)+ calcd. for C20H18N2O3 335.1389, found 335.1390.
3-(4-(4-Methyl-1H-indol-7-yl)-2-oxopyrrolidin-1-yl)-N-propylbenzamide (9): General Procedure B was applied using carboxylic acid 6 (47.0 mg, 100 mmol) to give the corresponding n-propylamide (17.0 mg, 73%) as a colorless oil. 1H NMR (400 MHz, Chloroform-d) δ 8.09 (t, J = 2.0 Hz, 1H), 7.89–7.83 (m, 1H), 7.61–7.55 (m, 1H), 7.47 (t, J = 8.0 Hz, 1H), 7.18–7.12 (m, 2H), 7.00 (d, J = 7.7 Hz, 1H), 6.58 (d, J = 3.3 Hz, 1H), 5.63–5.52 (m, 3H), 4.69 (p, J = 7.3 Hz, 2H), 4.36 (dd, J = 9.6, 7.8 Hz, 2H), 4.09 (dd, J = 9.6, 5.3 Hz, 1H), 3.55–3.49 (m, 2H), 3.50–3.43 (m, 2H), 3.16 (dd, J = 17.1, 8.9 Hz, 2H), 2.95 (dd, J = 17.1, 6.3 Hz, 1H), 2.57 (d, J = 0.8 Hz, 3H), 1.71–1.65 (m, 2H), 1.03 (t, J = 7.4 Hz, 3H), 0.92 (t, J = 1.2 Hz, 2H), 0.01 (s, 9H). 13C NMR (126 MHz, Chloroform-d) δ 173.6, 167.2, 162.6, 135.6, 133.1, 131.2, 130.5, 129.4, 129.1, 124.2, 122.8, 122.6, 121.2, 118.1, 100.9. General Procedure D was applied directly on the propylamide to give the corresponding unprotected indole 9 (6.1 mg, 55%) as colorless oil. 1H NMR (400 MHz, Chloroform-d) δ 9.38 (s, 1H), 8.08 (t, J = 2.0 Hz, 1H), 7.70 (ddd, J = 8.2, 2.3, 1.0 Hz, 1H), 7.56 (dt, J = 7.7, 1.3 Hz, 1H), 7.40 (d, J = 7.9 Hz, 1H), 7.03 (d, J = 7.3 Hz, 1H), 6.94 (d, J = 7.0 Hz, 1H), 6.64 (dd, J = 3.3, 1.9 Hz, 1H), 6.53 (d, J = 6.0 Hz, 1H), 4.26 (dd, J = 9.1, 7.6 Hz, 1H), 4.01 (dt, J = 24.9, 8.3 Hz, 2H), 3.46–3.37 (m, 2H), 3.08 (dd, J = 17.0, 9.1 Hz, 1H), 2.97 (dd, J = 17.1, 8.5 Hz, 1H), 2.59 (s, 3H), 1.66–1.59 (m, 2H), 0.99 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 173.8, 167.4, 139.3, 135.6, 133.8, 129.7, 129.2, 128.5, 124.2, 123.04, 122.4, 120.7, 120.1, 119.1, 118.1, 101.7, 54.4, 41.9, 38.9, 33.4, 29.7, 22.8, 18.6, 11.5; HRMS (M+H)+ calcd. for C23H26N3O2 376.2013, found 376.2019.
Methyl-3-((4-(4-methyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indol-7-yl)-2-oxopyrrolidin-1-yl)methyl) benzoate (10): Indole 4 (100 mg, 290 µmol) in DMF (1.45 mL) and 15-CROWN-5 (80.4 mL, 406 µmol) was treated with sodium hydride (23.2 mg, 581 µmol) at 0 °C, and the suspension was stirred at room temperature for 1 h. The solution as cooled, and methyl 4-(bromomethyl)-3-methoxybenzoate (160 mg, 598 µmol) in THF was added at 0 °C, and the solution was stirred for 6h at room temperature. EtOAc and water were added, and the organic phase was extracted three times with EtOAc and then washed with water and brine. The organic extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by chromatography (0–90% EtOAc in hexanes) to afford compound 10 (97.1 mg, 64%) as a colorless oil. 1H NMR (400 MHz, Chloroform-d) δ 7.96 (dt, J = 7.7, 1.7 Hz, 1H), 7.93 (d, J = 1.8 Hz, 1H), 7.48 (dd, J = 7.6, 1.7 Hz, 1H), 7.41 (t, J = 7.6 Hz, 1H), 7.05 (d, J = 3.3 Hz, 1H), 6.96 (d, J = 7.5 Hz, 1H), 6.89 (d, J = 7.5 Hz, 1H), 6.50 (d, J = 3.3 Hz, 1H), 5.52–5.39 (m, 2H), 4.64–4.54 (m, 2H), 4.48 (ddd, J = 8.7, 7.0, 4.3 Hz, 1H), 3.91 (s, 3H), 3.69 (dd, J = 9.9, 8.0 Hz, 1H), 3.42 (dd, J = 8.9, 7.6 Hz, 2H), 3.36 (dd, J = 9.8, 4.9 Hz, 1H), 2.98 (dd, J = 17.0, 9.2 Hz, 1H), 2.76–2.70 (m, 1H), 2.50 (s, 3H), 0.80 (td, J = 7.7, 1.6 Hz, 2H), 0.07 (s, 9H). 13C NMR (101 MHz, Chloroform-d) δ 136.8, 132.8, 130.9, 130.3, 129.3, 129.0, 128.9, 128.9, 125.3, 121.1, 119.8, 100.7, 65.4, 54.9, 52.2, 46.4, 30.4, 18.4, 17.7; HRMS (M+Na)+ calcd. for C28H36N2O4SiNa 515.2337, found 515.2327.
3-((4-(4-Methyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indol-7-yl)-2-oxopyrrolidin-1-yl)methyl)benzoic acid (11): Compound 11 was prepared following General Procedure A from methyl ester 10 (20.0 mg, 40.6 µmol) and was obtained as a colorless oil (17.7 mg, 91%). 1H NMR (400 MHz, Chloroform-d) δ 8.09–8.02 (m, 2H), 7.57 (dt, J = 7.7, 1.5 Hz, 1H), 7.48 (t, J = 7.7 Hz, 1H), 7.08 (d, J = 3.3 Hz, 1H), 7.02 (d, J = 7.6 Hz, 1H), 6.95 (dd, J = 7.5, 1.0 Hz, 1H), 6.53 (d, J = 3.3 Hz, 1H), 5.56–5.42 (m, 2H), 4.71–4.57 (m, 2H), 4.59–4.49 (m, 1H), 3.76 (dd, J = 9.8, 8.1 Hz, 1H), 3.44 (ddd, J = 14.6, 9.3, 6.3 Hz, 3H), 3.04 (dd, J = 17.1, 9.2 Hz, 1H), 2.80 (dd, J = 17.1, 5.8 Hz, 1H), 2.54 (d, J = 0.8 Hz, 3H), 0.88–0.82 (m, 2H), 0.04 (s, 9H). 13C NMR (101 MHz, Chloroform-d) δ 174.5, 170.7, 136.9, 133.4, 132.9, 130.9, 130.3, 130.0, 129.8, 129.6, 129.1, 125.2, 121.1, 119.9, 100.7, 65.4, 55.0, 46.4, 39.9, 30.5, 18.4, 17.7; HRMS (M+Na)+ calcd. for C27H34N2O4SiNa 501.2180, found 501.2182.
3-((4-(4-Methyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indol-7-yl)-2-oxopyrrolidin-1-yl)methyl)-N-propylbenzamide (12):
Compound 12 was prepared following General Procedure B from carboxylic acid 11 (10 mg, 50.6 mmol) and obtained as a colorless oil (10 mg, 95%). 1H NMR (500 MHz, Chloroform-d) δ 7.74 (d, J = 1.3 Hz, 1H), 7.66 (q, J = 1.3 Hz, 1H), 7.45–7.40 (m, 2H), 7.09 (d, J = 3.3 Hz, 1H), 6.99 (d, J = 7.5 Hz, 1H), 6.93 (dd, J = 7.5, 1.0 Hz, 1H), 6.54 (d, J = 3.3 Hz, 1H), 6.22 (s, 1H), 5.54–5.41 (m, 2H), 4.60 (s, 2H), 4.52 (s, 1H), 3.74 (dd, J = 9.8, 8.0 Hz, 1H), 3.50–3.43 (m, 4H), 3.41 (dd, J = 9.8, 4.8 Hz, 1H), 3.04–2.98 (m, 2H), 2.76 (dd, J = 17.1, 5.7 Hz, 1H), 2.54 (d, J = 0.8 Hz, 3H), 1.73–1.64 (m, 3H), 1.03 (t, J = 7.4 Hz, 3H), 0.84 (td, J = 7.8, 1.8 Hz, 2H), 0.04 (s, 9H). 13C NMR (126 MHz, Chloroform-d) δ 174.3, 136.9, 135.4, 133.0, 131.1, 130.9, 130.3, 129.1, 129.1, 126.5, 126.4, 120.9, 119.7, 100.8, 65.4, 54.9, 46.4, 41.8, 39.9, 38.6, 30.4, 22.9, 18.4, 17.7, 11.5; HRMS (M+Na)+ calcd. for C30H41N3O3SiNa 542.2818, found 542.2809.
Methyl 3-((4-(4-methyl-1H-indol-7-yl)-2-oxopyrrolidin-1-yl)methyl)benzoate (13):
Compound 13 was prepared following General Procedure D from the protected indole 10 (25.0 mg, 50.7 µmol) and obtained as a colorless oil (8 mg, 44%). 1H NMR (500 MHz, Chloroform-d) δ 8.13 (s, 1H), 8.00–7.94 (m, 2H), 7.52 (dt, J = 7.7, 1.5 Hz, 1H), 7.44 (t, J = 7.6 Hz, 1H), 7.15–7.12 (m, 1H), 6.92–6.81 (m, 2H), 6.57 (dd, J = 3.3, 1.9 Hz, 1H), 4.65–4.56 (m, 2H), 3.92 (s, 3H), 3.88–3.79 (m, 1H), 3.72 (dd, J = 9.9, 8.5 Hz, 1H), 3.48 (dd, J = 9.8, 6.4 Hz, 1H), 2.98 (dd, J = 17.2, 9.3 Hz, 1H), 2.83–2.73 (m, 1H), 2.52 (d, J = 0.8 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 174.1, 141.4, 130.2, 129.5, 128.2, 123.8, 120.1, 119.3, 101.9, 52.6, 52.2, 46.5, 37.7, 33.2, 18.6; HRMS (M+H)+ calcd. for C22H23N2O3 363.1694, found 363.1703.
3-((4-(4-Methyl-1H-indol-7-yl)-2-oxopyrrolidin-1-yl)methyl)benzoic acid (14): Compound 14 was synthesized following General Procedure D from the protected indole 11 (17.0 mg, 35.5 µmol) and obtained as a colorless oil (7 mg, 57%). 1H NMR (500 MHz, Methanol-d4) δ 7.91–7.88 (m, 2H), 7.38–7.35 (m, 2H), 7.23–7.20 (m, 1H), 6.87 (d, J = 7.3 Hz, 1H), 6.78 (d, J = 7.3 Hz, 1H), 6.49 (d, J = 3.2 Hz, 1H), 4.76 (d, J = 14.8 Hz, 1H), 4.43 (d, J = 14.8 Hz, 1H), 4.03 (s, 1H), 3.83 (dd, J = 9.9, 8.1 Hz, 1H), 3.47 (dd, J = 9.9, 6.8 Hz, 1H), 2.95 (dd, J = 16.8, 8.8 Hz, 1H), 2.79–2.71 (m, 1H), 2.48 (d, J = 0.9 Hz, 2H). 13C NMR (126 MHz, Methanol-d4) δ 132.3, 128.9, 128.7, 128.6, 119.1, 117.1, 52.8, 45.7, 37.5, 32.3, 31.4, 29.4, 22.3, 17.3, 13.0; HRMS (M+H)+ calcd. for C21H21N2O3 349.1546, found 349.1531.
3-((4-(4-Methyl-1H-indol-7-yl)-2-oxopyrrolidin-1-yl)methyl)-N-propylbenzamide (15): Compound 15 was prepared following General Procedure D from the protected indole 12 (10 mg, 26.9 µmol) and obtained as a colorless oil (5 mg, 67%). 1H NMR (400 MHz, Chloroform-d) δ 8.72 (s, 1H), 7.78–7.67 (m, 2H), 7.43 (dd, J = 4.4, 2.0 Hz, 2H), 7.21 (t, J = 2.7 Hz, 1H), 6.95–6.86 (m, 2H), 6.60 (dd, J = 3.4, 1.3 Hz, 1H), 6.42 (s, 1H), 4.69 (d, J = 14.8 Hz, 1H), 4.51 (d, J = 14.7 Hz, 1H), 3.90 (d, J = 8.1 Hz, 1H), 3.82–3.73 (m, 1H), 3.47 (dq, J = 23.6, 6.6 Hz, 3H), 2.97 (dd, J = 17.3, 9.1 Hz, 1H), 2.84 (dd, J = 17.1, 8.1 Hz, 1H), 2.56 (d, J = 0.8 Hz, 3H), 1.70–1.64 (m, 2H), 1.01 (d, J = 7.4 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 129.4, 129.2, 126.6, 119.9, 101.7, 41.9, 33.3, 22.9, 18.6; HRMS (M+H)+ calcd. for C24H28N3O2 390.2167, found 390.2176, (M+Na)+ calcd. for C24H27N3O2Na 407.2431, found 407.2442.
Methyl 3-(3-(4-methyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indol-7-yl)pyrrolidin-1-yl)benzoate (16): To a solution of compound 5 (70.0 mg, 146 µmol) in THF (439 mL), a solution of Borane-THF complex 1M in THF (190 mL, 190 µmol) was added. The reaction mixture was stirred at room temperature overnight and then quenched by slowly adding MeOH (1 mL) followed by H2O. The above mixture was then extracted with EtOAc, and the organic layer was washed with H2O and brine, dried over Na2SO4, filtered, and concentrated. Purification by chromatography (0–30% EtOAc/Hex) gave compound 16 (44.2 mg, 65%) as a clear, colorless oil. 1H NMR (400 MHz, Chloroform-d) δ 7.42–7.31 (m, 3H), 7.15 (d, J = 3.3 Hz, 1H), 7.06 (d, J = 7.5 Hz, 1H), 6.94 (d, J = 7.5 Hz, 1H), 6.82 (ddd, J = 8.1, 2.7, 1.1 Hz, 1H), 6.58 (d, J = 3.3 Hz, 1H), 5.71–5.53 (m, 2H), 4.54–4.44 (m, 1H), 3.94 (s, 3H), 3.83 (dd, J = 9.5, 7.2 Hz, 1H), 3.66–3.58 (m, 2H), 3.56–3.49 (m, 3H), 2.56 (s, 3H), 2.49 (dt, J = 13.0, 6.3 Hz, 1H), 2.28 (dq, J = 13.2, 6.8 Hz, 1H), 0.97–0.88 (m, 2H), 0.01 (s, 9H). 13C NMR (101 MHz, Chloroform-d) δ 167.89, 147.4, 130.9, 130.8, 130.3, 129.1, 128.7, 125.1, 120.9, 120.1, 116.6, 115.9, 112.4, 100.7, 65.4, 55.1, 52.0, 47.2, 37.6, 33.8, 18.6, 17.7; HRMS (M+H)+ calcd. for C21H23N2O2 335.1763, found 335.1754.
Methyl 3-(3-(4-methyl-1H-indol-7-yl)pyrrolidin-1-yl)benzoate (17): Compound 17 was prepared following General Procedure D from the protected indole 16 (100 mg, 209 µmol) and obtained as a colorless oil (46.6 mg, 48%). 1H NMR (400 MHz, Chloroform-d) δ 8.88 (s, 1H), 7.52–7.47 (m, 1H), 7.43 (dd, J = 2.6, 1.5 Hz, 1H), 7.37 (t, J = 7.9 Hz, 1H), 7.22 (t, J = 2.8 Hz, 1H), 7.00 (d, J = 7.3 Hz, 1H), 6.96–6.87 (m, 2H), 6.63 (dd, J = 3.2, 2.1 Hz, 1H), 3.95 (s, 3H), 3.89–3.75 (m, 3H), 3.70 (dd, J = 9.5, 8.0 Hz, 1H), 3.41 (dt, J = 9.3, 7.8 Hz, 1H), 2.59 (d, J = 0.8 Hz, 3H), 2.55 (p, J = 4.1, 3.6 Hz, 1H), 2.41–2.29 (m, 1H). 13C NMR (101 MHz, Chloroform-d) δ 148.1, 131.0, 129.3, 128.9, 124.1, 123.6, 120.1, 119.9, 118.4, 117.5, 114.0, 101.5, 54.0, 53.8, 52.1, 48.8, 40.5, 32.1, 20.8, 18.6, 14.1; HRMS (M+H)+ calcd. for C21H23N2O2 335.1763, found 335.1754.
3-(3-(4-Methyl-1H-indol-7-yl)pyrrolidin-1-yl)benzoic acid (18): General Procedure A was applied to methyl ester 16 (47.0 mg, 100 mmol) to give the corresponding carboxylic acid (30 mg, 66%) as a colorless oil: 1H NMR (400 MHz, Chloroform-d) δ 7.43 (d, J = 7.6 Hz, 1H), 7.36–7.28 (m, 2H), 7.10 (d, J = 3.3 Hz, 1H), 7.03 (d, J = 7.5 Hz, 1H), 6.90 (d, J = 7.5 Hz, 1H), 6.82 (dd, J = 8.1, 2.6 Hz, 1H), 6.54 (d, J = 3.3 Hz, 1H), 5.63 (dd, J = 11.6, 2.5 Hz, 1H), 5.52 (d, J = 11.6 Hz, 1H), 4.50–4.39 (m, 1H), 3.84–3.74 (m, 1H), 3.62–3.54 (m, 2H), 3.49 (t, J = 8.1 Hz, 3H), 2.52 (s, 3H), 2.47 (dd, J = 12.5, 5.9 Hz, 1H), 2.25 (dd, J = 12.4, 6.6 Hz, 1H), 0.93–0.84 (m, 3H), 0.06 (s, 9H). General Procedure D was applied directly to the carboxylic acid to give the corresponding unprotected indole 18 (10 mg, 48%) as a colorless oil. 1H NMR (400 MHz, Methanol-d4) δ 7.28 (d, J = 10.1 Hz, 2H), 7.23 (d, J = 3.2 Hz, 2H), 6.90 (s, 1H), 6.77 (d, J = 7.3 Hz, 1H), 6.72 (d, J = 7.5 Hz, 1H), 6.49 (d, J = 3.2 Hz, 1H), 3.91 (q, J = 6.8, 6.0 Hz, 1H), 3.83 (t, J = 8.3 Hz, 1H), 3.56 (s, 1H), 3.49 (d, J = 8.2 Hz, 2H), 2.49 (s, 4H), 2.27 (dd, J = 12.3, 8.0 Hz, 1H). 13C NMR (101 MHz, Methanol-d4) δ 127.9, 127.4, 123.5, 118.9, 117.6, 99.7, 39.3, 31.5, 17.3; HRMS (M+H)+ calcd. for C20H21N2O2 321.1582, found 321.1598.
3-(3-(4-Methyl-1H-indol-7-yl)pyrrolidin-1-yl)-N-propylbenzamide (19): General Procedure A was applied to methyl ester 16 (47.0 mg, 100 mmol) to give the corresponding carboxylic acid (30 mg, 66%) as a colorless oil which was directly used in General Procedure B to obtain the corresponding propylamide (30 mg, 68%) as a colorless oil. 1H NMR (400 MHz, Chloroform-d) δ 7.37 (q, J = 7.4, 6.9 Hz, 2H), 7.18 (d, J = 7.7 Hz, 1H), 7.14 (d, J = 3.3 Hz, 1H), 7.11 (d, J = 7.6 Hz, 1H), 7.04 (s, 1H), 6.96 (d, J = 7.5 Hz, 1H), 6.57 (d, J = 3.3 Hz, 1H), 6.21 (s, 1H), 5.70–5.55 (m, 2H), 4.62–4.52 (m, 1H), 3.92–3.85 (m, 1H), 3.74–3.67 (m, 2H), 3.62 (t, J = 7.5 Hz, 1H), 3.57–3.41 (m, 4H), 2.56 (s, 3H), 2.54 (d, J = 6.2 Hz, 1H), 2.36 (dd, J = 12.8, 6.9 Hz, 1H), 1.68 (q, J = 7.3 Hz, 2H), 1.03 (t, J = 7.4 Hz, 3H), 0.91 (dd, J = 9.3, 7.1 Hz, 2H), 0.02 (s, 9H). 13C NMR (101 MHz, Chloroform-d) δ 136.2, 133.4, 130.9, 130.4, 129.6, 121.0, 120.1, 100.7, 65.4, 41.8, 37.7, 33.8, 22.9, 18.5, 17.7, 11.5. General Procedure D was applied directly on the propylamide to give the corresponding unprotected indole 19 (5 mg, 68%) as a white solid. 1H NMR (400 MHz, Chloroform-d) δ 8.88 (s, 1H), 7.31 (d, J = 7.9 Hz, 1H), 7.20–7.16 (m, 2H), 7.04 (dt, J = 7.8, 1.1 Hz, 1H), 6.95 (d, J = 7.3 Hz, 1H), 6.86 (dd, J = 7.3, 0.9 Hz, 1H), 6.80 (ddd, J = 8.2, 2.6, 0.9 Hz, 1H), 6.59 (dd, J = 3.2, 2.0 Hz, 1H), 6.12 (s, 1H), 3.85–3.70 (m, 3H), 3.46–3.32 (m, 4H), 2.55 (d, J = 0.9 Hz, 3H), 2.52–2.47 (m, 1H), 2.29 (dd, J = 12.7, 7.8 Hz, 1H), 1.66–1.61 (m, 2H), 0.98 (d, J = 7.4 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 168.2, 148.3, 136.1, 133.5, 129.4, 128.2, 124.1, 123.7, 120.0, 119.8, 115.78, 114.6, 111.9, 101.5, 53.9, 48.7, 41.8, 40.5, 32.1, 22.9, 18.6, 11.5; HRMS (M+H)+ calcd. for C23H28N3O 362.2216, found 362.2227.
4-Methyl-7-(pyrrolidin-3-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indole (20): To a 1M solution of lithium aluminum hydride (755 µL, 755 µmol) in anhydrous THF (677 µL), a solution of indole 4 (130 mg, 377 µmol) in anhydrous THF (1.43 mL) was added dropwise at 0 °C for 1h. The reaction was refluxed for 2 h and then quenched with water at 0 °C. The resulting mixture was filtered, and the filter cake was washed with a mixture of DCM and MeOH (1/1 (v/v), 10 mL). The filtrate was concentrated under reduced pressure. The residue was purified by chromatography (10% MeOH in DCM) to give the free pyrrolidine 20 as a yellow solid (104 mg, 83%). 1H NMR (400 MHz, Chloroform-d) δ 7.08 (d, J = 3.3 Hz, 1H), 7.04 (d, J = 7.5 Hz, 1H), 6.93 (d, J = 7.5 Hz, 1H), 6.51 (d, J = 3.3 Hz, 1H), 5.55 (s, 2H), 4.13 (p, J = 6.8 Hz, 1H), 3.51–3.48 (m, 2H), 3.32 (dd, J = 11.1, 7.3 Hz, 1H), 3.24 (ddd, J = 11.2, 8.3, 5.7 Hz, 1H), 3.11 (ddd, J = 11.2, 8.2, 6.5 Hz, 1H), 3.01 (dd, J = 11.1, 6.1 Hz, 1H), 2.51 (s, 3H), 2.30–2.19 (m, 1H), 2.08–1.95 (m, 1H), 0.93–0.84 (m, 2H), 0.05 (s, 9H). 13C NMR (126 MHz, Chloroform-d) δ 140.0, 134.1, 131.0, 130.2, 128.2, 120.8, 120.0, 104.1, 100.6, 81.1, 78.1, 65.3, 29.9, 18.4, 17.7, 2.0, 1.4; HRMS (ESI) m/z: [M+H]+ calculated for C19H31N2OSi: 331.21941, found 331.22002.
Methyl 3-((3-(4-methyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indol-7-yl)pyrrolidin-1-yl)methyl)benzoate (21): To a stirred solution of pyrrolidine 20 (375 mg, 1.13 mmol) in CH2Cl2 (2.05 mL), Et3N (398 µL, 2.84 mmol) was added at 0 °C followed by methyl 3-(bromomethyl)benzoate (345 µL, 1.59 mmol) in CH2Cl2 (1.37 mL). The mixture was stirred for 30 min at 0 °C and then warmed to room temperature and stirred for an additional 2 h. The above mixture was washed with saturated NaHCO3 solution, and the organic layer was dried over Na2SO4, filtered, and concentrated. Purification by chromatography (0–10% EtOAc in Hexanes) gave 21 as a colorless oil (443 mg, 82%). 1H NMR (400 MHz, Chloroform-d) δ 8.04 (d, J = 1.8 Hz, 1H), 7.93 (dt, J = 7.9, 1.5 Hz, 1H), 7.65–7.59 (m, 1H), 7.40 (t, J = 7.7 Hz, 1H), 7.30 (d, J = 7.5 Hz, 1H), 7.05 (d, J = 3.3 Hz, 1H), 6.96 (dd, J = 7.5, 1.0 Hz, 1H), 6.49 (d, J = 3.3 Hz, 1H), 5.59–5.44 (m, 2H), 4.30–4.17 (m, 1H), 3.92 (s, 3H), 3.80–3.70 (m, 2H), 3.46 (dd, J = 8.8, 7.6 Hz, 2H), 2.99 (dd, J = 9.3, 7.9 Hz, 1H), 2.87 (dt, J = 8.5, 4.3 Hz, 1H), 2.77 (ddd, J = 8.5, 6.5, 2.8 Hz, 2H), 2.54–2.49 (m, 3H), 2.38 (dddd, J = 13.0, 9.7, 7.7, 5.4 Hz, 1H), 2.02 (dtd, J = 12.7, 6.4, 2.0 Hz, 1H), 0.85 (td, J = 7.8, 1.9 Hz, 2H), 0.06 (s, 9H). 13C NMR (126 MHz, Chloroform-d) δ 167.2, 140.0, 139.9, 133.5, 133.4, 130.4, 130.1, 129.8, 128.2, 127.8, 121,0, 120.9, 100.5, 78.1, 65.2, 63.0, 60.3, 54.8, 52.1, 36.6, 34.8, 18.4, 17.7, 1.4; HRMS (ESI) m/z: [M+H]+ calculated for C28H39N2O3Si: 479.27245, found 479.27164.
3-((3-(4-Methyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indol-7-yl)pyrrolidin-1-yl)methyl)benzoic acid (22): Compound 22 was synthesized following General Procedure A from methyl ester 21 (340 mg, 710 µmol). Compound 22 was obtained as a colorless oil (306 mg, 93%). 1H NMR (400 MHz, Chloroform-d): δ 8.08 (s, 1H), 7.96 (dt, J = 7.7, 1.5 Hz, 1H), 7.67 (s, 1H), 7.49–7.39 (m, 1H), 7.32 (d, J = 7.5 Hz, 1H), 6.52 (d, 1H), 5.59 (d, J = 11.5 Hz, 1H), 5.51 (d, J = 11.5 Hz, 1H), 4.35–4.17 (m, 1H), 3.95 (s, 3H), 3.80 (s, 2H), 3.50 (t, 2H), 2.98 (s, 1H), 2.91 (s, 1H), 2.54 (s, 3H), 2.48–2.37 (m, 1H), 1.29 (s, 1H), 0.96–0.82 (m, 2H), −0.03 (s, 9H); 13C NMR (101 MHz, Chloroform-d) δ 170. 5, 133.4, 132.6, 131.1, 130.6, 129.3, 128.6, 121.9, 121.2, 120.3, 100.6, 78.2, 77.4, 77.1, 76.7, 65.3, 59.6, 58.9, 53.5, 53.1, 36.6, 18.4, 17.7, −1.4.; HRMS (ESI) m/z: [M+H]+ calculated for C27H37N2O3Si: 465.25680, found 465.25658.
3-((3-(4-Methyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indol-7-yl)pyrrolidin-1-yl)methyl)-N-propylbenzamide (23): Compound 23 was synthesized following General Procedure B from carboxylic acid 22 (150 mg, 323 µmol). Compound 23 was obtained as a colorless oil (155 mg, 95%). 1H NMR (400 MHz, Chloroform-d) δ 7.89 (d, J = 1.8 Hz, 1H), 7.83 (dt, J = 7.8, 1.4 Hz, 1H), 7.49 (dt, J = 7.7, 1.5 Hz, 1H), 7.39 (t, J = 7.7 Hz, 1H), 7.15 (d, J = 7.5 Hz, 1H), 7.05 (d, J = 3.3 Hz, 1H), 6.72 (t, J = 5.7 Hz, 1H), 6.49 (d, J = 3.3 Hz, 1H), 5.56–5.46 (m, 2H), 4.46 (p, J = 8.5 Hz, 1H), 4.18 (s, 2H), 3.62–3.53 (m, 1H), 3.45–3.27 (m, 6H), 3.17 (t, J = 10.0 Hz, 1H), 2.50 (d, J = 0.8 Hz, 3H), 2.34–2.20 (m, 1H), 2.05 (d, J = 6.4 Hz, 1H), 1.65 (h, J = 7.4 Hz, 2H), 0.96 (t, J = 7.4 Hz, 3H), 0.85–0.70 (m, 2H), −0.07 (s, 9H); 13C NMR (101 MHz, Chloroform-d) δ 167.0, 135.9, 133.3, 132.4, 131.1, 130.6, 129.4, 129.3, 128.5, 127.8, 121.2, 120.3, 100.7, 78.2, 65.4, 61.6, 59.8, 54.9, 42.0, 36.6, 32.9, 22.6, 18.4, 17.7, 11.4, −1.5; HRMS (ESI) m/z: [M+H]+ calculated for C30H44N3O2Si: 506.31973, found 506.32176.
Methyl 3-((3-(4-methyl-1H-indol-7-yl)pyrrolidin-1-yl)methyl)benzoate (24): Compound 24 was prepared following General Procedure C from the protected indole 21 (50 mg, 104 µmol) and obtained as a white solid (20.7 mg, 57%). 1H NMR (400 MHz, Chloroform-d) δ 11.66 (s, 1H), 8.16–8.14 (m, 1H), 7.95 (dt, J = 7.8, 1.5 Hz, 1H), 7.56 (dt, J = 7.7, 1.5 Hz, 1H), 7.42 (t, J = 7.7 Hz, 1H), 7.28–7.25 (m, 1H), 6.79 (d, J = 7.1 Hz, 1H), 6.74–6.71 (m, 1H), 3.96 (s, 3H), 6.53 (dd, J = 3.1, 2.2 Hz, 1H), 3.87 (d, J = 12.6 Hz, 1H), 3.72 (d, J = 12.5 Hz, 1H), 3.59–3.48 (m, 1H), 3.35–3.27 (m, 1H), 3.16 (d, J = 10.1 Hz, 1H), 2.59 (t, J = 9.4 Hz, 1H), 2.52 (d, J = 0.9 Hz, 3H), 2.43–2.28 (m, 2H), 2.06–1.92 (m, 1H); 13C NMR (101 MHz, Chloroform-d) δ 167.0, 139.1, 133.4, 133.1, 130.7, 130.1, 128.8, 128.8, 128.5, 128.3, 128.1, 123.4, 120.7, 118.8, 100.6, 60.0, 59.9, 55.8, 52.3, 41.4, 32.6, 18.8; HRMS (M+H)+ calcd. for C22H25N2O2 349.19105, found 349.19110.
3-((3-(4-Methyl-1H-indol-7-yl)pyrrolidin-1-yl)methyl)benzoic acid (25): Compound 25 was synthesized following General Procedure C from the protected indole 22 (92 mg, 198 µmol). Compound 25 was obtained as a white solid (33 mg, 50%). 1H NMR (400 MHz, Chloroform-d) δ 11.40 (s, 1H), 8.90 (s, 1H), 8.21 (d, J = 7.5 Hz, 1H), 7.50–7.28 (m, 3H), 6.95 (d, J = 7.3 Hz, 1H), 6.85 (d, J = 7.2 Hz, 1H), 6.54 (t, J = 2.3 Hz, 1H), 4.61–3.86 (m, 4H), 3.33 (s, 2H), 2.71 (s, 2H), 2.56 (s, 3H), 2.35 (s, 1H); 13C NMR (101 MHz, Chloroform-d) δ 148.2, 130.7, 129.6, 128.3, 125.2, 119.1, 100.3, 59.6, 55.6, 18.8.; HRMS (M+H)+ calcd. for C21H23N2O2 335.17540, found 335.17611.
3-((3-(4-Methyl-1H-indol-7-yl) pyrrolidin-1-yl)methyl)-N-propylbenzamide (26): Compound 26 was synthesized following General Procedure C from the protected indole 23 (50 mg, 98.9 µmol). Compound 26 was obtained as a white solid (22.5 mg, 60%).
1H NMR (400 MHz, Chloroform-d) δ 11.66 (s, 1H), 8.18–8.12 (m, 1H), 7.95 (dt, J = 7.8, 1.5 Hz, 1H), 7.56 (dt, J = 7.7, 1.5 Hz, 1H), 7.42 (t, J = 7.7 Hz, 1H), 7.28–7.25 (m, 1H), 6.79 (d, J = 7.1 Hz, 1H), 6.72 (dq, J = 7.0, 0.8 Hz, 1H), 6.53 (dd, J = 3.1, 2.2 Hz, 1H), 3.96 (s, 3H), 3.87 (d, J = 12.6 Hz, 1H), 3.72 (d, J = 12.5 Hz, 1H), 3.59–3.47 (m, 1H), 3.37–3.27 (m, 1H), 3.16 (d, J = 10.1 Hz, 1H), 2.59 (t, J = 9.4 Hz, 1H), 2.52 (d, J = 0.9 Hz, 3H), 2.43–2.28 (m, 2H), 2.06–1.92 (m, 1H); 13C NMR (101 MHz, Chloroform-d) δ 167.0, 139.1, 133.4, 133.1, 130.7, 130.1, 128.8, 128.8, 128.5, 128.3, 128.1, 123.4, 120.7, 118.8, 100.6, 60.0, 59.9, 55.8, 52.3, 41.4, 32.6, 18.8; HRMS (M+H)+ calcd. for C24H30N3O 376.23834, found 376.23780.
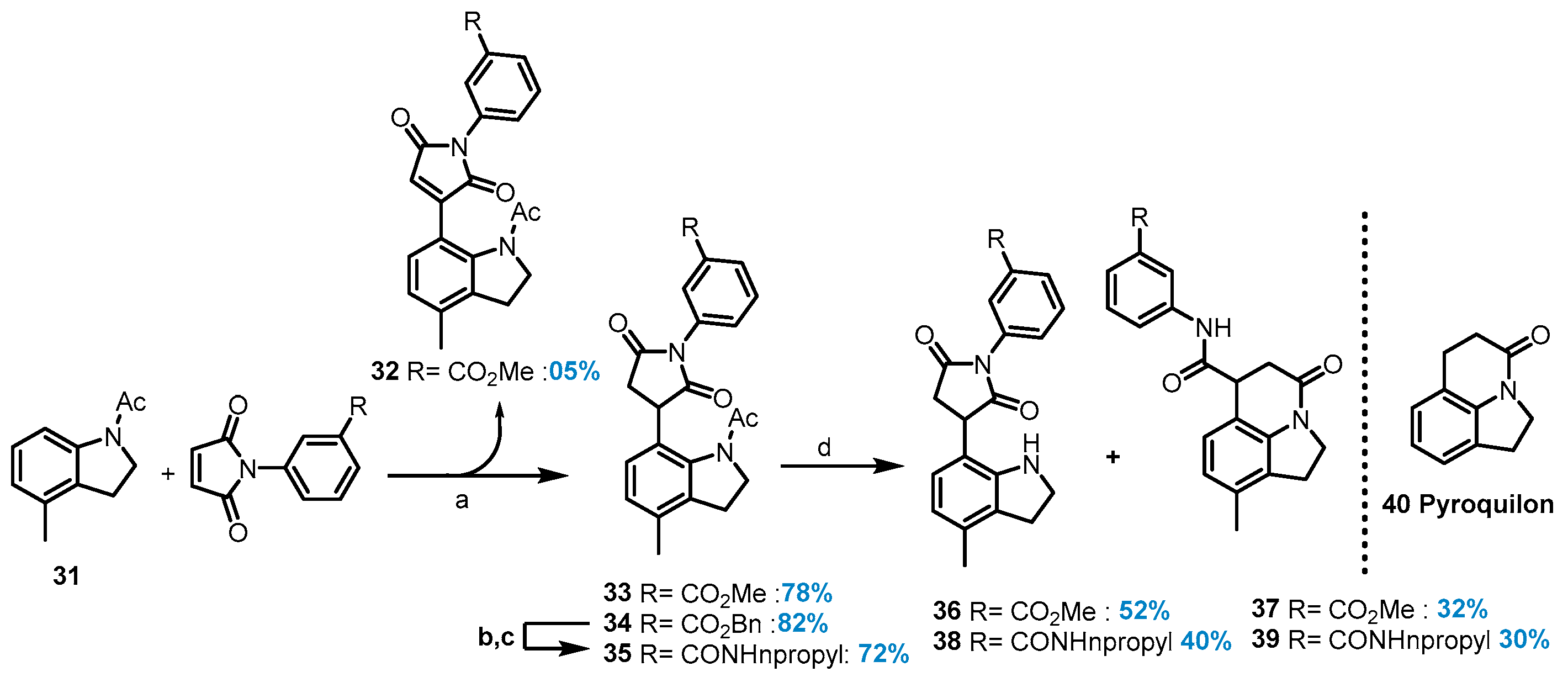
1-Ethyl-3-(4-methyl-1-pivaloyl-1H-indol-7-yl)pyrrolidine-2,5-dione (29) and 1-Ethyl-3-(4-methyl-1-pivaloyl-1H-indol-7-yl)-1H-pyrrole-2,5-dione (30): To a pre-dried sealed tube, 2,2-dimethyl-1-(4-methyl-1H-indol-1-yl)propan-1-one (0.2 mmol, 1.0 equiv.), N-ethyl maleimide (0.6 mmol), [RhCp*Cl2]2 (5 mol%), AgSbF6 (20 mol%), and Ag2O (2.0 equiv.) were added. To this mixture, TFE (2 mL) was added, and the vial was tightly capped and placed in a pre-heated (80 °C) oil bath. After 24h, the reaction mixture was cooled to room temperature and diluted with ethyl acetate (5 mL) and water (5 mL), and then aqueous layers were extracted three times with ethyl acetate (5 mL). The organic layer was concentrated under reduced pressure, and the crude product was purified on a silica gel column using an ethyl acetate/hexane mixture to afford the desired hydroarylation product (29, 10.0 mg, 15%) and the undesired Heck product (30, 6.5 mg, 10%).
1-Ethyl-3-(4-methyl-1-pivaloyl-1H-indol-7-yl)pyrrolidine-2,5-dione (29): This compound was obtained as a brownish liquid, 1H NMR (400 MHz, Chloroform-d) δ 7.64 (d, J = 3.9 Hz, 1H), 7.06 (dd, J = 7.6, 0.9 Hz, 1H), 6.94 (d, J = 7.7 Hz, 1H), 6.67 (d, J = 3.9 Hz, 1H), 4.52 (dd, J = 8.6, 5.0 Hz, 1H), 3.67–3.59 (m, 2H), 3.31 (dd, J = 18.4, 9.3 Hz, 1H), 2.87 (dd, J = 18.5, 5.1 Hz, 1H), 2.50 (d, J = 0.7 Hz, 3H), 1.23 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 179.4, 178.5, 176.9, 150.9, 134.9, 131.8, 130.2, 126.2, 124.7, 123.4, 106.4, 44.2, 41.9, 37.2, 34.0, 29.3, 18.4, 13.3. HRMS (M+H) + calcd. for C20H24N2O3 341.1859, found 341.1857.
1-Ethyl-3-(4-methyl-1-pivaloyl-1H-indol-7-yl)-1H-pyrrole-2,5-dione (30): This compound was obtained as yellowish liquid, 1H NMR (400 MHz, Chloroform-d) δ 7.70 (d, J = 3.9 Hz, 1H), 7.18 (d, J = 7.5 Hz, 1H), 7.09 (dd, J = 7.5, 0.8 Hz, 1H), 6.69–6.67 (m, 1H), 6.43 (s, 1H), 3.57 (q, J = 7.2 Hz, 2H), 2.55 (s, 3H), 1.22 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 179.0, 171.5, 170.8, 149.9, 134.3, 133.4, 131.1, 127.6, 126.1, 124.1, 120.5, 115.6, 105.9, 41.5, 32.9, 32.8, 28.7, 18.8, 14.2, 14.1. HRMS (M+H)+ calcd. for C20H22N2O3 339.1703, found 339.1699.
Methyl 3-(3-(1-acetyl-4-methylindolin-7-yl)-2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)benzoate (32): This compound was prepared following General Procedure E and was obtained as a yellow liquid (4.2 mg, 05%). 1H NMR (500 MHz, Chloroform-d) δ 8.08–8.04 (m, 1H), 8.00 (d, J = 7.9 Hz, 1H), 7.59 (d, J = 8.1 Hz, 1H), 7.56–7.48 (m, 1H), 7.18 (d, J = 7.9 Hz, 1H), 6.98 (d, J = 7.7 Hz, 1H), 6.47 (s, 1H), 4.16 (t, J = 8.0 Hz, 2H), 3.90 (s, 3H), 3.07 (t, J = 8.1 Hz, 2H), 2.29 (s, 3H), 2.20 (s, 3H). 13C NMR (126 MHz, Chloroform-d) δ 170.0, 168.9, 168.9, 166.4, 149.7, 140.0, 137.2, 134.4, 133.3, 131.2, 130.6, 129.6, 129.1, 128.5, 127.4, 126.2, 119.4, 115.7, 52.3, 49.7, 27.8, 24.1, 18.9. HRMS (M+H)+ calcd. for C23H20N2O5 405.1445, found 405.1443.
Methyl 3-(3-(1-acetyl-4-methylindolin-7-yl)-2,5-dioxopyrrolidin-1-yl)benzoate (33): This compound was prepared following General Procedure E and was obtained as a gum (63.3 mg, 78%). 1H NMR (400 MHz, Chloroform-d) δ 8.11–7.99 (m, 2H), 7.59–7.51 (m, 2H), 6.98 (s, 2H), 4.52 (dd, J = 9.6, 5.8 Hz, 1H), 4.15 (m,1H), 4.11–4.01 (m, 1H), 3.91 (s, 3H), 3.61–3.43 (m, 2H), 3.05 (m, 2H), 2.91 (m, 1H), 2.28 (s, 3H), 2.24 (s, 3H). 13C NMR (101 MHz, Chloroform-d) δ 177.4, 176.0, 169.5, 166.2, 140.6, 134.2, 133.8, 132.7, 131.4, 131.2, 129.6, 129.3, 128.0, 127.5, 127.2, 125.4, 52.4, 50.7, 45.0, 37.5, 28.5, 24.2, 18.6. HRMS (M+H)+ calcd. for C23H22N2O5 407.1604, found 407.1598.
Benzyl 3-(3-(1-acetyl-4-methylindolin-7-yl)-2,5-dioxopyrrolidin-1-yl) benzoate (34): This compound was prepared following General Procedure E and was obtained as a gel (79 mg, 82%). 1H NMR (400 MHz, Chloroform-d) δ 8.14–8.07 (m, 1H), 8.05 (q, J = 1.4 Hz, 1H), 7.59–7.52 (m, 2H), 7.46–7.31 (m, 5H), 6.98 (s, 2H), 5.37 (s, 2H), 4.52 (dd, J = 9.6, 5.9 Hz, 1H), 4.21–4.00 (m, 2H), 3.55 (dd, J = 18.7, 9.6 Hz, 1H), 3.06 (ddd, J = 21.7, 17.9, 7.3 Hz, 2H), 2.92 (ddd, J = 15.5, 8.7, 4.3 Hz, 1H), 2.27 (s, 3H), 2.25 (s, 3H). 13C NMR (101 MHz, Chloroform-d) δ 177.4, 176.1, 169.5, 165.6, 140.6, 135.9, 134.2, 133.9, 132.8, 131.5, 129.9, 129.4, 128.8, 128.5, 128.2, 127.5, 127.3, 125.4, 67.1, 50.7, 45.1, 37.5, 28.5, 24.2, 18.6. HRMS (M+H)+ calcd. for C29H26N2O5 483.1914, found 483.1910.
3-(3-(1-Acetyl-4-methylindolin-7-yl)-2,5-dioxopyrrolidin-1-yl)-N-propylbenzamide (35): To a stirred solution of benzyl 3-(3-(1-acetyl-4-methylindolin-7-yl)-2,5-dioxopyrrolidin-1-yl)benzoate 34 (62.7 mg, 0.130 mmol) in EtOAc/MeOH (3:1, 2 mL), Pd/C (15 mol%) was added under argon. The reaction mixture was hydrogenated (balloon) for 24h, and the reaction mixture was passed through a Celite pad and washed with methanol. The solvent was evaporated, and the crude product was subjected to the next step without purification. To a solution containing HATU (1.3 equiv.), 4-dimethylaminopyridine (20 mol%) and N, N-diisopropylethylamine (2.2 equiv.) in dry DMF (2.0 mL). was added to the acid (0.127 mmol) under a N2 atmosphere. After 1h, LCMS showed the formation of the activated ester intermediate. Propylamine (2.0 equiv.) was added, and the resulting mixture was stirred at room temperature overnight. The solution was diluted with ethyl acetate, a saturated solution of aqueous sodium bicarbonate was added, and the organic layer was dried with MgSO4, filtered, and evaporated in vacuo. The crude was purified by chromatography (80% EtOAc in Hexanes) to give amide 36 (30 mg, 72%). 1H NMR (400 MHz, Chloroform-d) δ 7.81 (dt, J = 7.7, 1.5 Hz, 1H), 7.73 (t, J = 1.9 Hz, 1H), 7.54 (t, J = 7.8 Hz, 1H), 7.48 (dt, J = 8.2, 1.5 Hz, 1H), 6.99 (s, 2H), 6.17 (s, 1H), 4.52 (dd, J = 9.6, 5.8 Hz, 1H), 4.21–4.13 (m, 1H), 4.11–4.01 (m, 1H), 3.55 (dd, J = 18.7, 9.6 Hz, 1H), 3.44–3.36 (m, 2H), 3.13–3.00 (m, 2H), 2.97–2.86 (m, 1H), 2.29 (s, 3H), 2.25 (s, 3H), 1.62 (m, 2H), 0.98 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 177.4, 176.0, 169.5, 166.5, 136.0, 134.1, 133.8, 132.6, 129.4, 127.5, 127.1, 125.3, 125.0, 77.4, 77.0, 76.7, 50.7, 44.9, 41.9, 37.3, 28.4, 24.1, 22.9, 18.5, 11.5. HRMS (M+H)+ calcd. for C25H27N3O4 434.2079, found 434.2071.
Methyl 3-(3-(4-methylindolin-7-yl)-2,5-dioxopyrrolidin-1-yl)benzoate (36): This compound was prepared following General Procedure F and was obtained as a gum (47 mg, 52%). 1H NMR (400 MHz, Acetone-d6) δ 8.12–7.98 (m, 2H), 7.72–7.59 (m, 2H), 6.93 (d, J = 7.8 Hz, 1H), 6.54 (dd, J = 7.7, 0.8 Hz, 1H), 4.37 (dd, J = 9.6, 5.5 Hz, 1H), 3.92 (s, 3H), 3.65–3.56 (m, 2H), 3.38 (dd, J = 18.1, 9.6 Hz, 1H), 3.09–2.87 (m, 3H), 2.19 (s, 3H). 13C NMR (101 MHz, Acetone-d6) δ 205.3, 177.1, 174.9, 165.6, 150.2, 133.5, 133.1, 131.5, 131.0, 129.1, 128.8, 127.9, 125.5, 120.0, 116.5, 51.7, 46.3, 42.2, 35.3, 29.5, 29.4, 29.2, 29.0, 28.8, 28.6, 28.5, 28.4, 17.8. HRMS (M+H)+ calcd. for C21H20N2O4 365.1495, found 365.1498.
Methyl 3-(9-methyl-4-oxo-1,2,5,6-tetrahydro-4H-pyrrolo[3,2,1-ij] quinoline-6-carboxamido) benzoate (37): This compound was prepared following General Procedure F and was obtained as a gel (31.4 mg, 35%). 1H NMR (400 MHz, DMSO-d6) δ 10.46 (s, 1H), 8.27 (t, J = 1.9 Hz, 1H), 7.82 (ddd, J = 8.2, 2.3, 1.1 Hz, 1H), 7.64 (dt, J = 7.8, 1.3 Hz, 1H), 7.45 (t, J = 7.9 Hz, 1H), 7.15 (d, J = 7.7 Hz, 1H), 6.78–6.66 (m, 1H), 4.03 (td, J = 7.2, 5.0 Hz, 1H), 3.99–3.86 (m, 2H), 3.84 (s, 3H), 3.05 (t, J = 8.4 Hz, 2H), 2.83–2.67 (m, 2H), 2.16 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 171.0, 166.0, 165.5, 140.9, 139.3, 133.7, 130.1, 129.3, 128.2, 128.1, 125.1, 124.0, 123.6, 119.7, 115.6, 59.7, 52.2, 44.8, 42.7, 33.9, 26.1, 18.1, 17.9, 14.1. HRMS (M+H)+ calcd. for C21H20N2O4 365.1495, found 365.1504.
3-(3-(4-Methylindolin-7-yl)-2,5-dioxopyrrolidin-1-yl)-N-propylbenzamide (38): This compound was prepared following General Procedure F and was obtained as a sticky liquid (18 mg, 40%). 1H NMR (400 MHz, Acetone-d6) δ 7.96–7.81 (m, 3H), 7.60–7.53 (m, 1H), 7.50 (ddd, J = 7.9, 2.0, 1.2 Hz, 1H), 6.89 (d, J = 7.8 Hz, 1H), 6.59–6.42 (m, 1H), 4.33 (dd, J = 9.6, 5.3 Hz, 1H), 3.58 (dd, J = 9.2, 8.1 Hz, 2H), 3.42–3.32 (m, 3H), 3.06–2.89 (m, 3H), 2.18 (s, 3H), 1.68–1.58 (m, 2H), 0.95 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, Acetone-d6) δ 177.1, 175.0, 165.4, 150.7, 136.1, 133.8, 132.9, 129.5, 128.8, 128.6, 126.5, 126.1, 125.3, 119.7, 116.3, 46.2, 42.1, 41.4, 35.3, 28.5, 22.6, 17.8, 10.8. HRMS (M+H)+ calcd. for C23H25N3O3 392.1968, found 392.19830.
9-Methyl-4-oxo-N-(3-(propylcarbamoyl)phenyl)-1,2,5,6-tetrahydro-4H-pyrrolo[3,2,1-ij] quinoline-6-carboxamide (39): This compound was prepared following General Procedure F and was obtained as a brown solid (14mg, 30%). 1H NMR (400 MHz, Methanol-d4) δ 8.01 (t, J = 1.9 Hz, 1H), 7.70 (ddd, J = 8.1, 2.2, 1.1 Hz, 1H), 7.58–7.49 (m, 1H), 7.40 (t, J = 7.9 Hz, 1H), 7.13 (d, J = 7.7 Hz, 1H), 6.86–6.80 (m, 1H), 4.16–3.99 (m, 3H), 3.38–3.31 (m, 4H), 3.15 (t, J = 8.3 Hz, 2H), 2.92–2.88 (m, 2H), 2.25 (s, 3H), 1.63 (h, J = 7.4, 6.9 Hz, 2H), 0.98 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 173.3, 169.9, 168.7, 141.6, 139.8, 136.8, 135.9, 130.1, 130.1, 126.3, 126.0, 124.1, 124.0, 120.2, 117.2, 46.4, 44.6, 42.8, 35.3, 27.6, 23.7, 18.2, 11.8. HRMS (M+H)+ calcd. for C23H25N3O3 392.1968, found 392.1973.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}