
The Miracle of Vitamin B12 Biochemistry

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
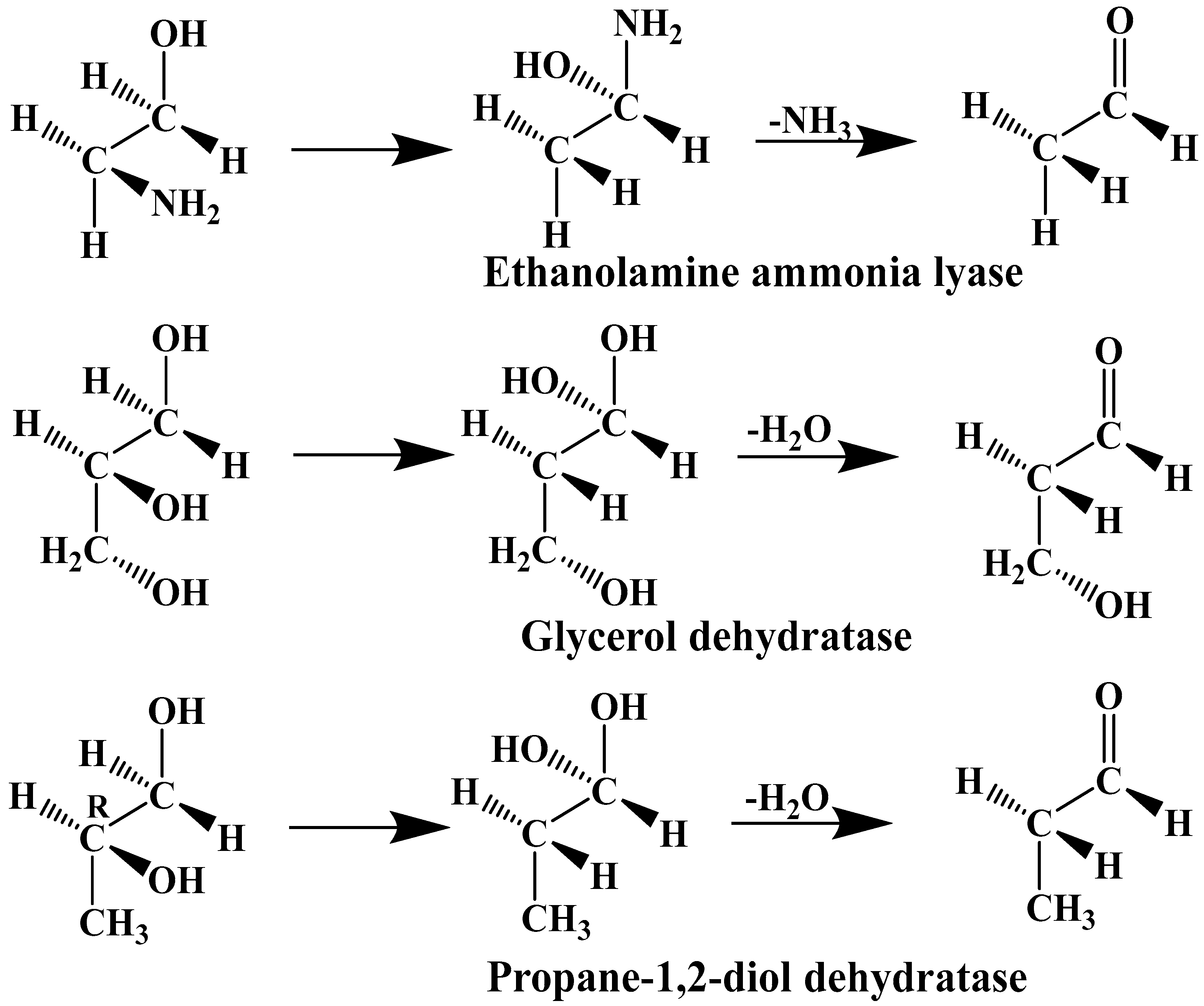
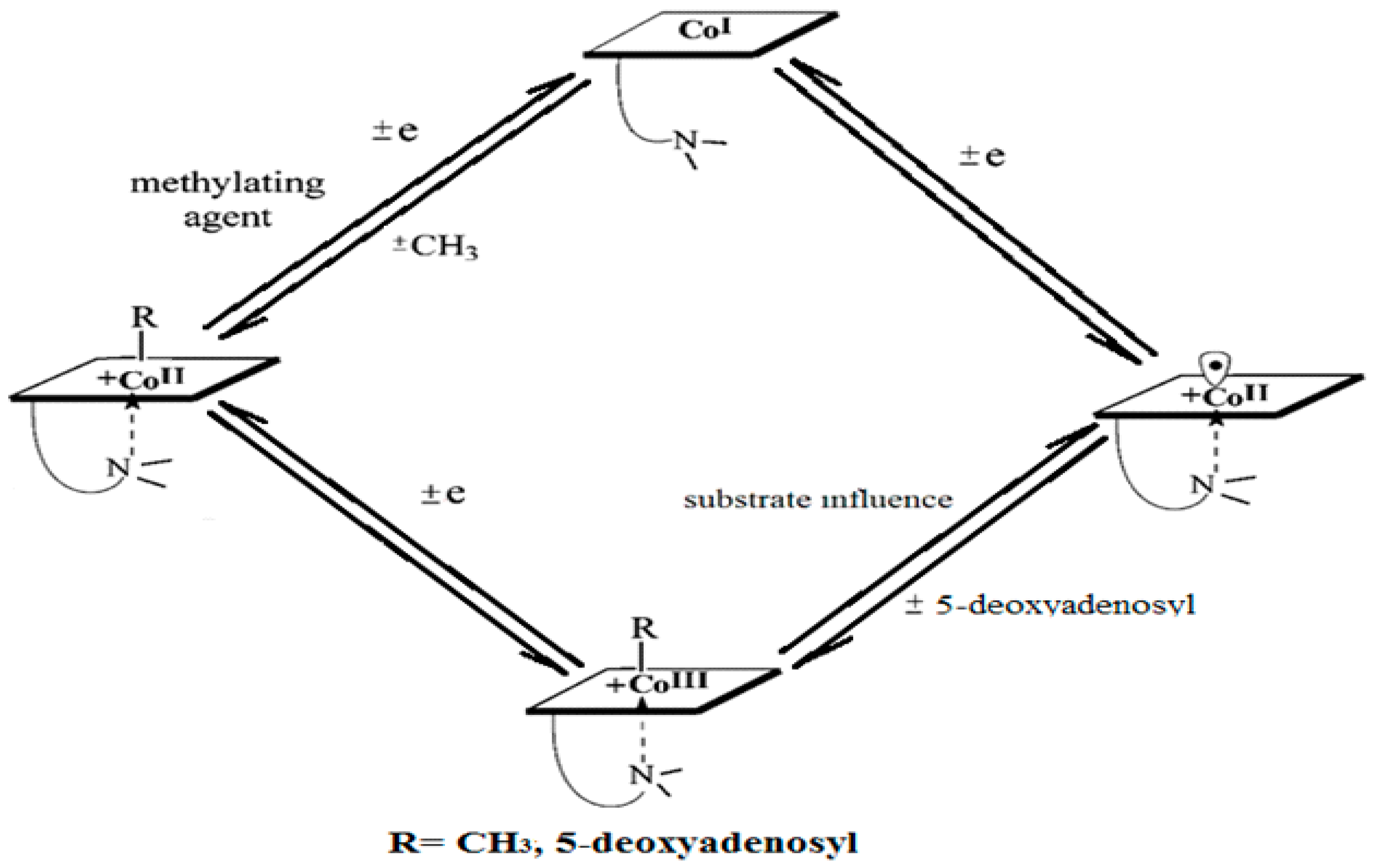
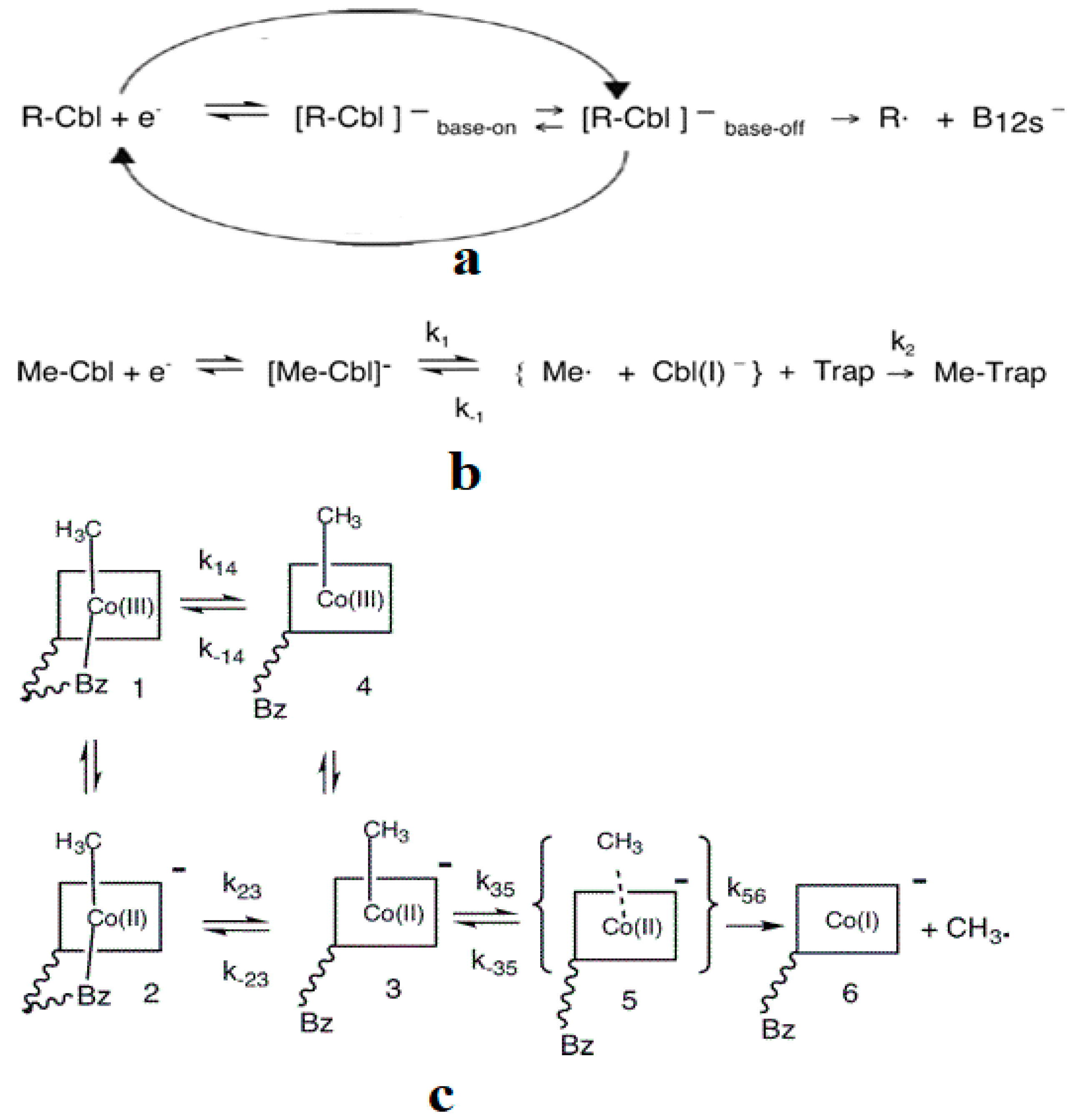
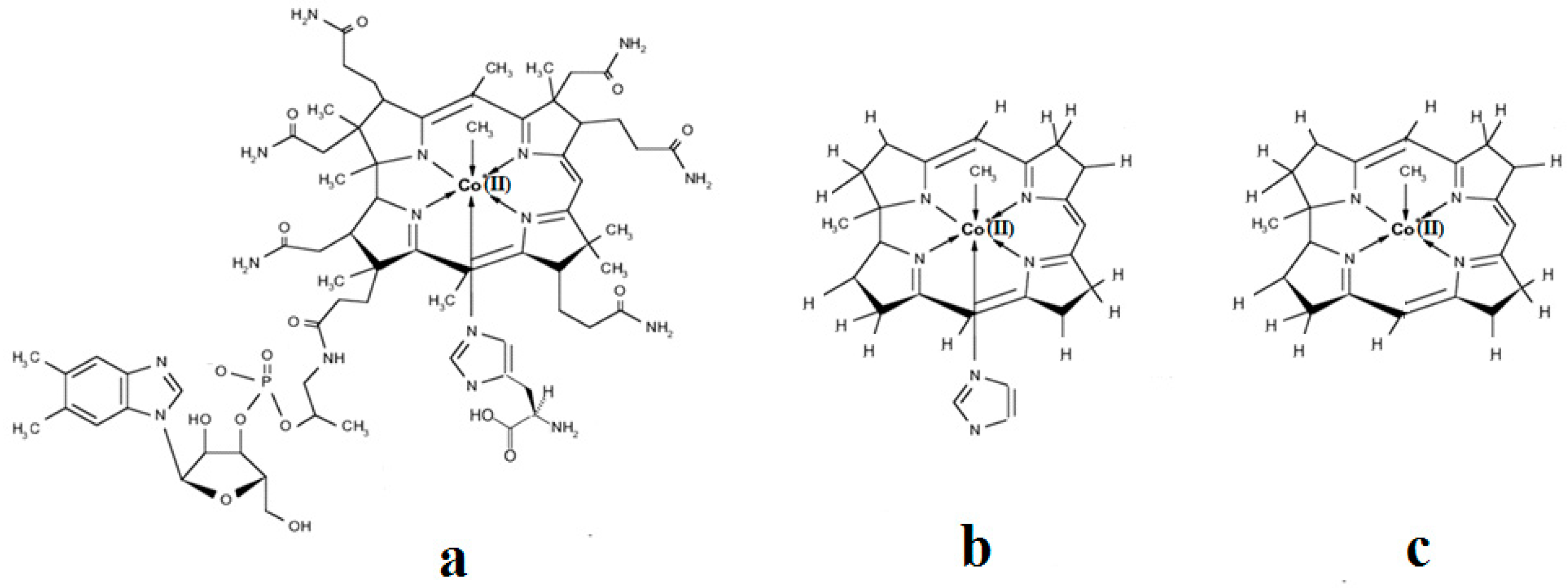
2. Experimental Mechanistic Evidence
3. DFT and QM/MM (Based on DFT) Calculations
3.1. General Considerations
“Density functional theory (DFT) finds increasing use in applications related to biological systems. Advancements in methodology and implementations have reached a point where predicted properties of reasonable to high quality can be obtained. Thus, DFT studies can complement experimental investigations, or even venture with some confidence into experimentally unexplored territory. Many properties can be calculated with DFT, such as geometries, energies, reaction mechanisms, and spectroscopic properties. A wide range of spectroscopic parameters is nowadays accessible with DFT, including quantities related to infrared and optical spectra, X-ray absorption, and Mössbauer, as well as all of the magnetic properties connected with electron paramagnetic resonance spectroscopy except relaxation times. Density functional theory is considered an extremely successful approach for the description of ground-state properties of metals, semiconductors, and insulators. The success of DFT not only encompasses standard bulk materials but also complex materials such as proteins and carbon nanotubes”[161]
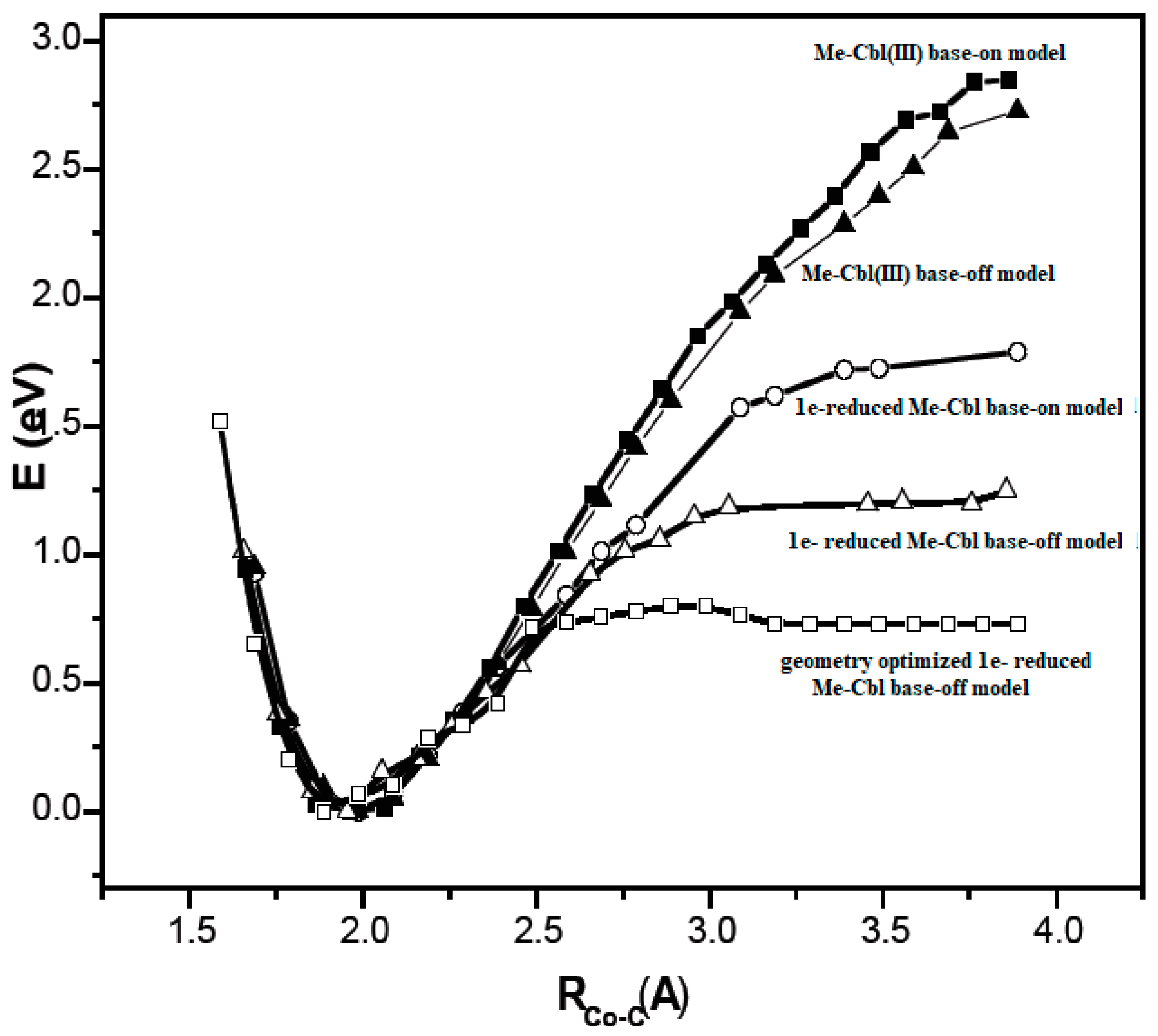
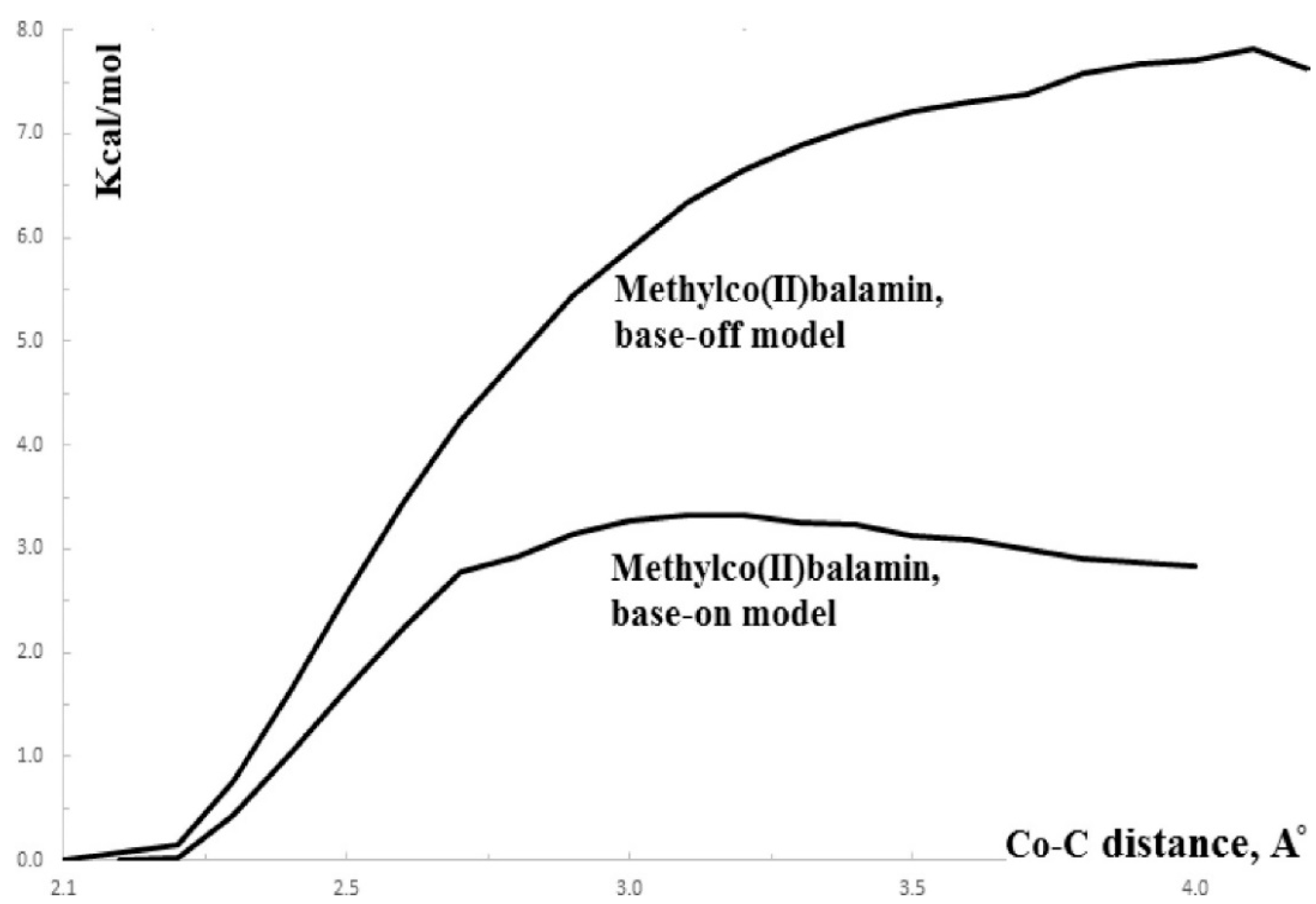
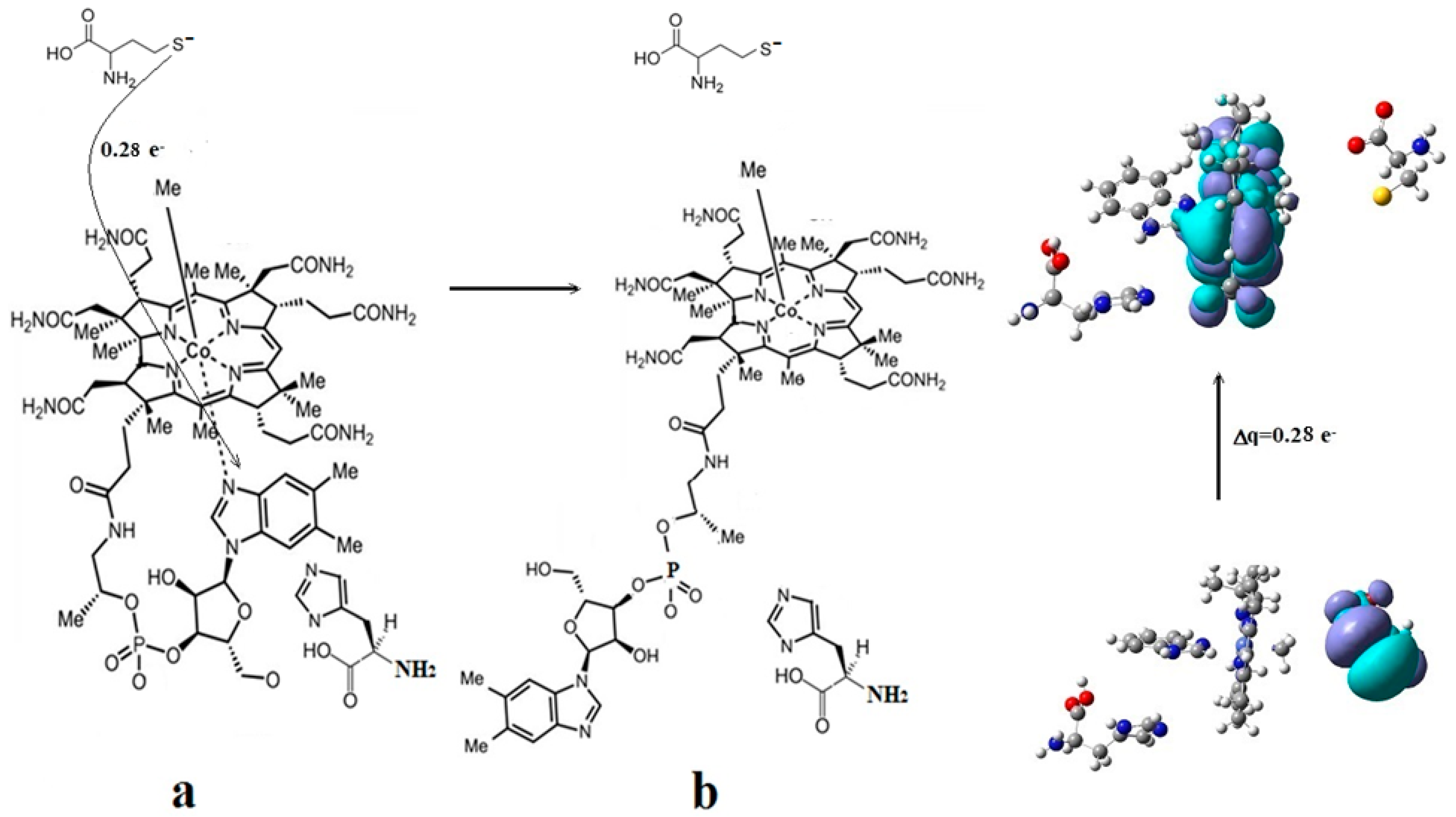
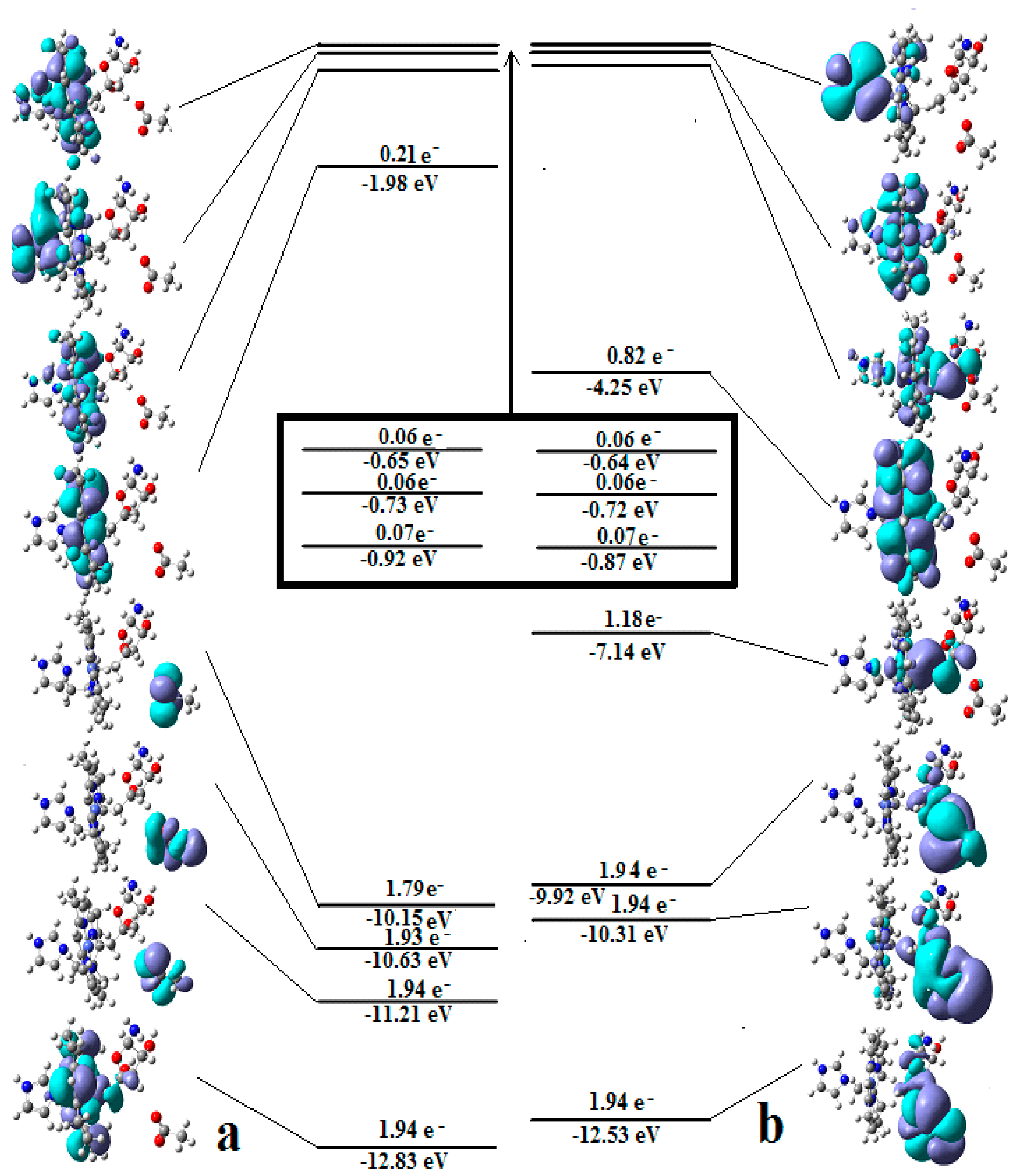
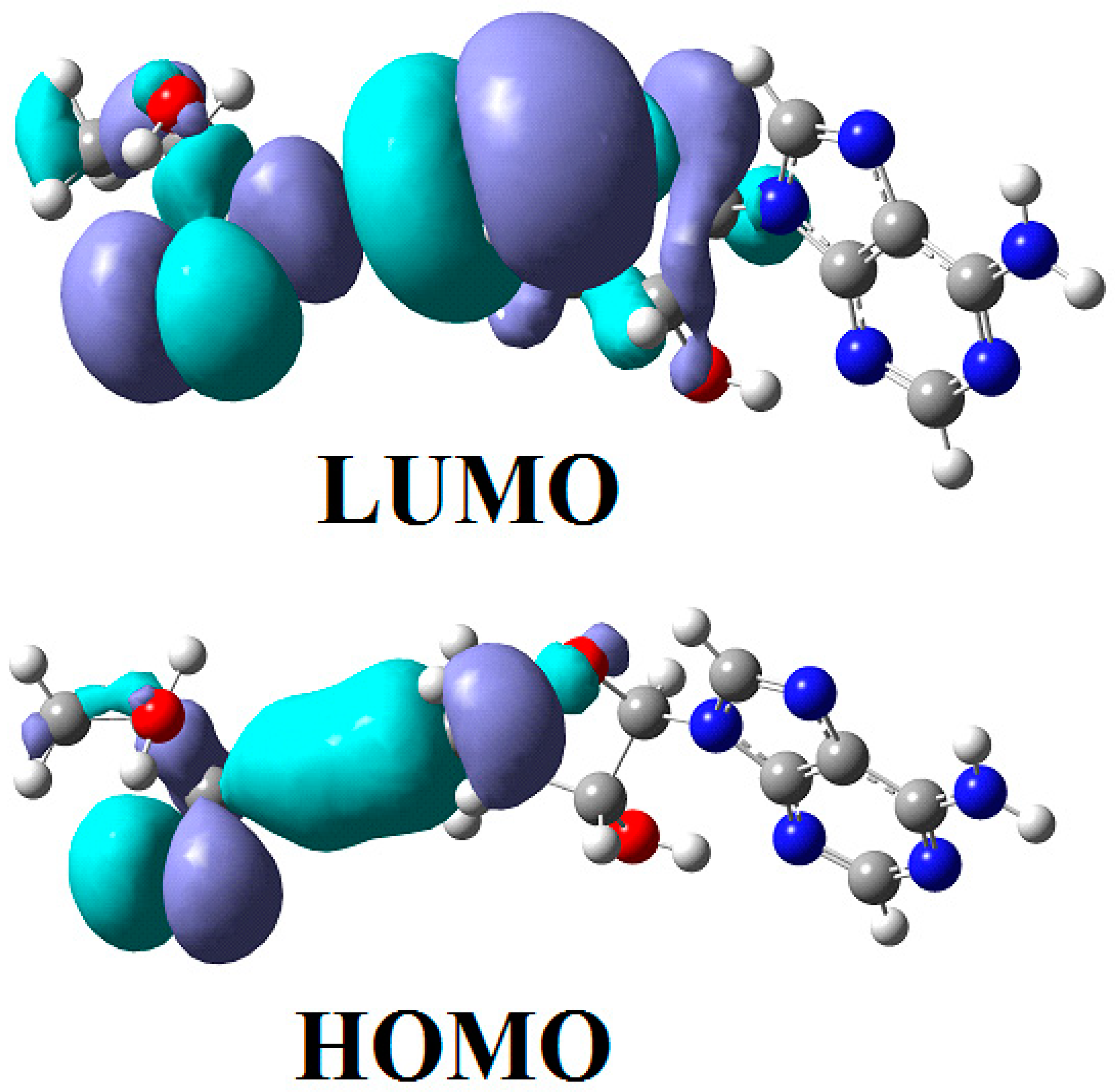
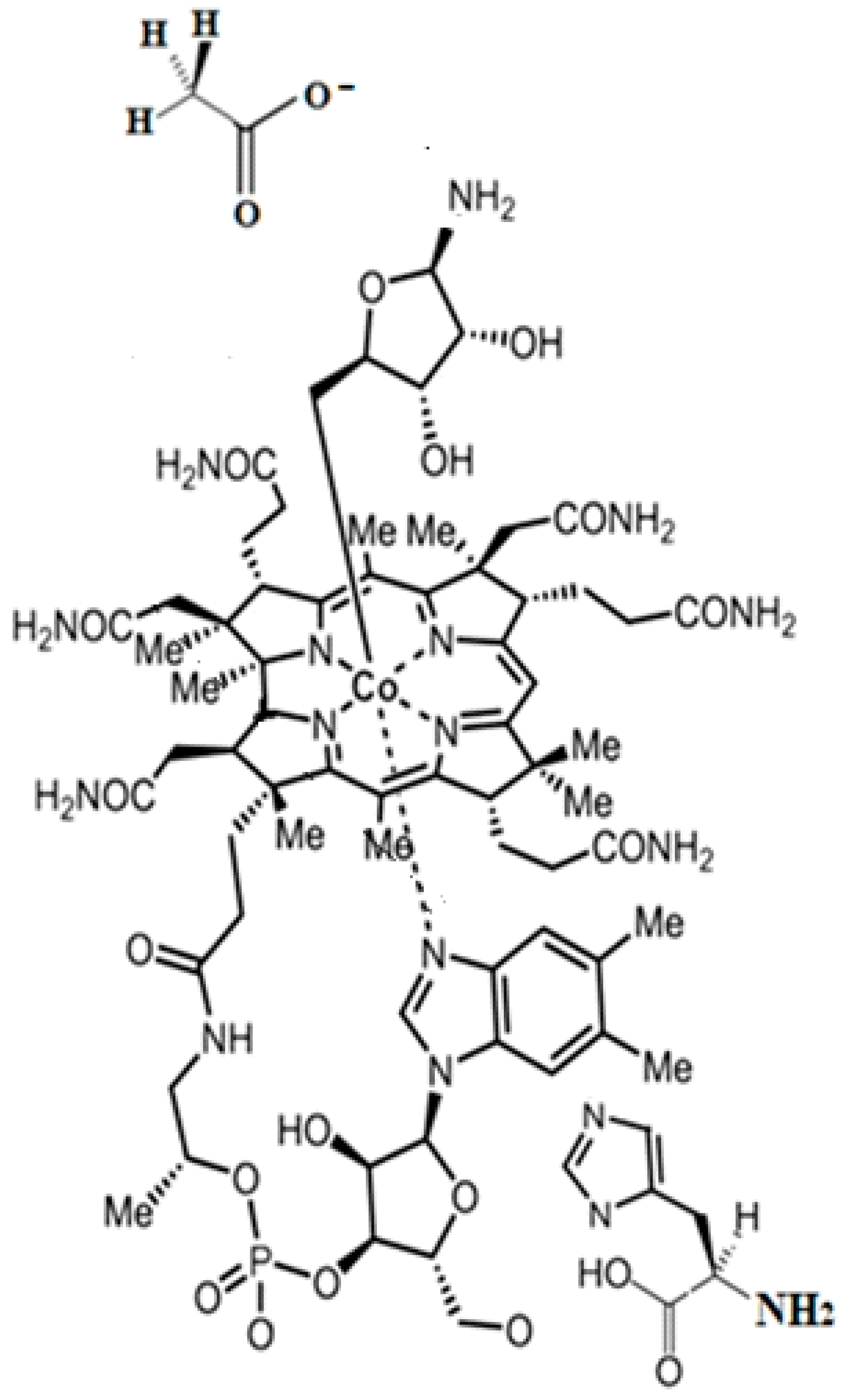
3.2. Calculations Involving Methylcobalamin
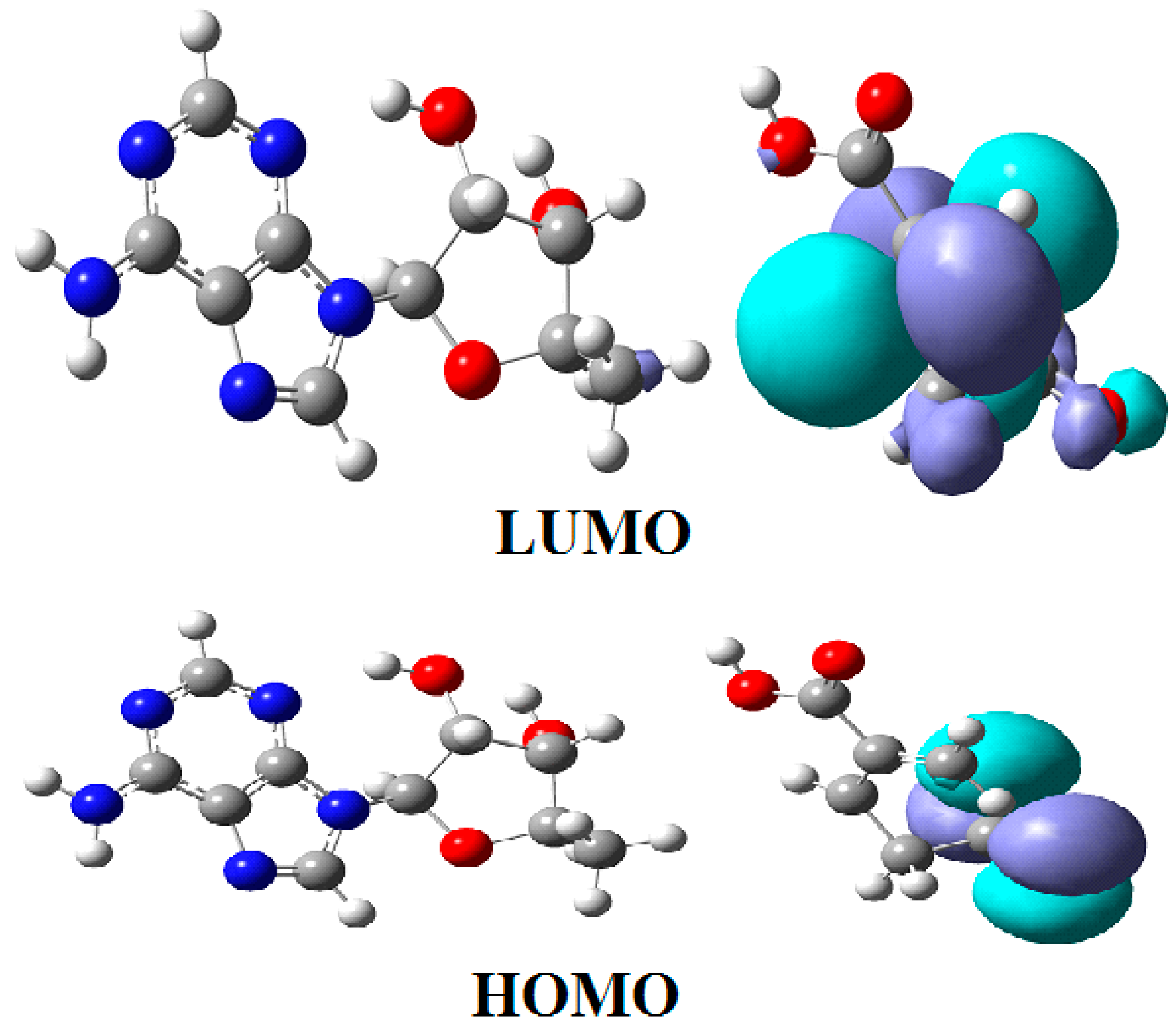
3.3. Calculations Involving Adenosylcobalamin
3.4. Summary Considerations
4. Pseudo-Jahn–Teller Effect and MCSCF Calculations: Methionine Synthase Process
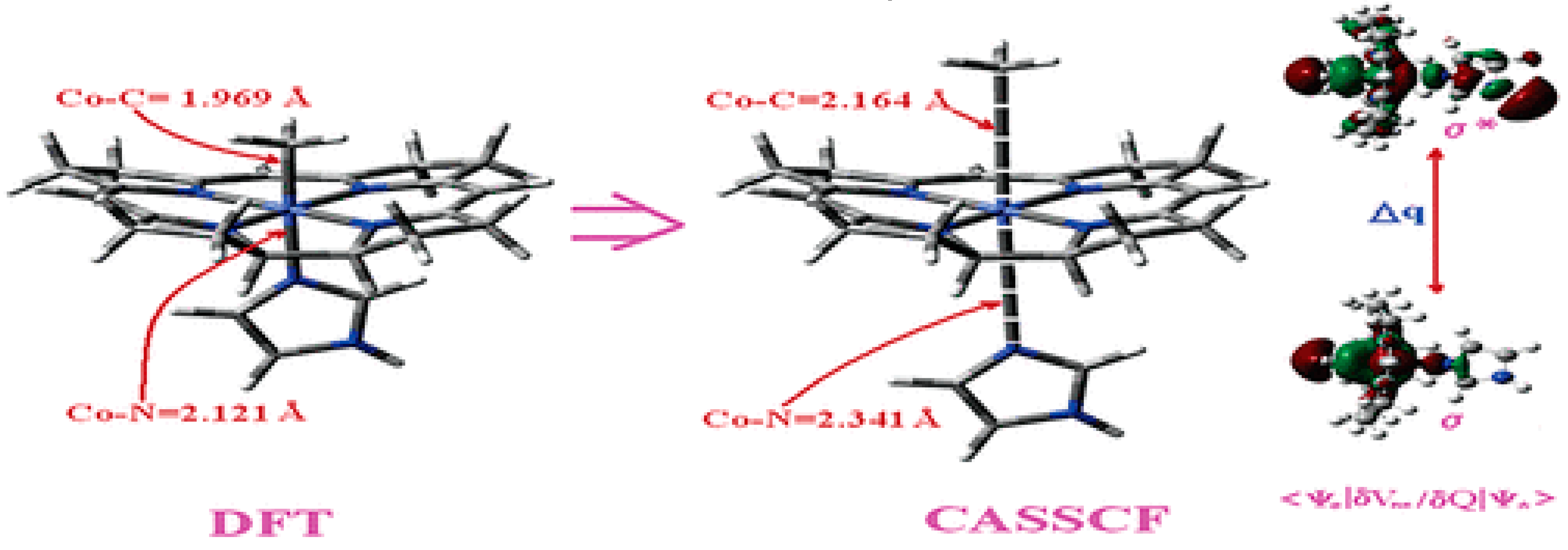
4.1. Pseudo-Jahn–Teller Effect and MCSCF Method: General Considerations
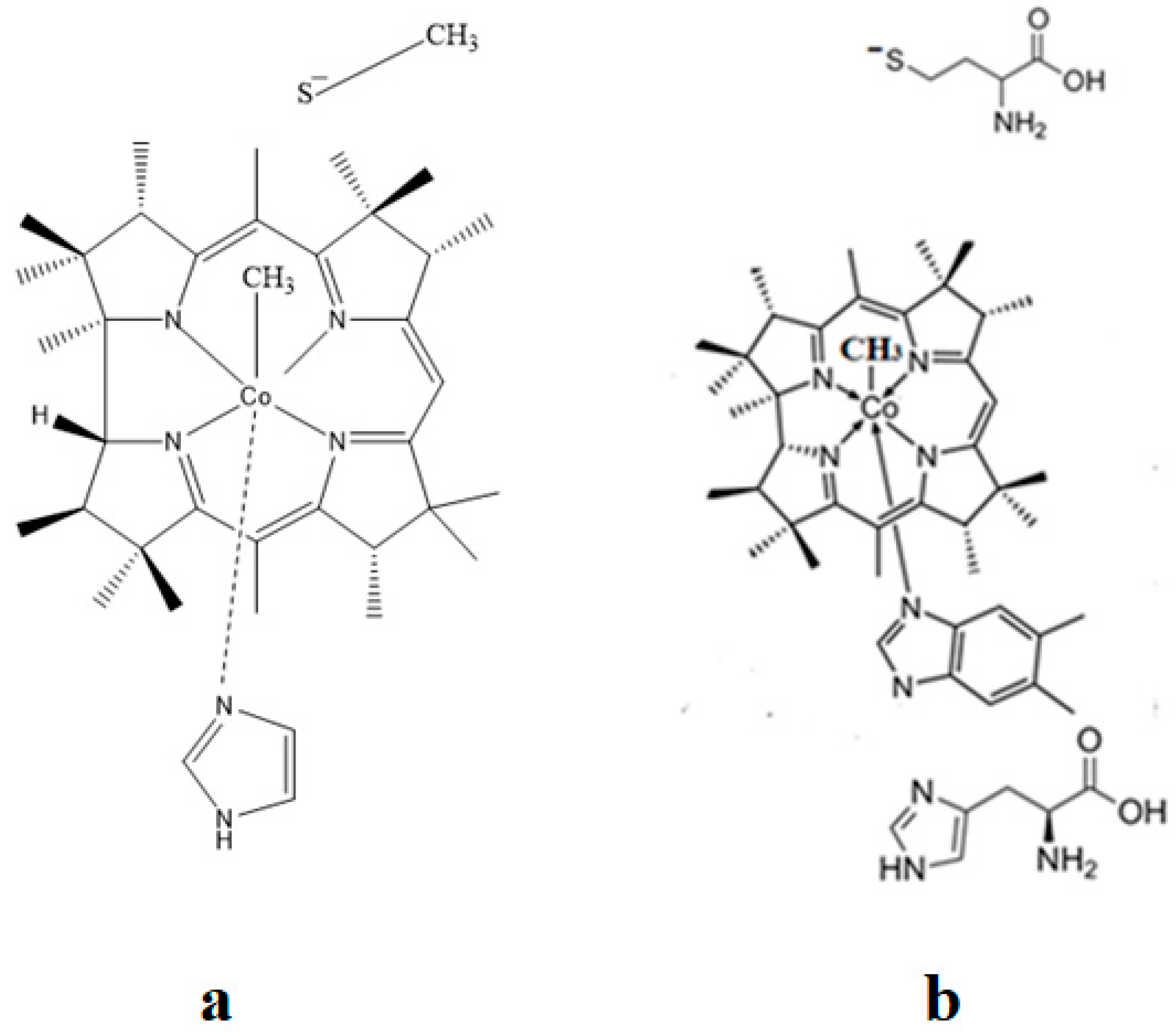
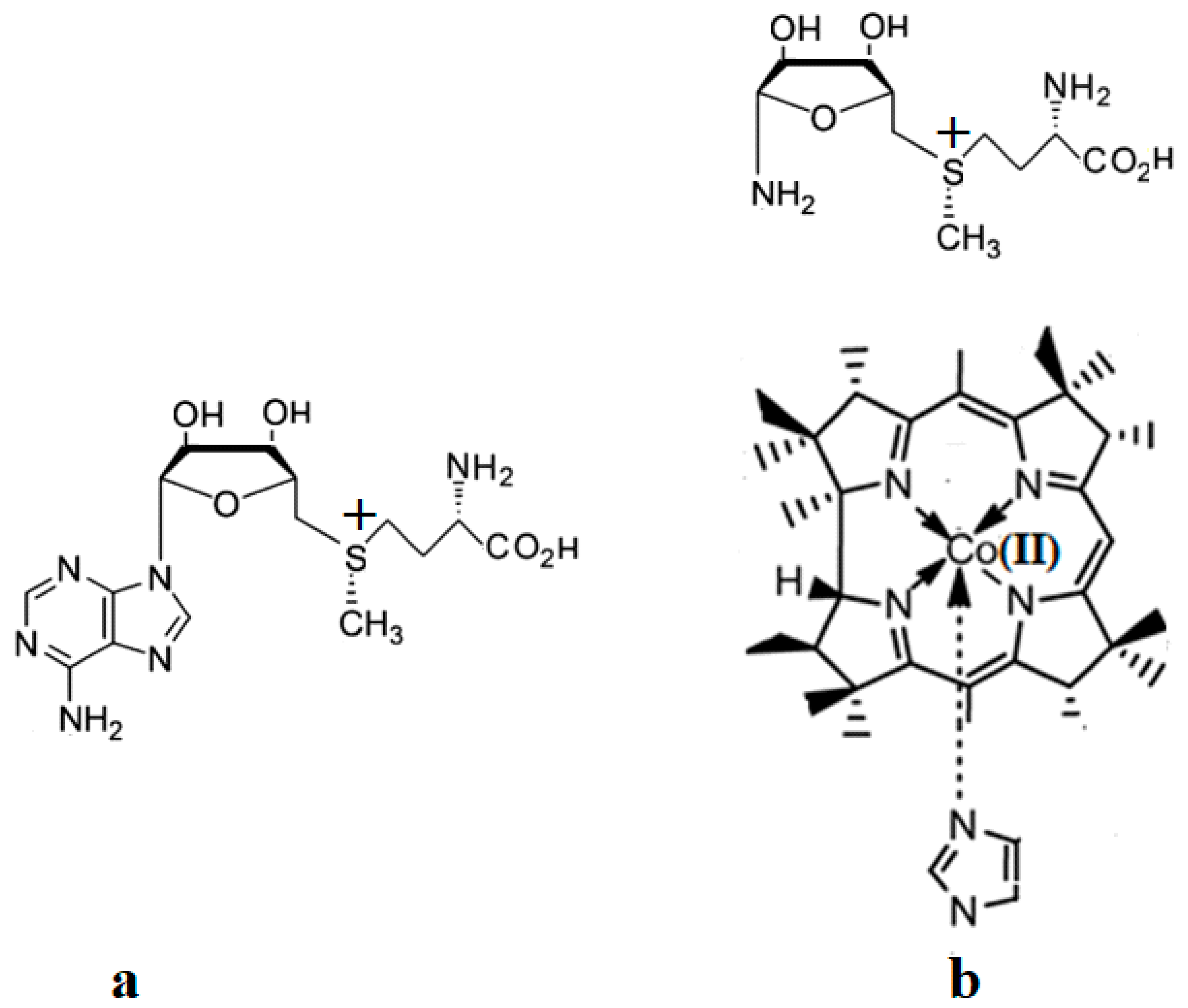
4.2. MCSCF: Methylcobalamin and Methionine Synthase Process
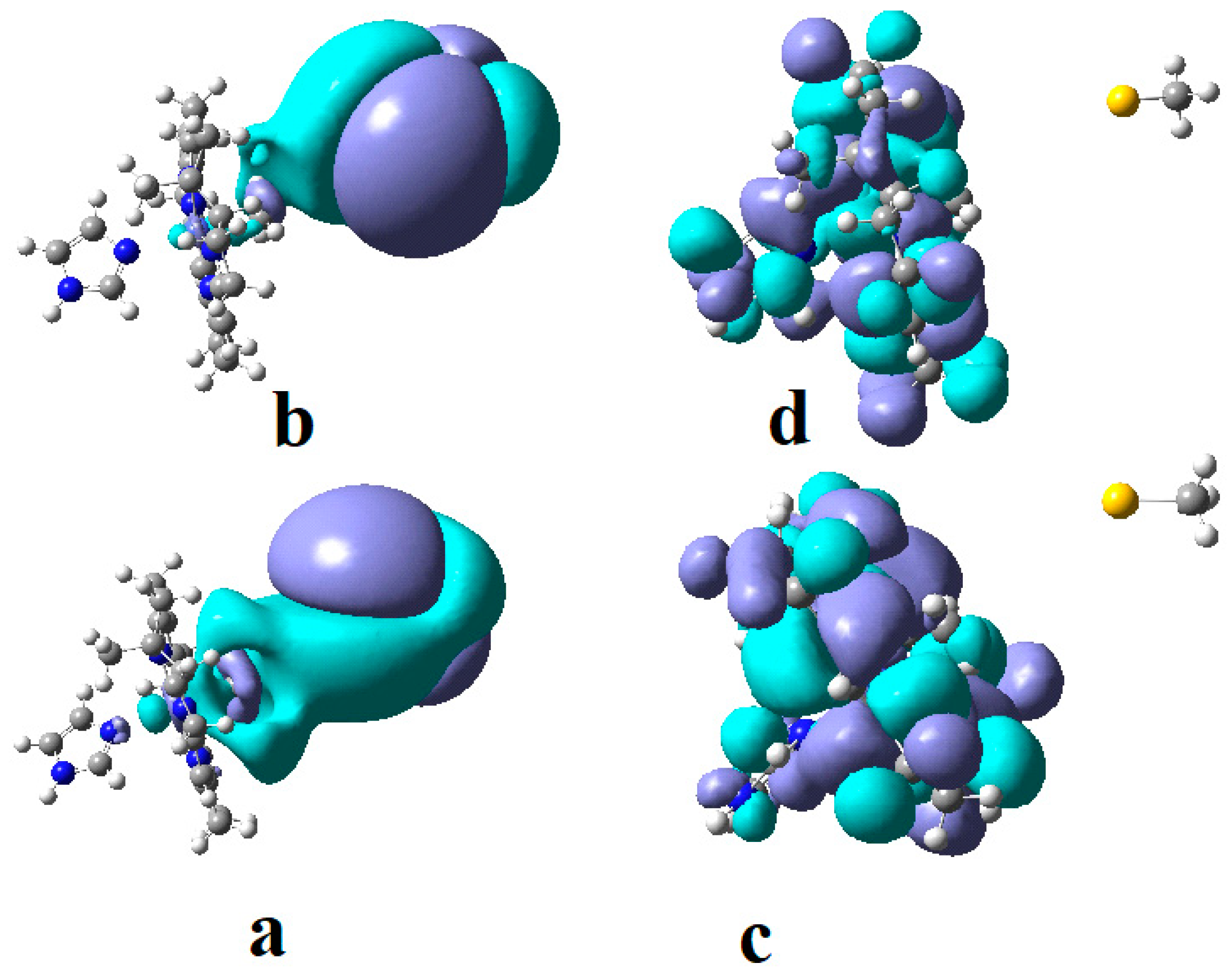
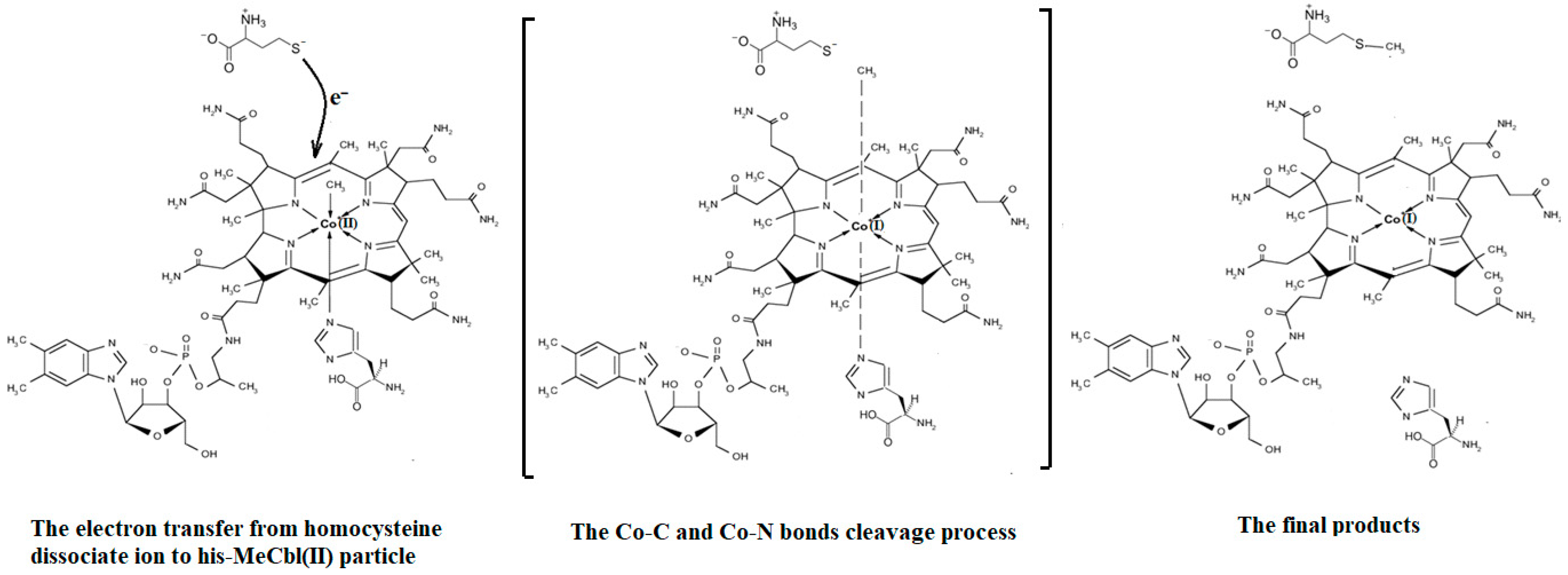
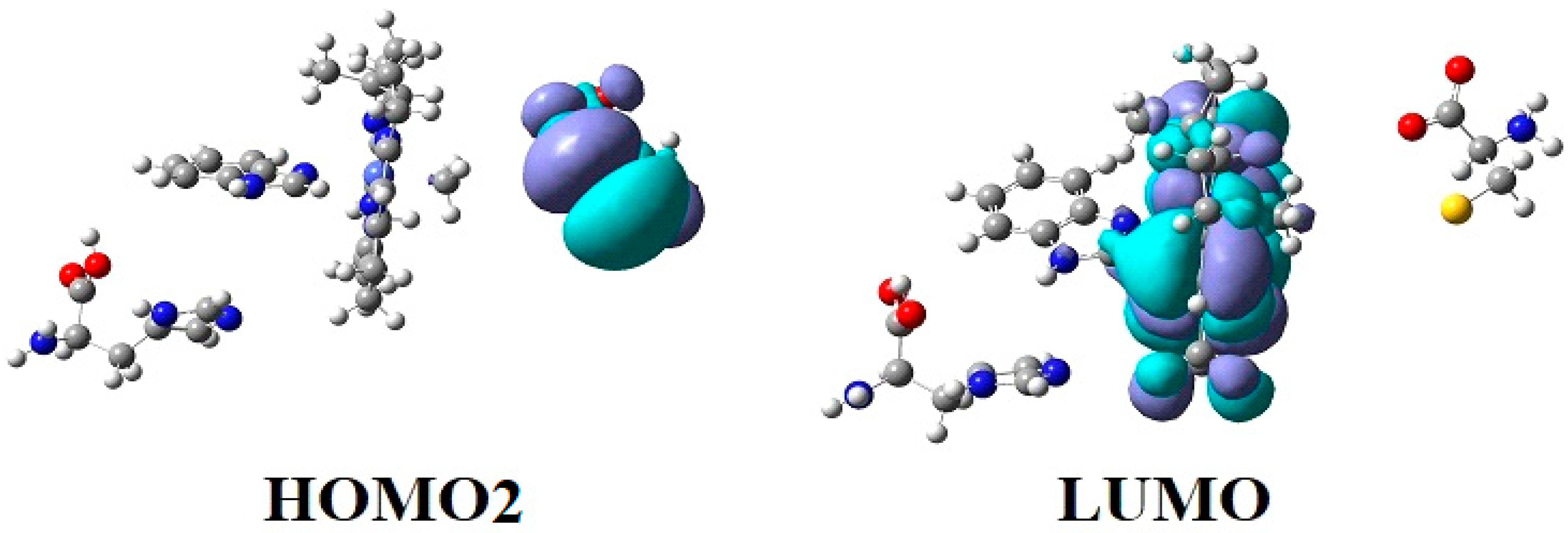
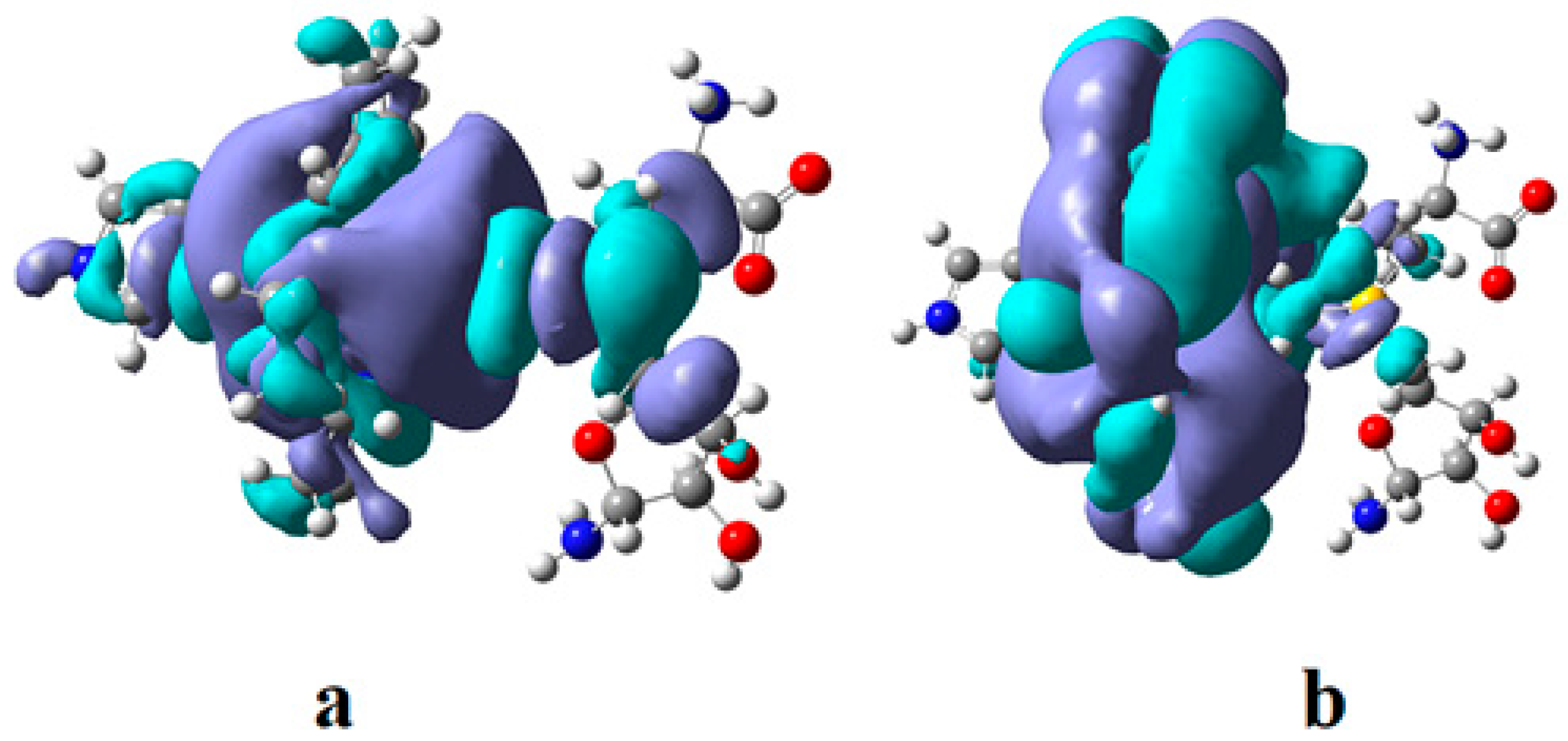
4.3. The Methyl Radical Transfer from Methylcob(II)alamin to Homocysteine (SN1 Reaction)
4.4. The Methyl Radical Transfer from Methylcob(II)alamin to Homocysteine, SN2 Reaction
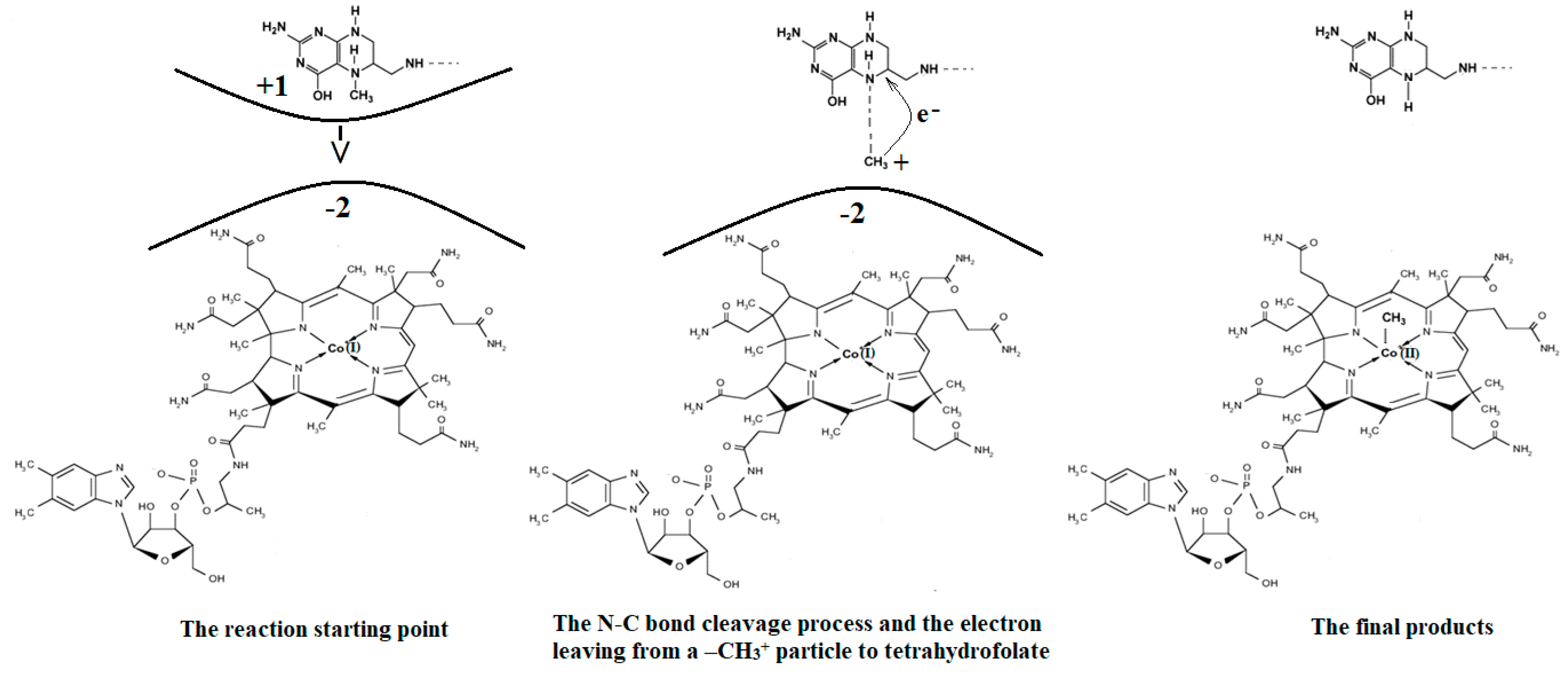
4.5. The N-C Bond Cleavage and Methyl Radical Transfer from 5-methyltetrahydrofolate to Cob(I)alamin
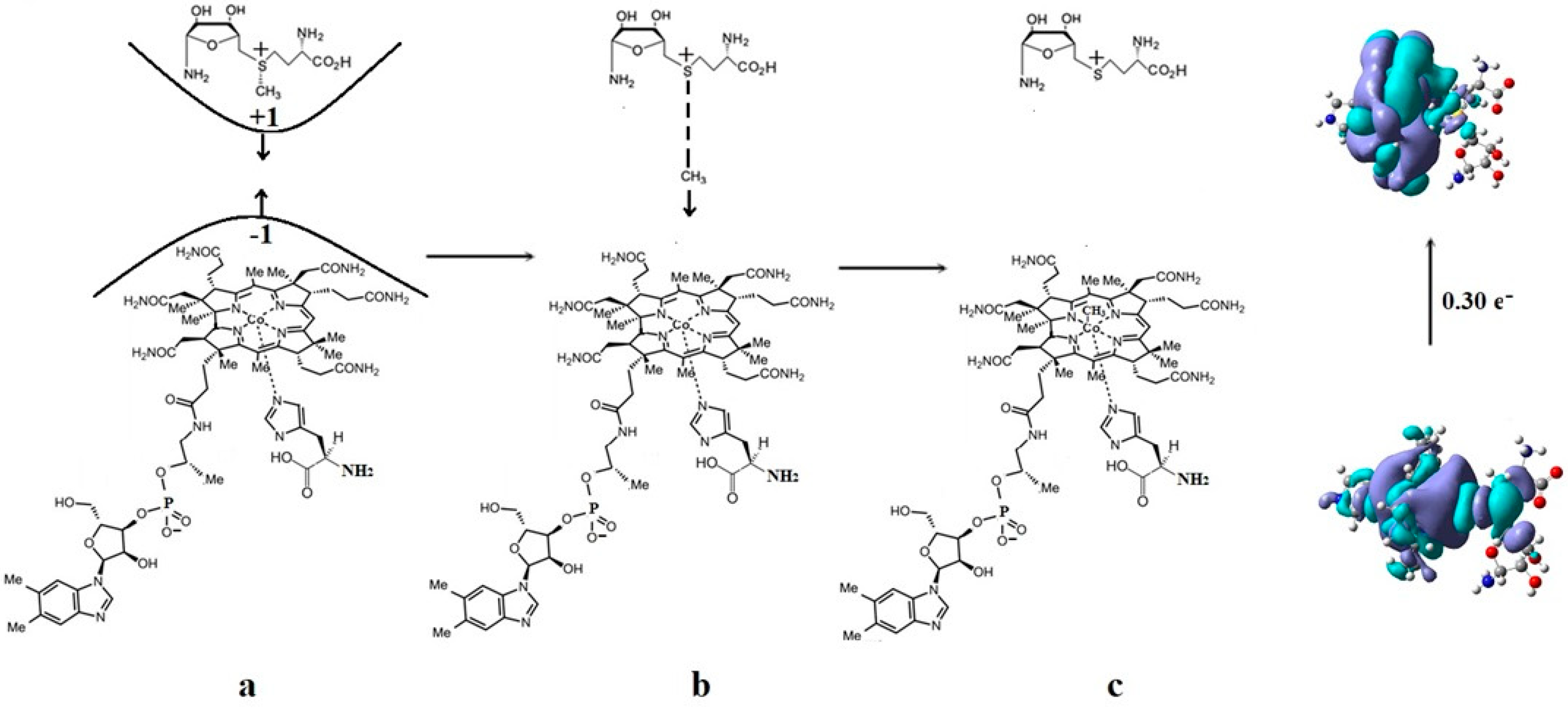
4.6. The Co-N Bond Cleavage and the Role of the Dimethylbenzimidazole Ligand in the Methionine Synthase Process
4.7. The Role of S-adenosyl-L-methionine (AdoMet) in the Methionine Synthase Process
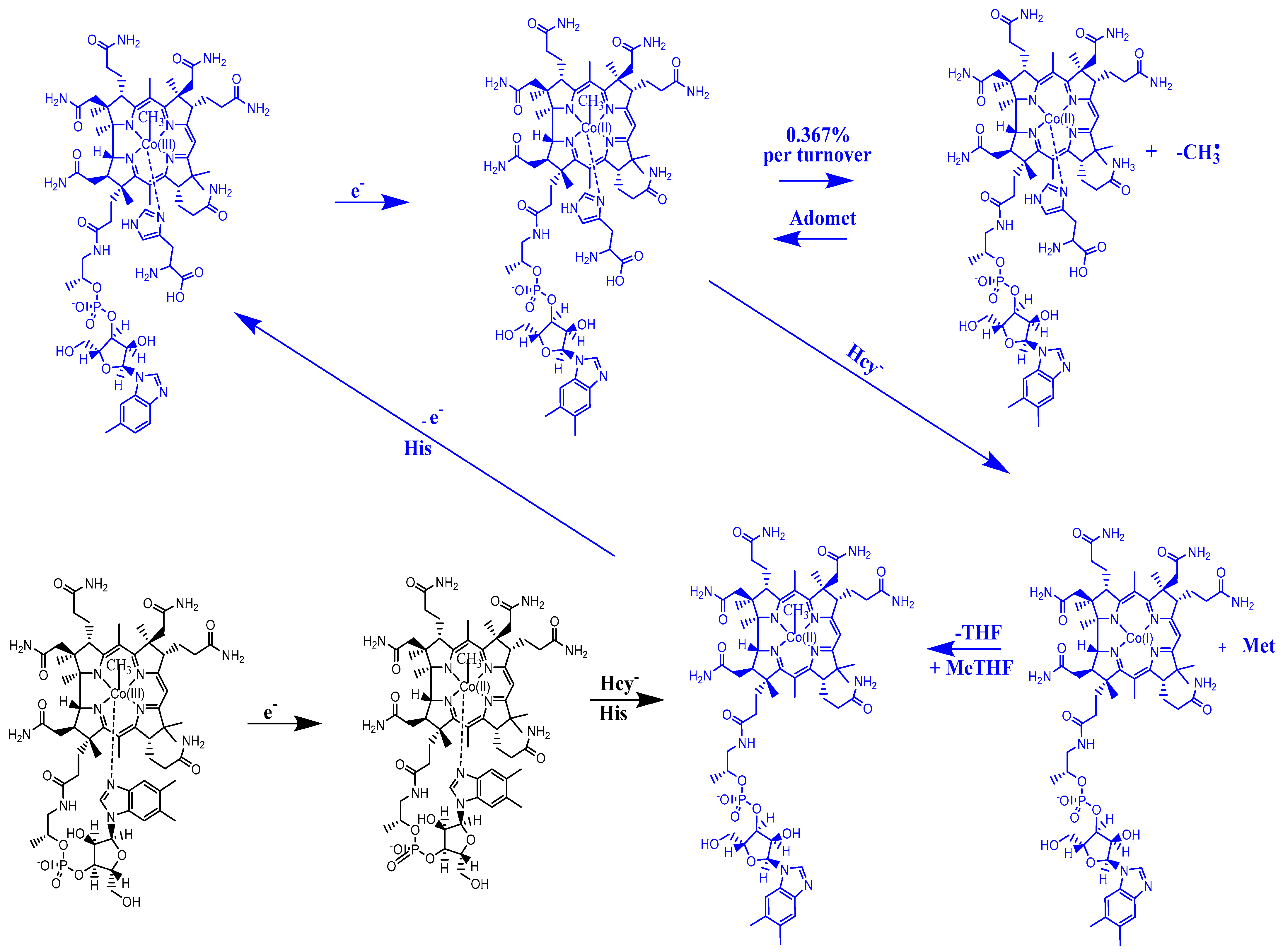
4.8. The Complete Mechanism of the Methionine Synthase Process
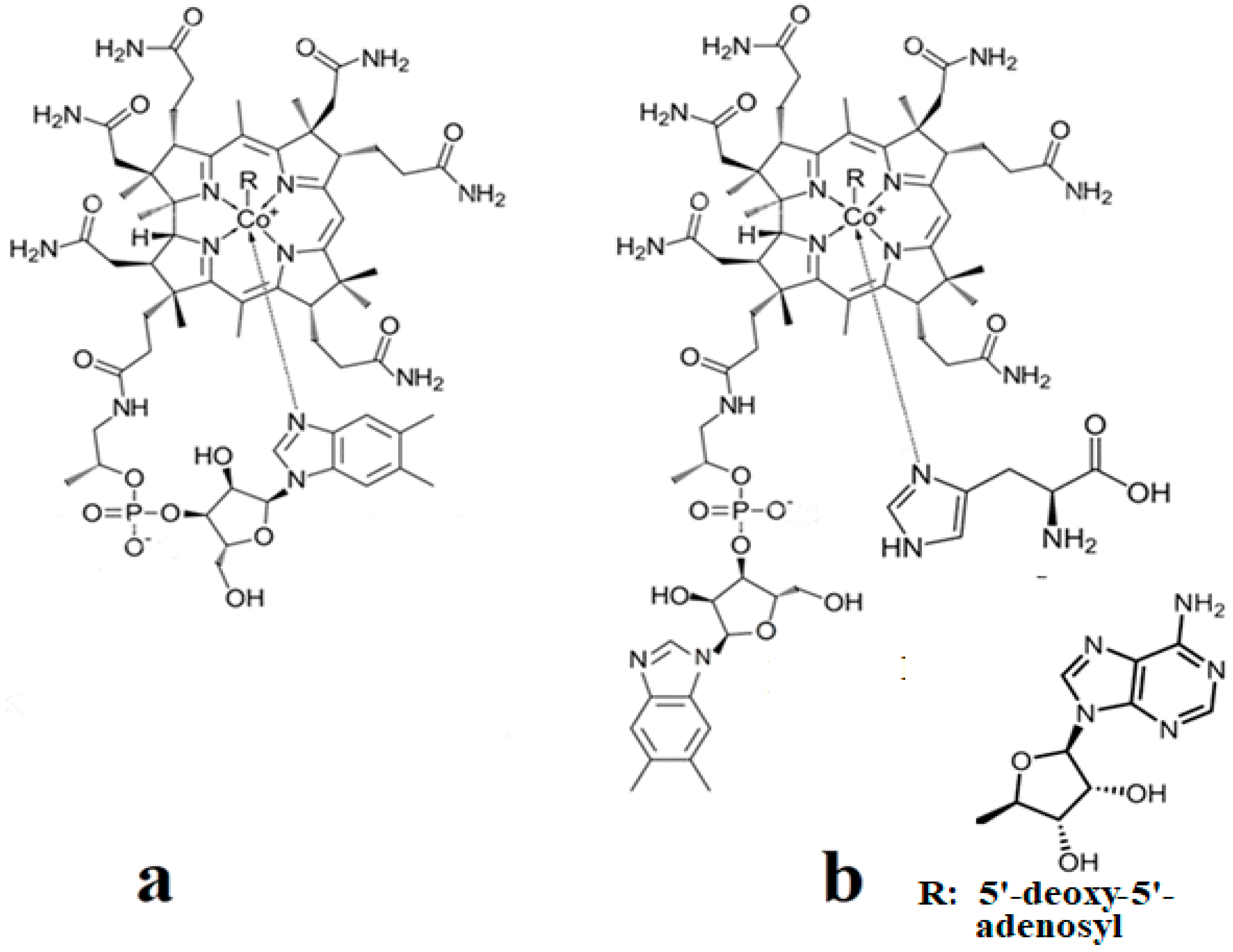
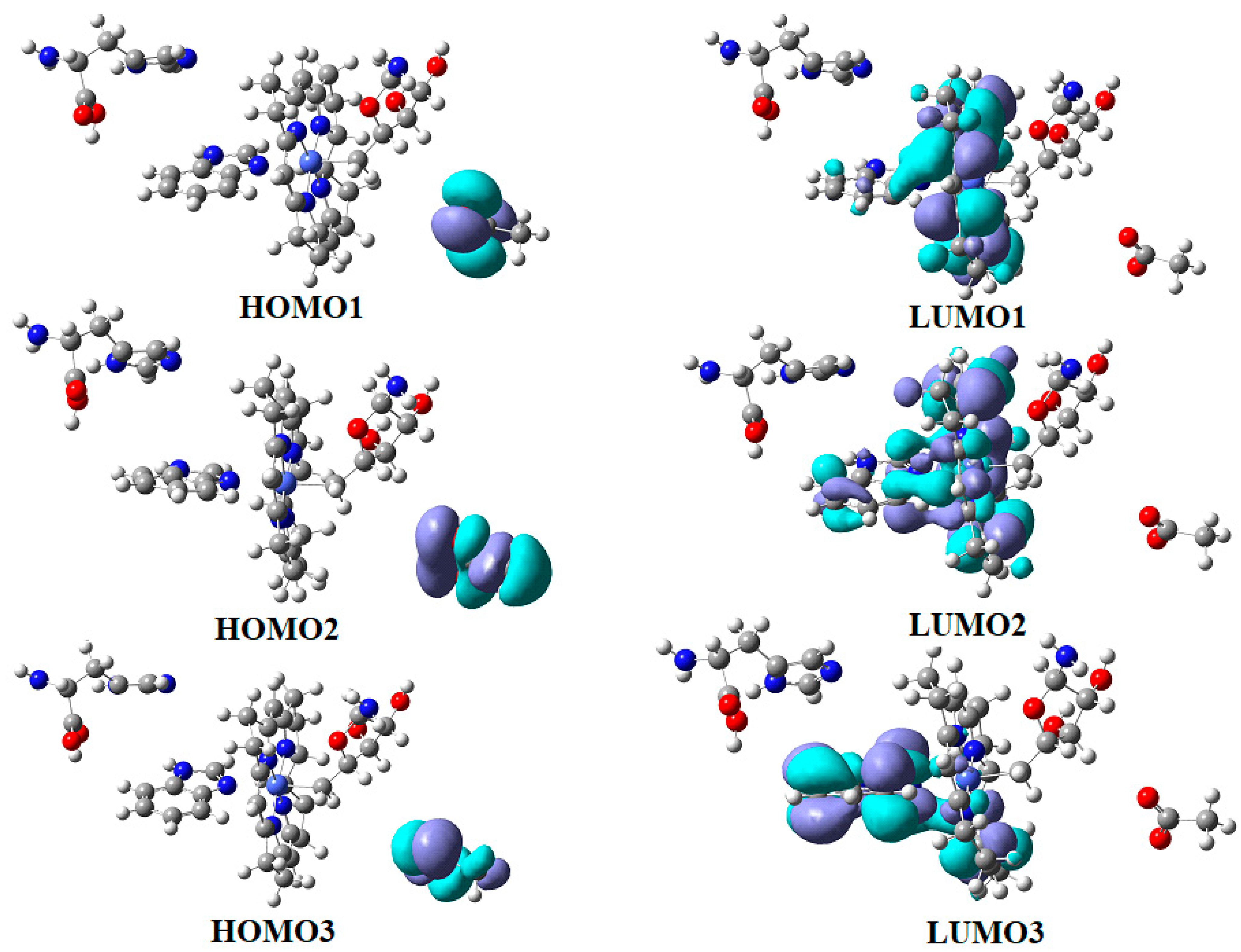
5. MCSCF Calculations: Adenosylcobalamin Cofactor-Dependent Bioprocesses
5.1. General Considerations
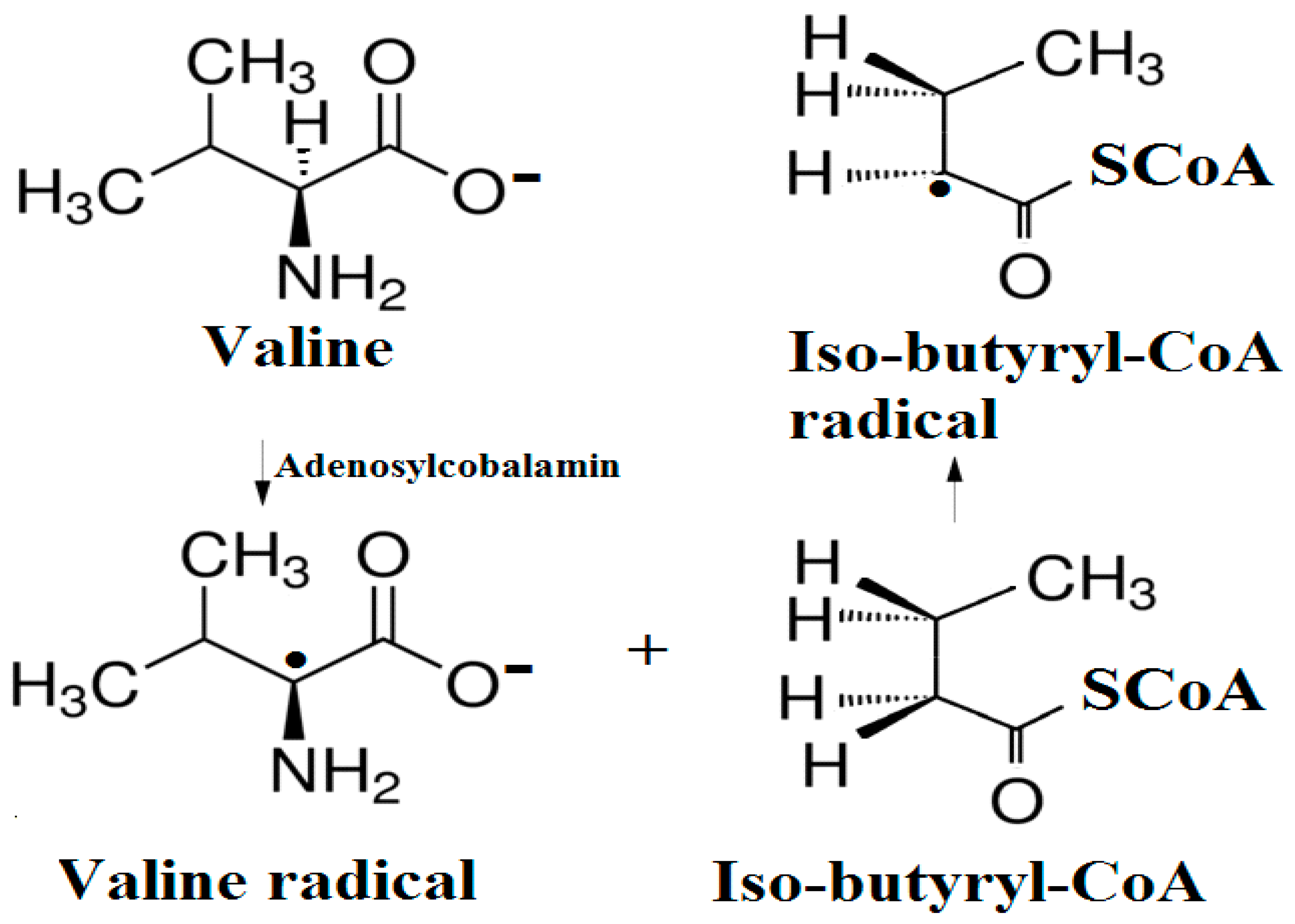
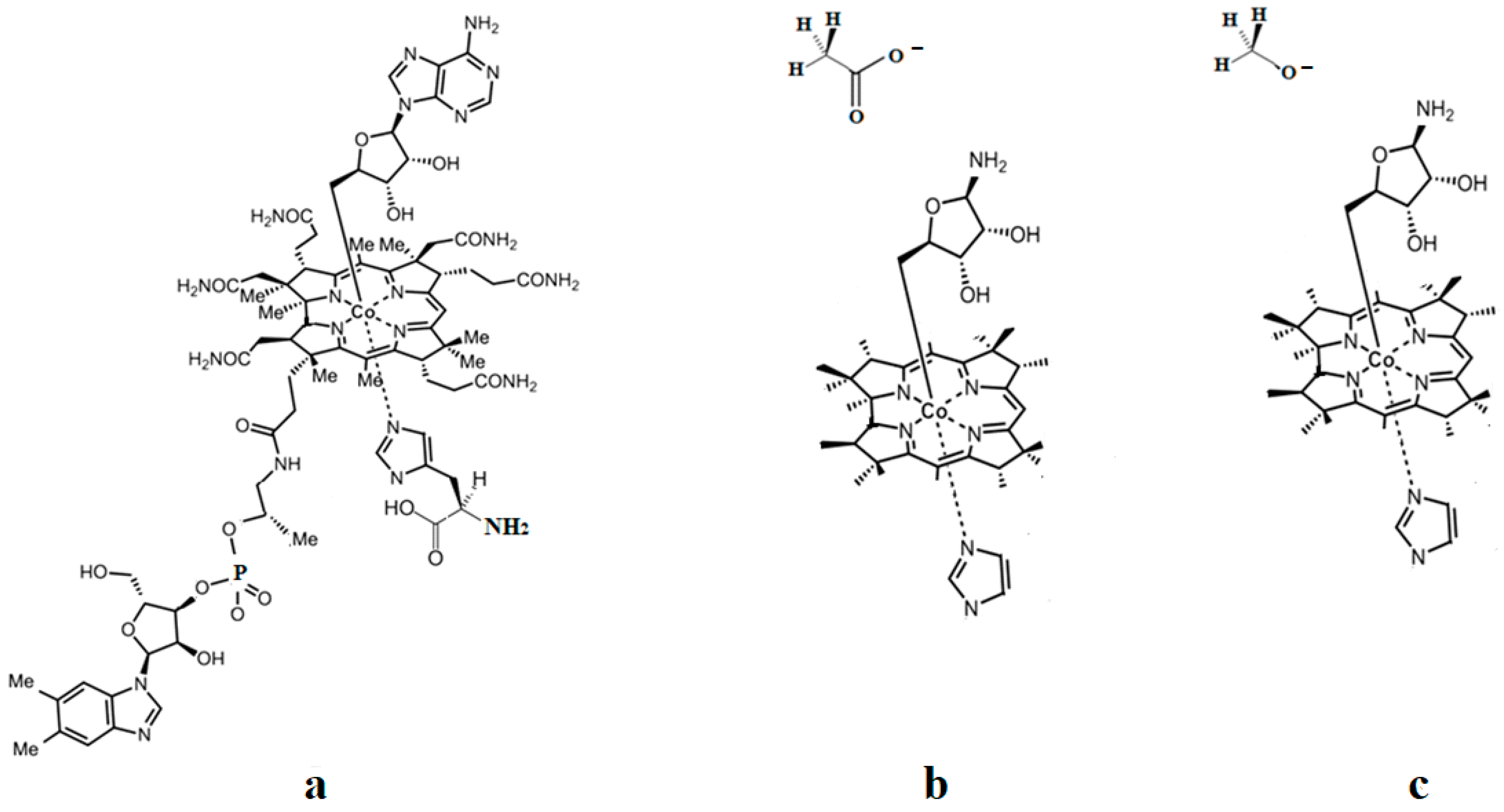
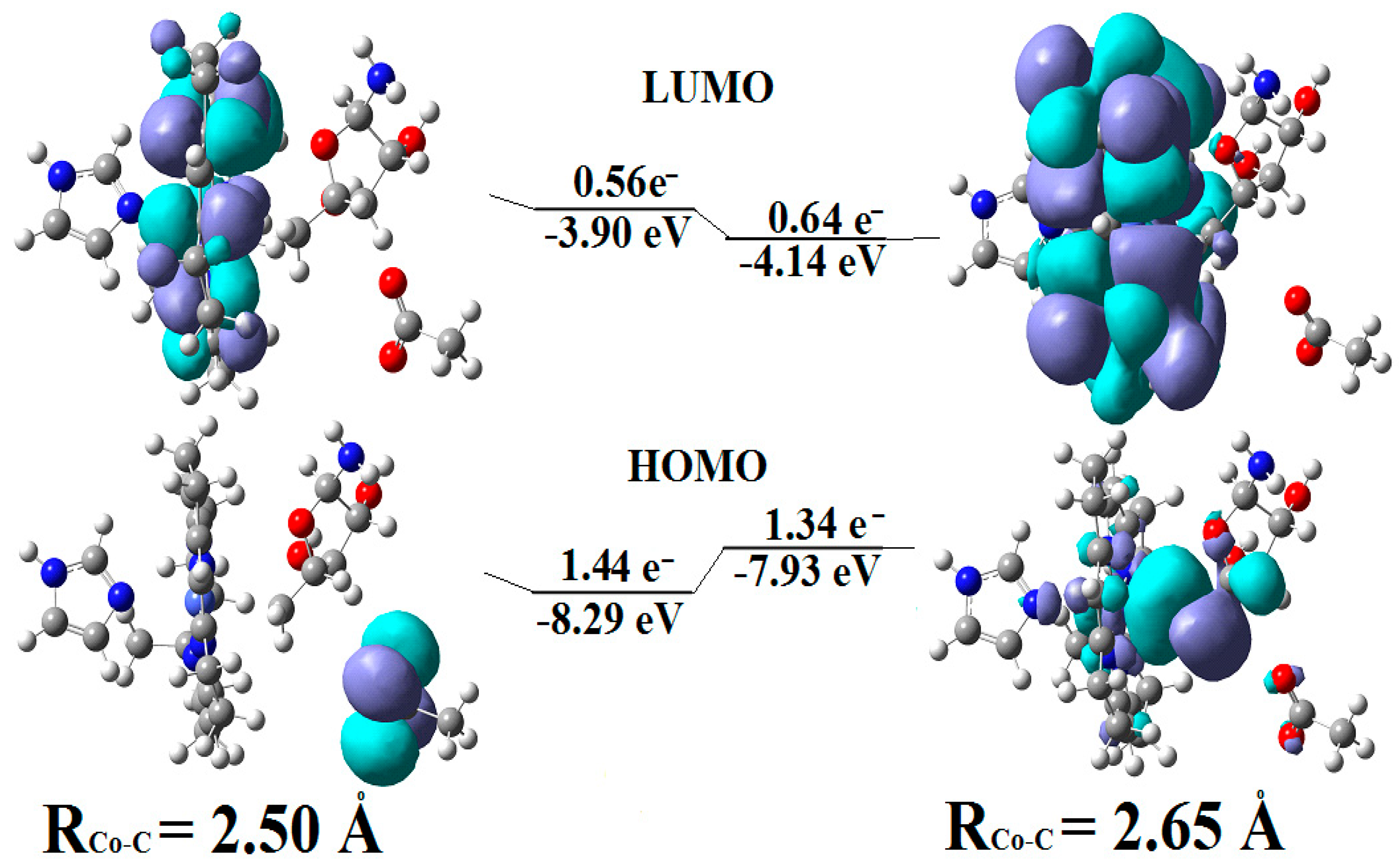
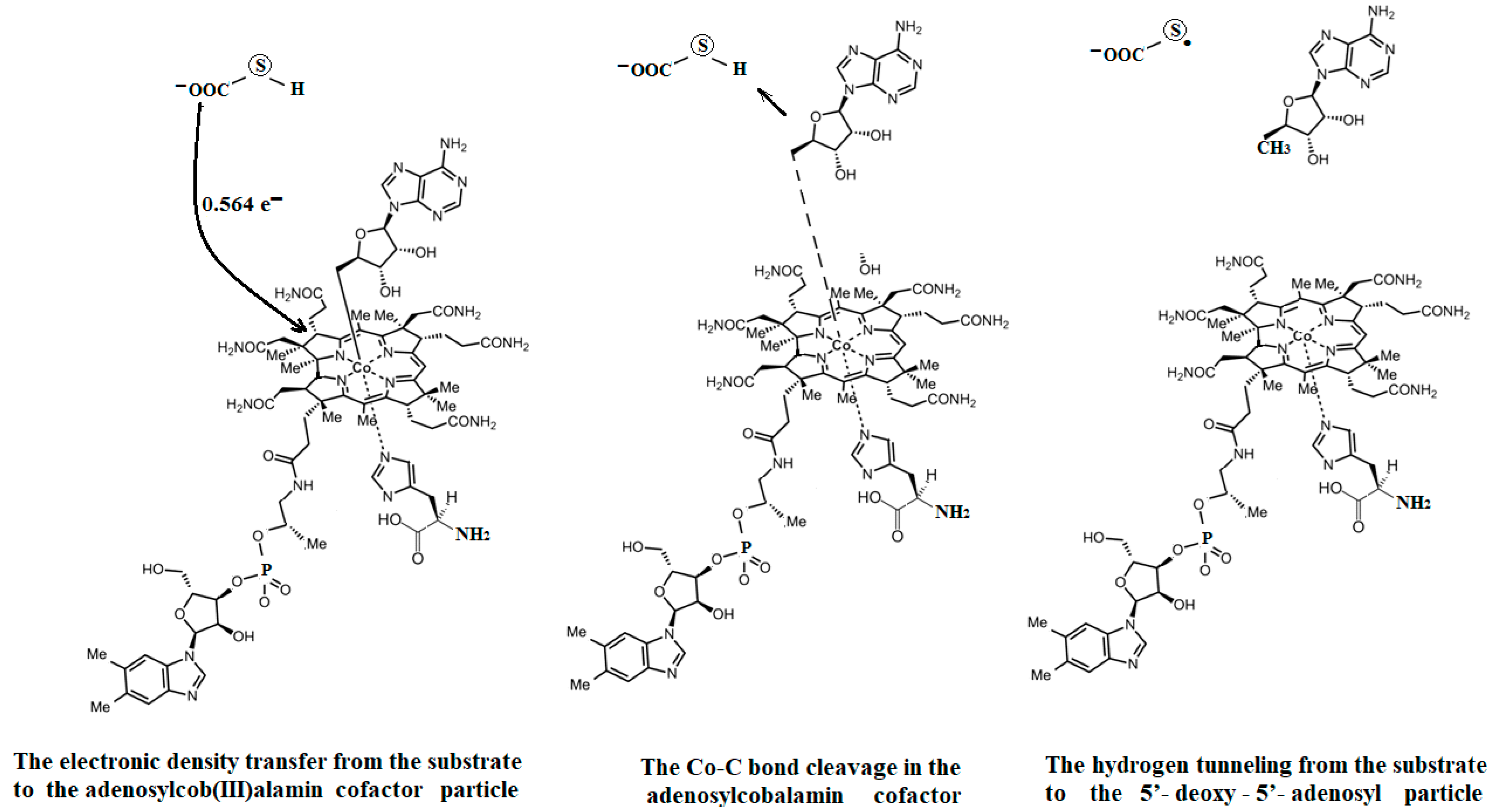
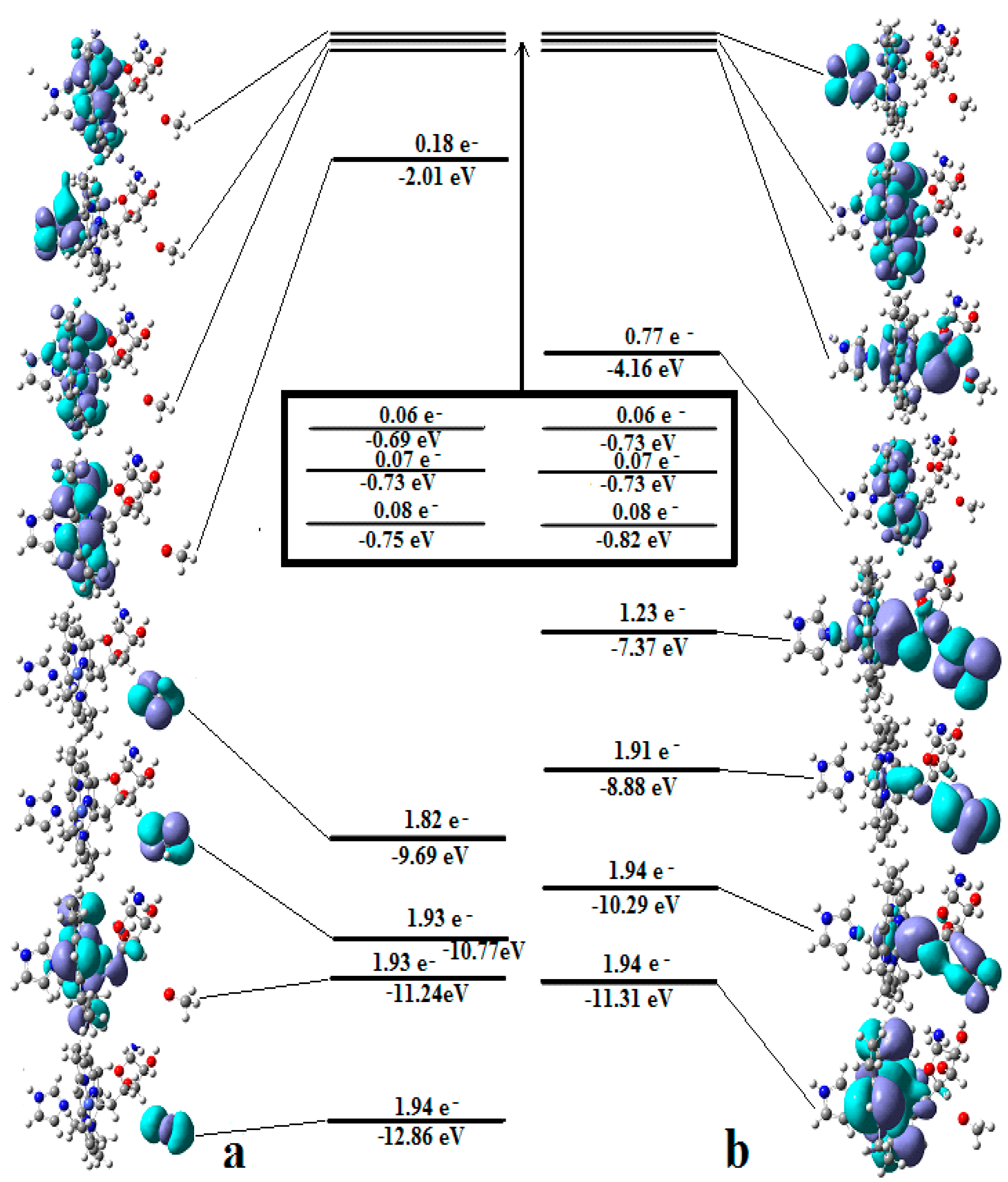
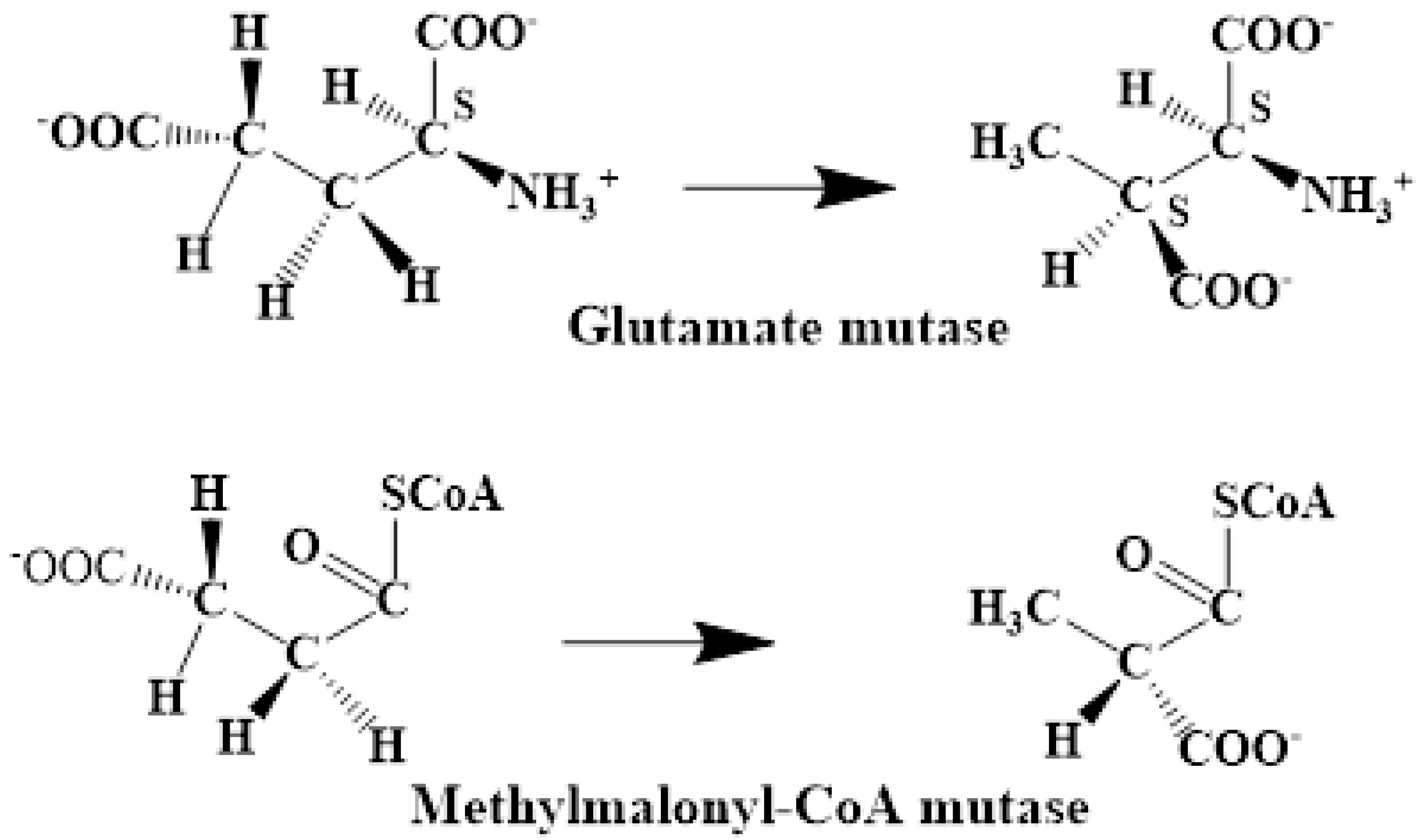
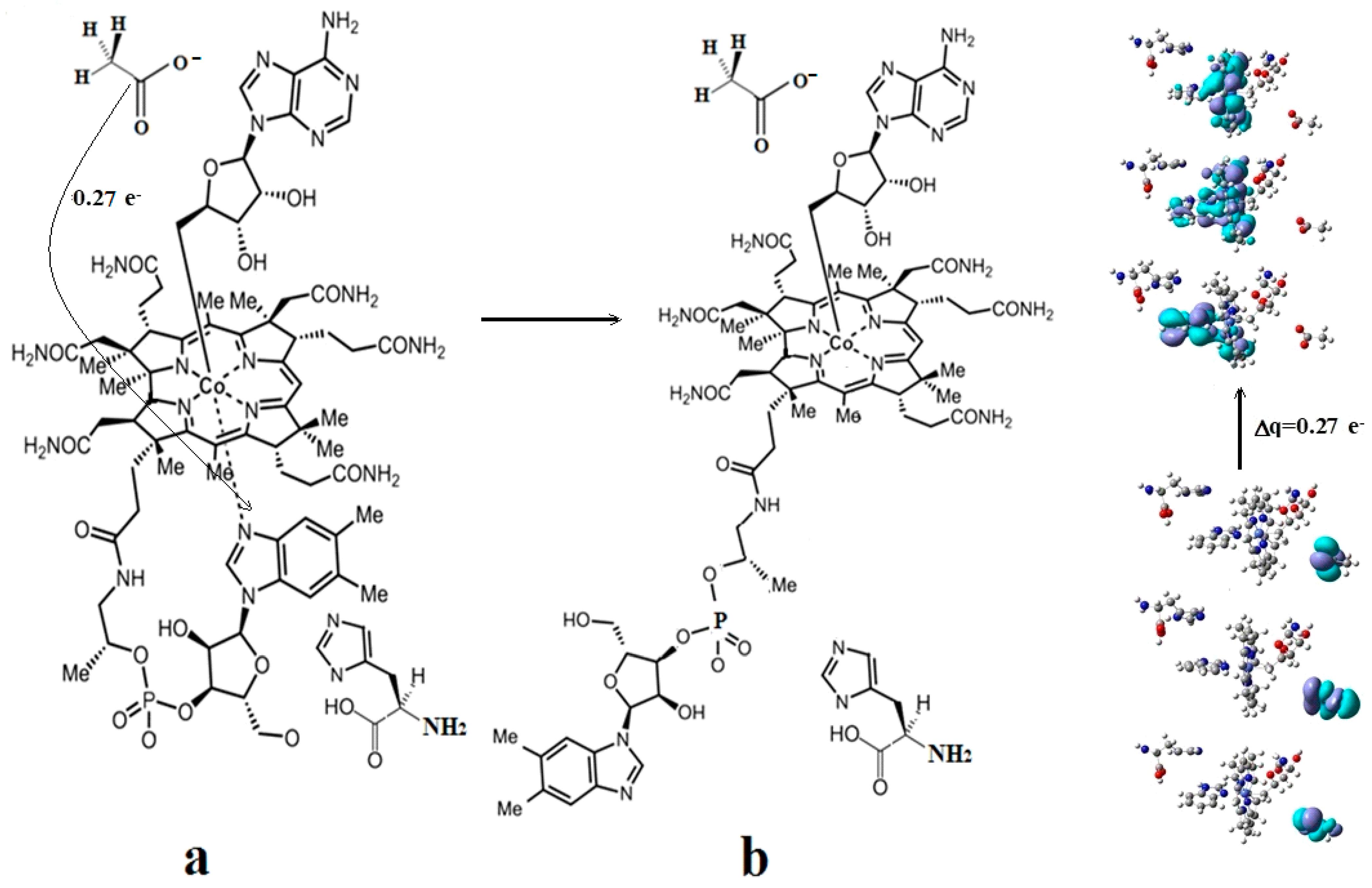
5.2. The Adenosylcobalamin Cofactor-Dependent Bioprocesses with Active Substrates Modeled with CH3COO−
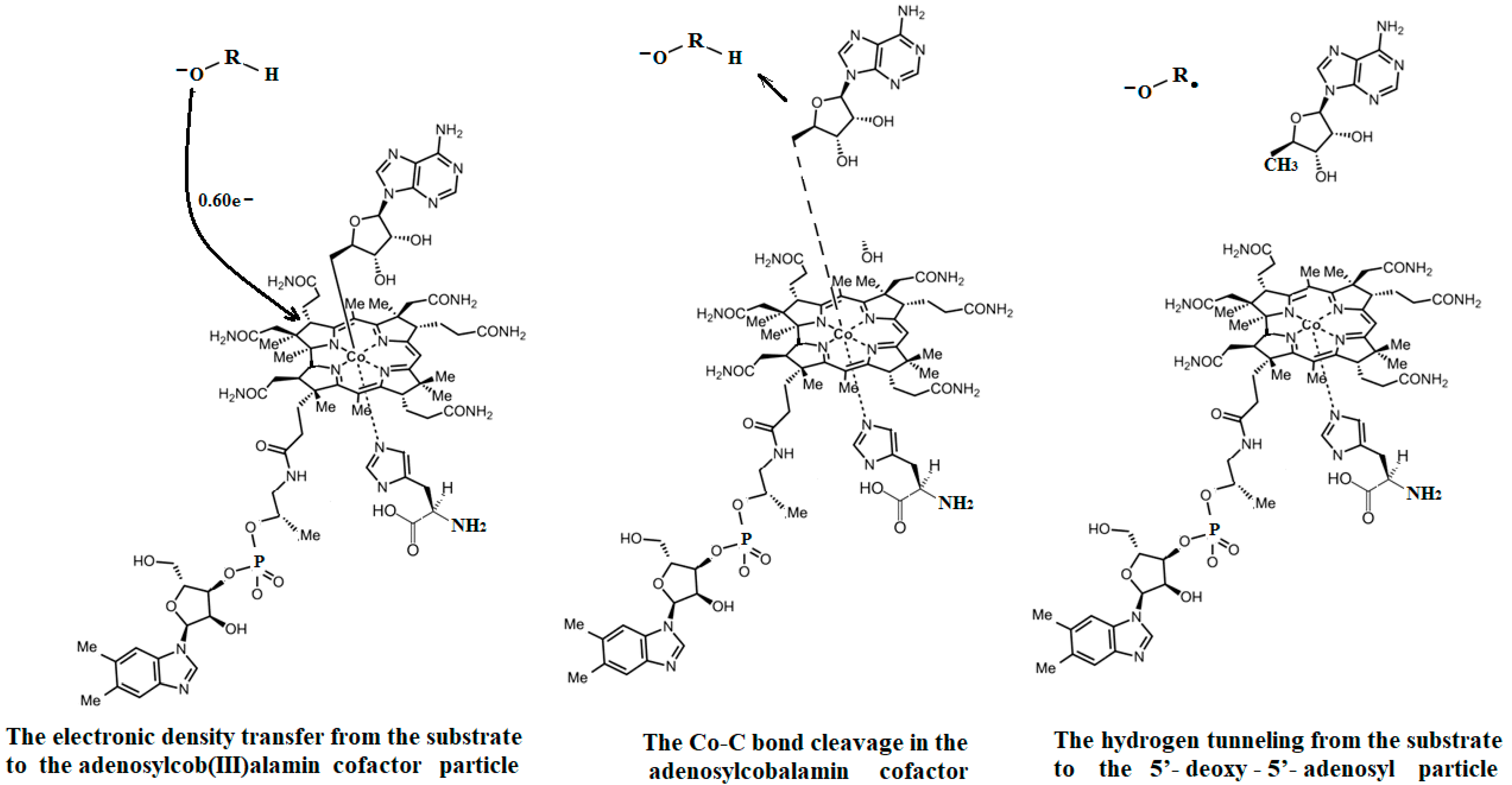
5.3. The Adenosylcobalamin Cofactor-Dependent Bioprocesses with Active Substrates Modeled with CH3-O−
5.4. The Nature of the Hydrogen Transfer from the Substrates to the 5′-Deoxy-5′-Adenosyl Radical
5.5. The Co-N Bond Cleavage in the Adenosylcobalamin Cofactor
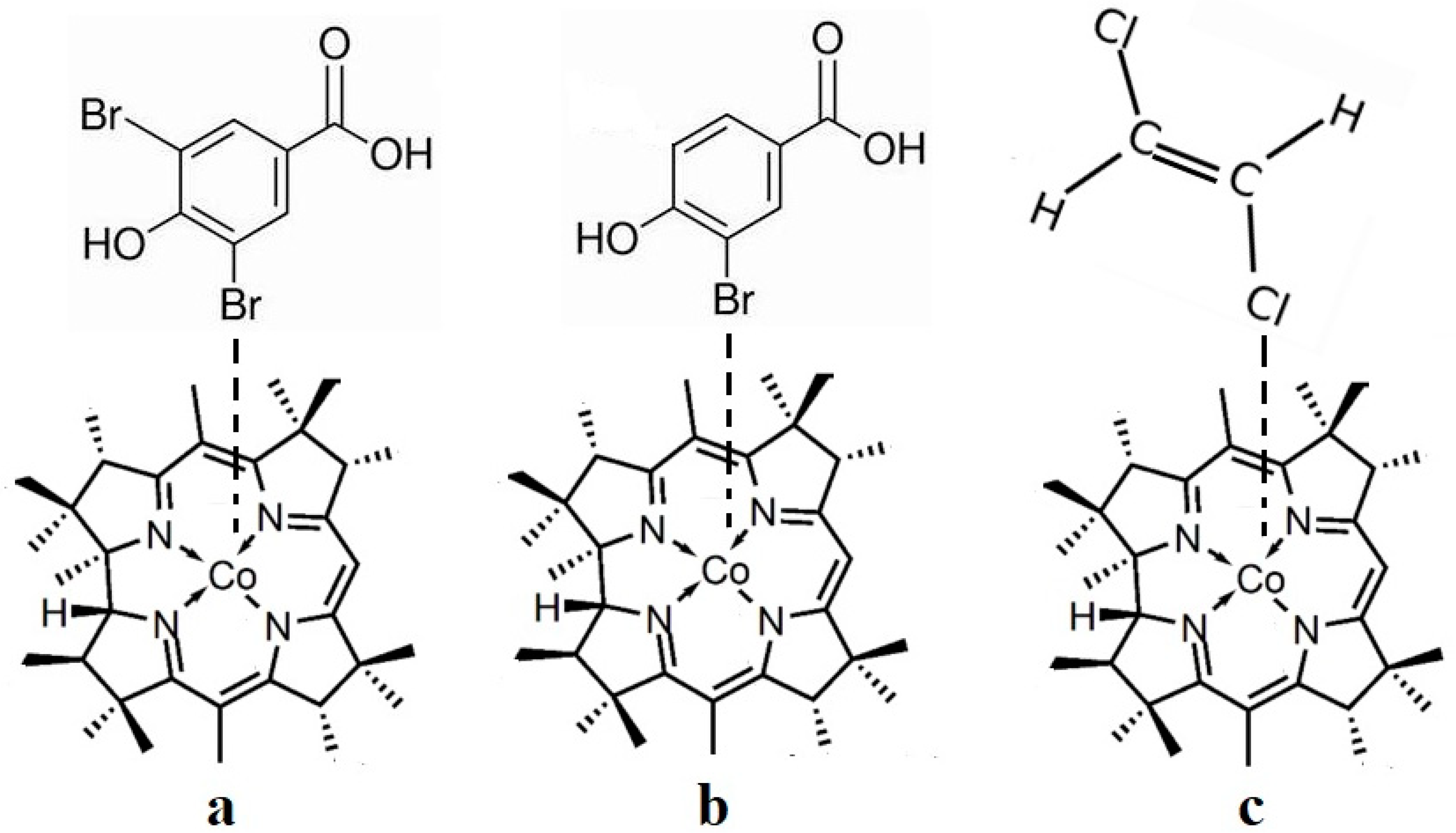
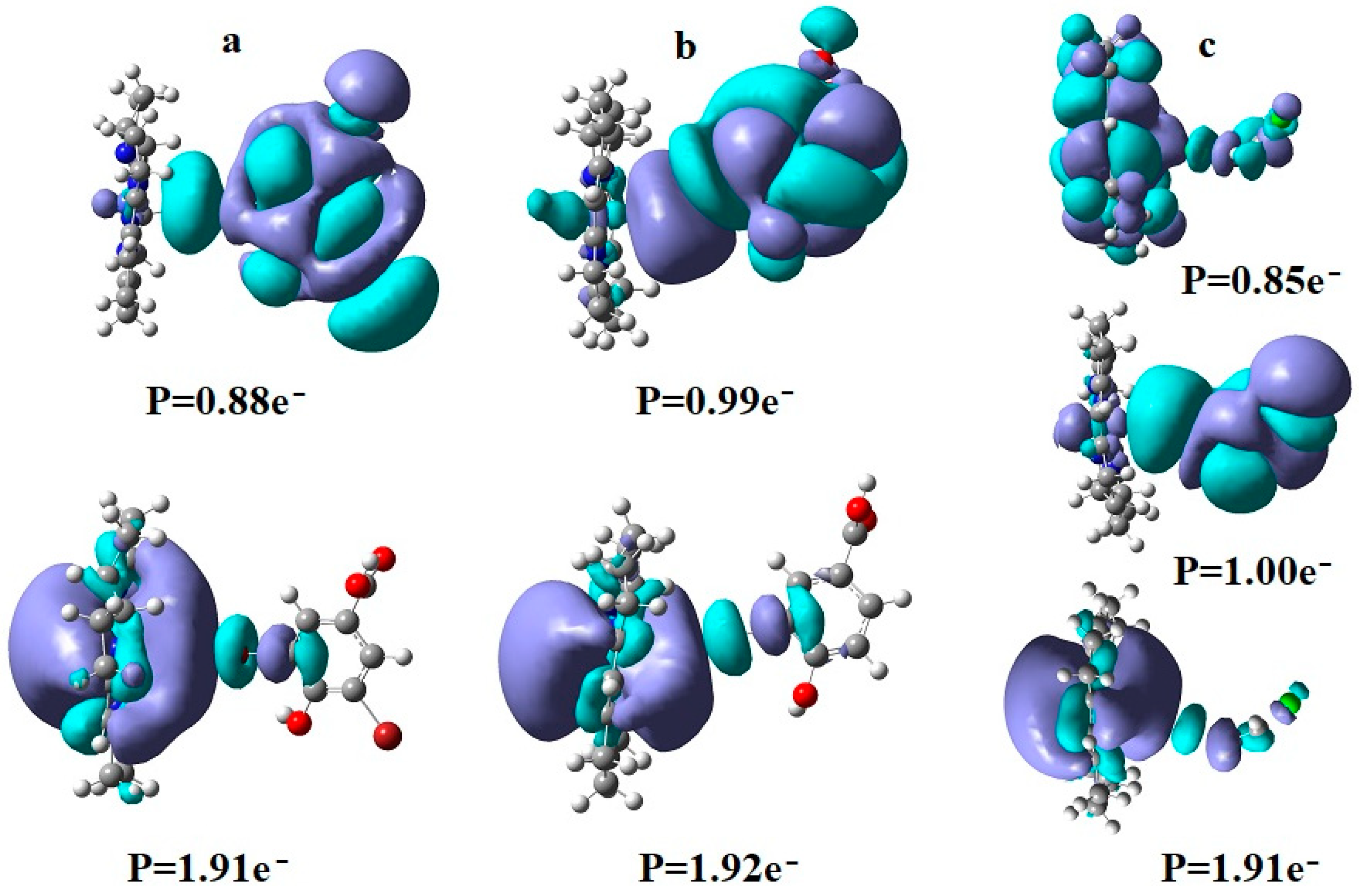
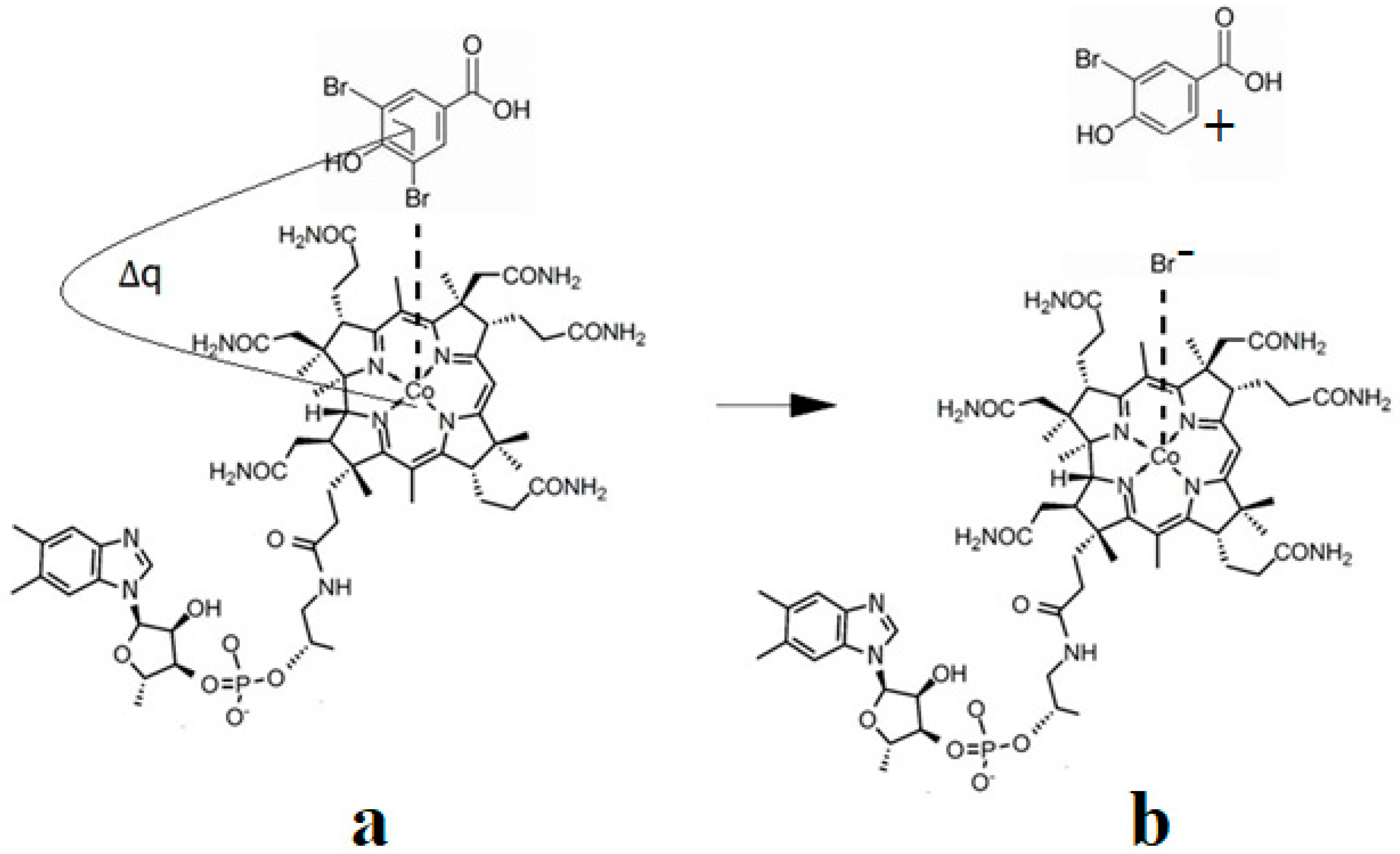
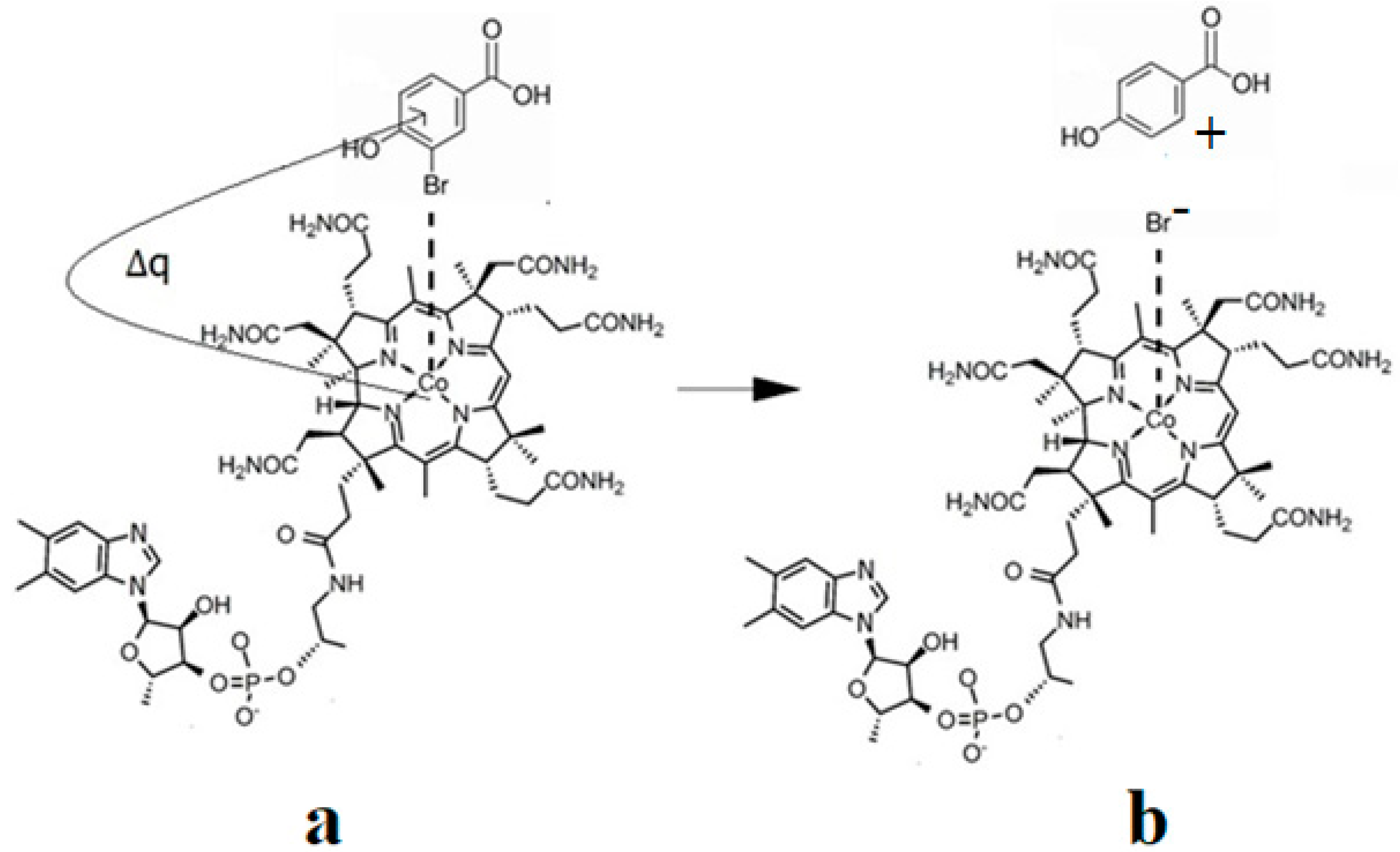
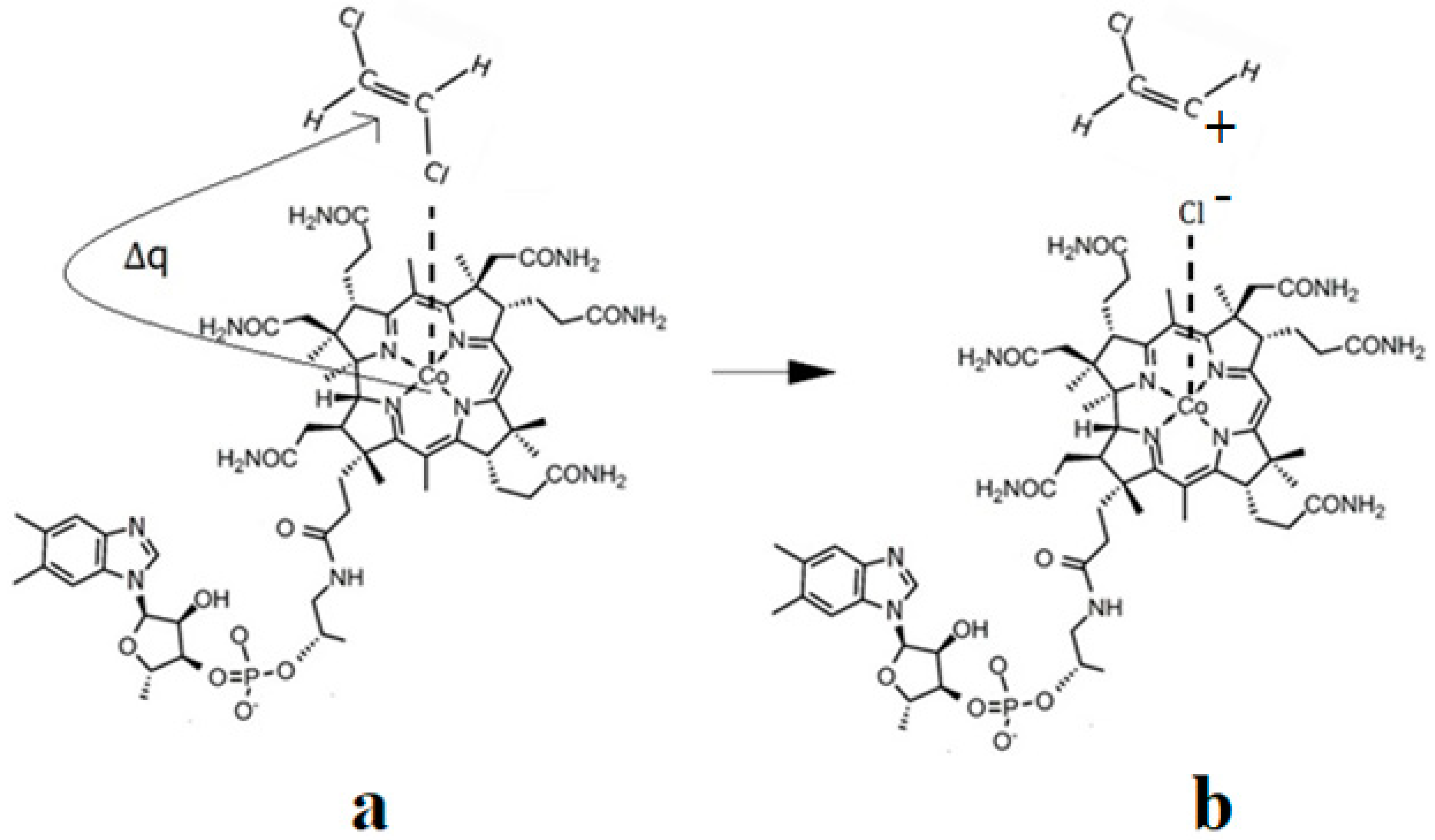
6. The Disposal of the Toxic Organic Halides under the Catalytic Influence of Vitamin B12
7. Concluding Remarks
Funding
Conflicts of Interest
References
- Carme, R. Chapter 36: Megaloblastic anemias: Disorders of impaired DNA synthesis. In Wintrobe’s Clinical Hematology, 13th ed.; Greer, J.P., Ed.; Wolters Kluwer Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2014; pp. 468–470. [Google Scholar]
- Cohn, E.J.; Minot, G.R.; Alles, G.A.; Salter, W.T. The nature of the material in liver effective in pernicious anemia. J. Biol. Chem. 1938, 77, 325–358. [Google Scholar] [CrossRef]
- Elrod, J.M.; Karnad, A.B. William Bosworth Castle: Pioneer of haematological clinical investigation. Br. J. Haematol. 2003, 121, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Minot, G.R.; Murphy, W.P. Treatment of pernicious anemia by a special diet. J. Am. Med. Assoc. 1926, 87, 470–476. [Google Scholar] [CrossRef]
- Shorb, M.S. Annual Lecture for the Department of Animal & Avian Sciences; Archived from the original on December 12; University of Maryland: College Park, MD, USA, 2012. [Google Scholar]
- Hodgkin, D.C.; Kamper, J.; Mackay, M.; Pickworth, J.; Trueblood, K.N.; White, J.G. Structure of vitamin B12. Nature 1956, 178, 64–66. [Google Scholar] [CrossRef]
- Dodson, G. Dorothy Mary Crowfoot Hodgkin, 12 May 1910–29 July 1994. Biogr. Mem. Fellows R. Soc. 2002, 48, 181–219. [Google Scholar] [CrossRef]
- Khan, A.G.; Eswaran, S.V. Woodward’s synthesis of vitamin B12. Resonance 2003, 8, 8–16. [Google Scholar] [CrossRef]
- Eschenmoser, A.; Wintner, C.E. Natural product synthesis and vitamin B12. Science 1977, 196, 1410–1420. [Google Scholar]
- Drennanenan, C.L.; Huang, S.; Drummond, J.T.; Mathews, R.G.; Ludwig, M.L. How a protein binds B12: A 3.0 Å X-ray structure of B12-binding domains of methionine synthase. Science 1994, 266, 1669–1680. [Google Scholar] [CrossRef]
- Mancia, F.; Keep, N.M.; Nakagawa, A.; Leadlay, P.F.; McSweeney, S.; Rasmussen, B.; Bosecke, P.; Diat, O.; Evans, P.F. How coenzyme B12 radicals are generated: The crystal structure of methylmalonyl-coenzyme A mutase at 2 Å resolution. Structure 1996, 4, 339–350. [Google Scholar] [CrossRef]
- Koutmos, M.; Datta, S.; Pattridge, K.A.; Janet, L.S.; Matthews, R.G. Insights into the reactivation of cobalamin-dependent methionine synthase. Proc. Natl. Acad. Sci. USA 2009, 106, 8527–18532. [Google Scholar] [CrossRef]
- Hagemeier, C.H.; Kruer, M.; Rudolf, K.; Thauer, R.K.; Eberhard, W.; Ermler, U. Insight into the mechanism of biological methanol activation based on the crystal structure of the methyl-cobalamin methyltransferase complex. Proc. Natl. Acad. Sci. USA 2006, 103, 18917–18922. [Google Scholar] [CrossRef]
- Reitzer, R.; Gruber, K.; Jogl, G.; Wagner, U.G.; Bothe, H.; Buckel, W.; Kratky, C. Glutamate mutase from clostridium cochlearium: The structure of a coenzyme B12-dependent enzyme provides new mechanistic insights. Structure 1999, 7, 891–902. [Google Scholar] [CrossRef]
- Gruber, K.; Reitzer, R.; Kratky, C. Radical shuttling in a protein: Ribose pseudorotation controls alkyl-radical transfer in the coenzyme B12 dependent enzyme glutamate mutase. Angew. Chem. Int. Ed. 2001, 40, 3377–3380. [Google Scholar] [CrossRef]
- Banerjee, R. Chapter 11: Spectroscopic and Molecular Genetic Characterization of the Two Mammalian Blz-dependent Enzymes. In Vitamin B12 and the B12 Proteins; Kräutler, B., Arigoni, D., Golding, B.T., Eds.; Wiley-VCH: Weinheim, Germany, 1998; pp. 189–197. [Google Scholar]
- Yamanishi, M.; Labunska, T.; Banerjee, R. Mirror base-off conformation of coenzyme B12 in human adenosyltransferase and its downstream target, methylmalonyl-CoA mutase. J. Am. Chem. Soc. 2004, 127, 526–527. [Google Scholar] [CrossRef]
- Schrauzer, G.N.; Lee, L.P. The molecular and electronic structure of vitamin B12r, cobaloximes(II), and related compounds. J. Am. Chem. Soc. 1968, 90, 6541–6543. [Google Scholar] [CrossRef]
- Schrauzer, G.N. Recent advances in the chemistry of vitamin B12 and vitamin B12 model compounds: Reductive cobalt-carbon bond cleavage reaction. Pure Appl. Chem. 1973, 33, 545–566. [Google Scholar] [CrossRef]
- Schrauzer, G.N. Cobaloximes give insight into reactions of B12. Chem. Eng. News 1968, 46, 42–44. [Google Scholar]
- Hogenkamp, H.P.C.; Bratt, G.T.; Sun, S. Methyl transfer from methylcobalamin to thiols: A reinvestigation. Biochemistry 1985, 24, 6428–6432. [Google Scholar] [CrossRef]
- Hogenkamp, H.P.C.; Bratt, G.T.; Kotchevar, A.T. Reaction of alkylcobalamins with thiols. Biochemistry 1987, 26, 4723–4727. [Google Scholar] [CrossRef]
- Pratt, J.M. Inorganic Chemistry of Vitamin B12; Academic Press: Cambridge, MA, USA, 1972. [Google Scholar]
- Banerjee, R.V.; Harder, S.R.; Ragsdale, S.W.; Matthews, R.G. Mechanism of reductive activation of cobalamin-dependent methionine synthase: An electron paramagnetic resonance spectroelectrochemical study. Biochemistry 1990, 29, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.V.; Frasca, V.; Ballou, D.P.; Matthews, R.G. Participation of cob(I)alamin in the reaction catalyzed by methionine synthase from escherichia coli: A steady-state and a rapid reaction kinetic analysis. Biochemistry 1990, 29, 11101–11109. [Google Scholar] [CrossRef]
- Gonzales, J.C.; Banerjee, R.V.; Huang, S.; Summer, J.S.; Matthews, R.G. Comparison of cobalamin-independent and cobalamin-dependent methionine synthase from escherichia coli: Two solutions to the same chemical problem. Biochemistry 1992, 31, 6045–6056. [Google Scholar] [CrossRef]
- Banerjee, R.V.; Matthews, R.G. Cobalamin–dependent methionine synthase. Fed. Am. Soc. Exp. Biol. J. 1990, 4, 1450–1459. [Google Scholar] [CrossRef]
- Retey, J. Enzymic reaction selectivity by negative catalysis or how do enzymes deal with highly reactive intermediates. Angew. Chem. Int. Ed. 1990, 29, 355–361. [Google Scholar] [CrossRef]
- Leutbecher, U.; Albracht SP, J.; Buckel, W. Identification of a paramagnetic species as an early intermediate in the coenzyme B12-dependent glutamate mutase reaction: A cob(II)amide. Fed. Eur. Biochem. Soc. Lett. 1992, 307, 144–146. [Google Scholar] [CrossRef]
- Michel, C.; Albracht, S.P.J.; Buckel, W. Adenosylcobalamin and cob(II)alamin as prosthetic groups of 2-methyleneglutarate mutase from clostridium barkeri. Eur. J. Biochem. 1992, 205, 767–773. [Google Scholar] [CrossRef]
- Stubbe, J. Radicals in biological catalysis. Biochemistry 1988, 27, 3893–3900. [Google Scholar] [CrossRef]
- Stubbe, J. Protein radical involvement in biological catalysis. Annu. Rev. Biochem. 1989, 58, 257–285. [Google Scholar] [CrossRef] [PubMed]
- Frey, P.A. Importance of organic radicals in enzymatic cleavage of unactivated C-H bonds. Chem. Rev. 1990, 90, 1343–1357. [Google Scholar] [CrossRef]
- Zhao, Y.; Such, P.; Retey, J. Radical intermediates in the coenzyme B12 dependent methyl-malonyl CoA mutase reaction shown by ESR spectroscopy. Angew. Chem. Int. Ed. 1992, 31, 215–216. [Google Scholar] [CrossRef]
- Stupperich, E. Recent advances in elucidation of biological corrinoid functions. Fed. Eur. Biochem. Soc. Microbiol. Rev. 1993, 12, 349–366. [Google Scholar] [CrossRef] [PubMed]
- Matthews, R.G. Cobalamin-Dependent Methionine Synthase. In Chemistry and Biochemistry of B12; Banerjee, R., Ed.; John Wiley: New York, NY, USA, 1999; pp. 681–706. [Google Scholar]
- Matthews, R.G. Cobalamin-dependent methyltransferases. Acc. Chem. Res. 2001, 34, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Koutmos, M.; Pejchal, R.; Bomer, T.M.; Matthews, R.G.; Smith, J.L.; Ludwig, M.L. Metal active site elasticity linked to activation of homocysteine in methionine synthases. Proc. Natl. Acad. Sci. USA 2005, 105, 3286–3291. [Google Scholar] [CrossRef] [PubMed]
- Matthews, R.G.; Koutmos, M.; Datta, S. Cobalamin-dependent and cobamide-dependent methyltransferases. Curr. Opin. Struct. Biol. 2008, 18, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Olteanu, H.; Banerjee, R. Human methionine synthase reductase, a soluble P-450 reductase-like dual-flavoprotein, is sufficient for NADPH-dependent methionine synthase activation. J. Biol. Chem. 2001, 276, 35558–35563. [Google Scholar] [CrossRef] [PubMed]
- James, T.; Drummond, J.T.; Sha, H.; Blumenthal, R.M.; Matthews, R.G. Assignment of enzymatic function to specific protein regions of cobalamin-dependent methionine synthase from escherichia coli. Biochemistry 1993, 32, 9290–9295. [Google Scholar]
- Hondorp, E.R.; Matthews, R.G. Oxidation of cysteine 645 of cobalamin-independent methionine synthase causes a methionine limitation in Escherichia coli. J. Bacteriol. 2009, 191, 3407–3410. [Google Scholar] [CrossRef]
- Smith, A.E.; Matthews, R.G. The protonation state of methyltetrahydrofolate in a binary complex with cobalamin-dependent methionine synthase. Biochemistry 2000, 39, 13880–13890. [Google Scholar] [CrossRef]
- Goulding, C.W.; Matthews, R.G. Cobalamin-dependent methionine synthase from escherichia coli: involvement of zinc in homocysteine activation. Biochemistry 1997, 36, 15749–15757. [Google Scholar] [CrossRef]
- Goulding, C.W.; Postigo, D.; Matthews, R.G. Cobalamin-dependent methionine synthase is a modular protein with distinct regions for binding homocysteine, methyltetrahydrofolate, cobalamin, and adenosylmethionine. Biochemistry 1997, 36, 8082–8091. [Google Scholar] [CrossRef]
- Bandarian, V.; Matthews, R.G. Quantitation of rate enhancements attained by the binding of cobalamin to methionine synthase. Biochemistry 2001, 40, 5056–5064. [Google Scholar] [CrossRef] [PubMed]
- Peariso, K.; Goulding, C.W.; Matthews, R.G.; Penner-Hahn, J. The role of zinc in the binding of homocysteine to cobalamin-dependent methionine synthase. J. Am. Chem. Soc. 1998, 120, 8410–8416. [Google Scholar] [CrossRef]
- Jarrett, J.T.; Choi, C.Y.; Matthews, R.G. Changes in protonation associated with substrate binding and cob(I)alamin formation in cobalamin-dependent methionine synthase. Biochemistry 1997, 36, 15739–15748. [Google Scholar] [CrossRef] [PubMed]
- Matthews, R.G. Activation of methyltetrahydrofolate by cobalamin-independent methionine synthase. Biochemistry 2006, 45, 5092–5102. [Google Scholar]
- Pratt, J.M. The Roles of Co, Corrin, and Protein. I. Co-Ligand Bonding and the Trans Effect. In Chemistry and Biochemistry of B12; Banerjee, R., Ed.; John Wiley & Sons: New York, NY, USA, 1999; pp. 73–112. [Google Scholar]
- Marsh, E.N.G. Review article coenzyme-B12-dependent glutamate mutase. Bioorganic Chem. 2000, 28, 176–189. [Google Scholar] [CrossRef]
- Verroust, P.J.; Christensent, E.I.; Moestrup, S.K.; Hammond, T.G.; Seetharam, B. Chapter 32: The Intrinsic Factor-Cobalamin Receptor Expressed by Yolk Sac and Proximal Tubule Epithelial Cells is the Target of Teratogenic Antibodies. In Vitamin B12 and the B12 Proteins; Kräutler, B., Arigoni, D., Golding, B.T., Eds.; Wiley-VCH: Weinheim, Germany, 1998; pp. 491–504. [Google Scholar]
- Babior, B.M.; Carty, T.J.; Abeles, R.H. The mechanism of action of ethanolamine ammonia-lyase, a B12-dependent enzyme: The reversible formation of 5′-deoxyadenosine from adenosylcobalamin during the catalytic process. J. Biol. Chem. 1974, 249, 1689–1695. [Google Scholar] [CrossRef]
- Toraya, T. Radical catalysis of B12 enzymes: Structure, mechanism, inactivation, and reactivation of diol and glycerol dehydratases. Cell. Mol. Life Sci. 2000, 57, 106–127. [Google Scholar] [CrossRef]
- Marsh, E.N.G.; Ballou, D.P. Coupling of cobalt−carbon bond homolysis and hydrogen atom abstraction in adenosylcobalamin-dependent glutamate mutase. Biochemistry 1998, 37, 11864–11872. [Google Scholar] [CrossRef]
- Chowdhury, S.; Banerjee, R. Thermodynamic and kinetic characterization of Co−C bond homolysis catalyzed by coenzyme B12-dependent methyl malonyl-CoA mutase. Biochemistry 2000, 39, 7998–8006. [Google Scholar] [CrossRef]
- Buckel, W.; Golding, B.T. Wiley Encyclopedia of Chemical Biology; Wiley & Sons: Hoboken, NJ, USA, 2008; pp. 1–9. [Google Scholar]
- Banerjee, R.; Ragsdale, S.W. The many faces of vitamin B12: Catalysis by cobalamin-dependent enzymes. Annu. Rev. Biochem. 2003, 72, 209–247. [Google Scholar] [CrossRef]
- Brooks, A.J.; Vlasie, M.; Banerjee, R.; Brunold, T.C. Co−C bond activation in methylmalonyl-CoA mutase by stabilization of the post-homolysis product Co2+cobalamin. J. Am. Chem. Soc. 2005, 127, 16522–16528. [Google Scholar] [CrossRef] [PubMed]
- Buckel, W.; Kratky, C.; Golding, B.T. Stabilization of methylene radicals by cob(II)alamin in coenzyme B12 dependent mutases. Chem. A Eur. J. 2006, 12, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Madhavapeddi, P.; Ballou, D.P.; Marsh, E.N.G. Pre-steady-state kinetic studies on the Glu171Gln active site mutant of adenosylcobalamin-dependent glutamate mutase. Biochemistry 2002, 41, 15803–15809. [Google Scholar] [CrossRef] [PubMed]
- Pallares, I.G.; Moore, T.C.; Escalante-Semerena, J.C.; Brunold, T.C. Spectroscopic studies of the salmonella enterica adenosyltransferase enzyme SeCobA: Molecular-level insight into the mechanism of substrate cob(II)alamin activation. Biochemistry 2014, 53, 7969–7982. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.L.; Li, J. Activation parameters for the carbon−cobalt bond homolysis of coenzyme B12 induced by the B12-dependent ribonucleotide reductase from lactobacillus leishmanial. J. Am. Chem. Soc. 1998, 120, 9466–9474. [Google Scholar] [CrossRef]
- Moore, T.C.; Newmister, S.A.; Rayment, I.; Escalante-Semerena, J.C. Structural insights into the mechanism of four-coordinate cob(II)alamin formation in the active site of the salmonella enterica ATP: Co(I)crinoid adenosyltransferase enzyme: Critical role of residues Phe91 and Trp93. Biochemistry 2012, 51, 9647–9657. [Google Scholar] [CrossRef] [PubMed]
- Marsh, E.N.G.; Meléndez, G.D.R. Adenosylcobalamin enzymes: Theory and experiment begin to converge. Biochim. Biophys. Acta 2012, 1824, 1154–1164. [Google Scholar] [CrossRef]
- Padmakumar, R.; Banerjee, R. Evidence that cobalt−carbon bond homolysis is coupled to hydrogen atom abstraction from substrate in methylmalonyl-CoA mutase. Biochemistry 1997, 36, 3713–3718. [Google Scholar] [CrossRef]
- Licht, S.S.; Booker, S.; Stubbe, J. Studies on the catalysis of carbon−cobalt bond homolysis by ribonucleoside triphosphate reductase: Evidence for concerted carbon−cobalt bond homolysis and thiyl radical formation. Biochemistry 1999, 38, 1221–1233. [Google Scholar] [CrossRef]
- Meier, T.W.; Thomä, N.H.; Evans, P.R.; Leadlay, P.F. Tritium isotope effects in adenosylcobalamin-dependent methylmalonyl-CoA mutase. Biochemistry 1996, 35, 11791–11796. [Google Scholar] [CrossRef]
- Chih, H.W.; Marsh, E.N.G. Pre-steady-state kinetic investigation of intermediates in the reaction catalyzed by adenosylcobalamin-dependent glutamate mutase. Biochemistry 1999, 38, 13684–13691. [Google Scholar] [CrossRef] [PubMed]
- Bandarian, V.; Reed, G.H. Isotope effects in the transient phases of the reaction catalyzed by ethanolamine ammonia-lyase: Determination of the number of exchangeable hydrogens in the enzyme−cofactor complex. Biochemistry 2000, 39, 12069–12075. [Google Scholar] [CrossRef] [PubMed]
- Fink, R.G. Chapter 25: Coenzyme B12-Based Chemical Precedent for Co-C Bond Homolysis and Other Key Elementary Steps. In Vitamin B12 and the B12 Proteins; Kräutler, B., Arigoni, D., Golding, B.T., Eds.; Wiley-VCH: Weinheim, Germany, 1998; pp. 383–402. [Google Scholar]
- Banerjee, R. The Yin-yang of cobalamin biochemistry. Chem. Biol. 1997, 4, 175–186. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yoon, M.; Song, H.; Håkansson, K.; Neil, E.; Marsh, G. Hydrogen tunneling in adenosylcobalamin-dependent glutamate mutase: Evidence from intrinsic kinetic isotope effects measured by intra-molecular competition. Biochemistry 2010, 49, 3168–3173. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hayward, G.C.; Hill, H.A.O.; Pratt, J.M.; Vanston, N.J.; Williams, R.J.P. The chemistry of vitamin B12: Part IV. the thermodynamic trans-effect. J. Chem. Soc. 1965, 6485–6493. [Google Scholar] [CrossRef]
- Grate, J.H.; Schrauzer, G.N. Chemistry of cobalamins and related compounds. 48. sterically induced, spontaneous dealkylation of secondary alkylcobalamins due to axial base coordination and conformational changes of the corrin ligand. J. Am. Chem. Soc. 1979, 101, 4601–4611. [Google Scholar] [CrossRef]
- Halpern, J. Mechanisms of coenzyme B12-dependent rearrangements. Science 1985, 227, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Marsh, E.N.G.; Drennan, C.L. Adenosylcobalamin-dependent isomerases: New insights into structure and mechanism. Curr. Opin. Chem. Biol. 2001, 5, 499–505. [Google Scholar] [CrossRef]
- Brown, K.L.; Zou, X.J. Thermolysis of coenzymes B12 at physiological temperatures: Activation parameters for cobalt-carbon bond homolysis and quantitative analysis of the perturbation of the homolysis equilibrium by the ribonucleoside triphosphate reductase from lactobacillus leishmania. Inorg. Biochem. 1999, 77, 185–195. [Google Scholar]
- Dong, S.L.; Padmakumar, R.; Banerjee, R.; Spiro, T.G. Co−C bond activation in B12-dependent enzymes: Cryogenic resonance raman studies of methylmalonyl-coenzyme A mutase. J. Am. Chem. Soc. 1999, 121, 7063–7070. [Google Scholar] [CrossRef]
- Bresciana-Pahor, N.; Forcolin, M.; Marzilli, L.G.; Randaccio, L.; Summers, M.F.; Toscano, P.J. Organocobalt B12 models: Axial ligand effects on the structural and coordination chemistry of cobaloximes. Coord. Chem. Rev. 1985, 63, 1–125. [Google Scholar] [CrossRef]
- Mancia, F.; Evans, P.R. Conformational changes on substrate binding to methyl malonyl CoA mutase and new insights into the free radical mechanism. Structure 1998, 6, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Vlasie, M.D.; Banerjee, R. Tyrosine 89 accelerates Co−C bond homolysis in methylmalonyl-CoA mutase. J. Am. Chem. Soc. 2003, 125, 5431–5435. [Google Scholar] [CrossRef] [PubMed]
- Thomä, N.H.; Meier, T.W.; Evans, P.R.; Leadlay, P.F. Stabilization of radical intermediates by an active-site tyrosine residue in methylmalonyl-CoA mutase. Biochemistry 1998, 37, 14386–14393. [Google Scholar] [CrossRef] [PubMed]
- Toraya, T.; Ishida, A. Acceleration of cleavage of the carbon-cobalt bond of sterically hindered alkylcobalamins by binding to apoprotein of diol dehydrase. Biochemistry 1988, 27, 7677–7682. [Google Scholar] [CrossRef]
- Zhu, L.; Kostic, N.M. Molecular orbital study of coenzyme B12: Activation of the cobalt-carbon bond by angular distortions. Inorg. Chem. 1987, 26, 4194–4197. [Google Scholar] [CrossRef]
- Stuart, S.L.; Lawrence, C.C.; Stubbe, J. Thermodynamic and kinetic studies on carbon−cobalt bond homolysis by ribonucleoside triphosphate reductase: the importance of entropy in catalysis. Biochemistry 1999, 38, 1234–1242. [Google Scholar]
- Vrijbloed, J.W.; Zerbe-Burkhardt, K.; Ratnatilleke, A.; Grubelnik-Leiser, A.; Robinson, J.A. Insertional inactivation of methylmalonyl coenzyme A (CoA) mutase and isobutyryl-CoA mutase genes in streptomyces cinnamonensis: Influence on polyketide antibiotic biosynthesis. J. Bacteriol. 1999, 181, 5600–5605. [Google Scholar] [CrossRef]
- Lexa, D.; Savéant, J.M. Electrochemistry of vitamin B12: Part 3: One-electron intermediates in the reduction of methylcobalamin and methylcobinamide. J. Am. Chem. Soc. 1978, 100, 3220–3222. [Google Scholar] [CrossRef]
- Spataru, T.; Birke, R.L. The effect of solvent on the electrode process of methylcobalamin as studied by cyclic voltammetry. J. Electroanal. Chem. 2006, 593, 74–86. [Google Scholar] [CrossRef]
- Birke, R.L.; Huang, Q.; Spataru, T.; Gosser Jr, D.K. Electroreduction of an alkylcobalamin: Mechanism of stepwise reductive cleavage of the Co-C bond. J. Am. Chem. Soc. 2006, 128, 1922–1936. [Google Scholar] [CrossRef] [PubMed]
- Scheffold, R.; Abrecht, S.; Ruf, H.R.; Stamouli, P.; Tinembart, O.; Walder, L.; Weymuth, C. Vitamin B12-mediated electrochemical reactions in the synthesis of natural products. Pure Appl. Chem. 1987, 59, 363–372. [Google Scholar] [CrossRef]
- Ogoshi, H.; Kikuchi, Y.; Yamaguchi, T.; Toi, H.; Aoyama, Y. Asymmetric induction in the nucleophilic cyclopropane ring cleavage reaction with vitamin B12s. Organometallics 1987, 6, 2175–2178. [Google Scholar] [CrossRef]
- Pattenden, G. Simonsen lecture. cobalt-mediated radical reactions in organic synthesis. Chem. Soc. Rev. 1988, 17, 361–382. [Google Scholar] [CrossRef]
- Baldwin, D.A.; Betterton, E.A.; Chemaly, S.M.; Pratt, J.M.J. The chemistry of vitamin B12.: Part 25: Mechanism of the β-elimination of olefins from alkylcorrinoids; evidence for initial homolytic fission of the Co–C bond. J. Chem. Soc. Dalton Trans. 1985, 8, 1613–1618. [Google Scholar] [CrossRef]
- Baldwin, J.E.; Adlington, R.M.; Kang, T.W. Direct ring expansion of penicillins to 3-exomethylene cephalosporins. Tetrahedron Lett. 1991, 48, 7093–7096. [Google Scholar] [CrossRef]
- Paquette, L. (Ed.) Encyclopedia of Reagents for Organic Synthesis; Wiley: New York, NY, USA, 1995; pp. 5511–5514. [Google Scholar]
- Lee, E.R.; Lakomy, I.; Bigler, P.; Scheffold, R. Reductive radical cyclisations of bromo acetals and (bromomethyl) silyl ethers of terpenoid alcohols. Helv. Chim. Acta 1991, 74, 146–162. [Google Scholar] [CrossRef]
- Shey, J.; McGinley, C.M.; McCauley, K.M.; Dearth, A.S.; Young, B.T.; van der Donk, W.A. Mechanistic investigation of a novel vitamin B12-catalyzed carbon−carbon bond forming reaction, the reductive dimerization of arylalkenes. J. Org. Chem. 2002, 67, 837–846. [Google Scholar] [CrossRef]
- McGinley, C.M.; Relyea, H.A.; van der Donk, W.A. Vitamin B12 catalyzed radical cyclizations of arylalkenes. Synlett 2006, 2, 211–214. [Google Scholar] [CrossRef]
- Kumar, M.; Kozlowski, P.M. Electronic and structural properties of cob(I)alamin: Ramifications for B12-dependent processes. Coord. Chem. Rev. 2017, 333, 71–81. [Google Scholar] [CrossRef]
- Andruniow, T.; Zagierski, M.Z.; Kozlowski, P.M. Density functional theory analysis of stereoelectronic properties of cobalamins. J. Phys. Chem. B 2000, 104, 10921–10927. [Google Scholar] [CrossRef]
- Andruniow, T.; Zagierski, M.Z.; Kozlowski, P.M. Theoretical determination of the Co-C bond energy dissociation in cobalamins. J. Am. Chem. Soc. 2001, 123, 2679–2680. [Google Scholar] [CrossRef]
- Kumar, M.; Kozlowski, P.M. The cobalt-methyl bond dissociation: New benchmark analysis based on density functional theory and completely renormalized coupled-cluster calculations. J. Chem. Theory Comput. 2012, 8, 1870–1894. [Google Scholar]
- Dolker, N.; Maseras, F.; Liedos, A. A density functional study on the effect of the trans axial ligand of cobalamin on the homolytic cleavage of the Co−C bond. J. Phys. Chem. B 2001, 105, 7564–7571. [Google Scholar] [CrossRef]
- Jensen, P.; Ryde, U. The axial N-base has minor influence on Co-C bond cleavage in cobalamins. J. Mol. Struct. 2002, 585, 239–255. [Google Scholar] [CrossRef]
- Kozlowski, P.M.; Zagierski, M.Z. Electronic and steric influence of trans axial base on the stereoelectronic properties of cobalamins. J. Phys. Chem. B 2004, 108, 14163–14170. [Google Scholar] [CrossRef]
- Andruniow, T.; Zagierski, M.Z.; Kozlowski, P.M. DFT–SQM force field for cobalt corrinoids. Chem. Phys. Lett. 2000, 331, 502–518. [Google Scholar] [CrossRef]
- Andruniow, T.; Zagierski, M.Z.; Kozlowski, P.M. Vibrational analysis of methylcobalamin. J. Phys. Chem. A 2002, 106, 1365–1373. [Google Scholar] [CrossRef]
- Andruniow, T.; Kozlowski, P.M.; Zagierski, M.Z. Theoretical analysis of electronic absorption spectra of vitamin B12 models. J. Chem. Phys. 2001, 115, 7522–7533. [Google Scholar] [CrossRef]
- Kozlowski, P.M. Quantum chemical modeling of Co-C bond activation in B12-dependent enzymes. Curr. Opin. Chem. Biol. 2001, 5, 736–743. [Google Scholar] [CrossRef]
- Mealli, C.; Sabat, M.; Marzilli, L. Coenzyme B12 cobalt-carbon bond homolysis: Insights from qualitative molecular orbital theory. J. Am. Chem. Soc. 1987, 109, 1593–1594. [Google Scholar] [CrossRef]
- Andruniow, T.; Kuta, J.; Zgierski, M.Z.; Kozlowski, P.M. Molecular orbital analysis of anomalous trans effect in cobalamins. Chem. Phys. Lett. 2005, 410, 410–416. [Google Scholar] [CrossRef]
- Kozlowski, P.M.; Kamachi, T. Reductive elimination pathway for homocysteine to methionine conversion in cobalamin-dependent methionine synthase. J. Biol. Inorg. Chem. 2012, 17, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-L.; Blomberg MR, A.; Siegbahn, P.E.M. How Is a Co-methyl intermediate formed in the reaction of cobalamin-dependent methionine synthase? theoretical evidence for a two-step methyl cation transfer mechanism. J. Phys. Chem. B 2011, 115, 4066–4077. [Google Scholar] [CrossRef]
- Reig, A.J.; Conrad, K.S.; Brunold, T.S. Combined spectroscopic/computational studies of vitamin B12 precursors: Geometric and electronic structures of cobinamides. Inorg. Chem. 2012, 51, 2867–2879. [Google Scholar] [CrossRef] [PubMed]
- Liptak, M.D.; Fleischhacker, A.S.; Matthews, R.G.; Teiser, J.; Brunold, T.C. Spectroscopic and computational characterization of the base-off forms of cob(II)alamin. J. Phys. Chem. B 2009, 113, 5245–5254. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Brunold, T. Combined spectroscopic and computational analysis of the vibrational properties of vitamin B12 in its Co3+, Co2+, and Co1+. J. Phys. Chem. B 2013, 117, 5397–5410. [Google Scholar] [CrossRef]
- Govender, P.P.; Navizet, I.; Perry, C.B.; Marcus, H.M. DFT studies of trans and cis influences in the homolysis of the Co-C bond in models of the alkylcobalamins. J. Phys. Chem. A 2013, 117, 3057–3068. [Google Scholar] [CrossRef]
- Kornobis HS, K.; Ruud, K.; Kozlowski, P.M. Electronically excited states of vitamin B12 and methylcobalamin: Theoretical analysis of absorption, CD, and MCD data. J. Phys. Chem. B 2011, 115, 737–748. [Google Scholar]
- Ghosh, A.P.; Al Mamun, A.; Kozlowski, P.M. Light-induced activation of organo-metallic Co-C bond in MeCbl-dependent methionine synthase- QM/MM study. Biophys. J. 2019, 116, 67–68. [Google Scholar] [CrossRef]
- Ghosha, A.P.; AlMamuna, A.; Lodowski, P.; Jaworska, M.; Kozlowski, P.M. Mechanism of the photo-induced activation of Co-C bond in methylcobalamin-dependent methionine synthase. J. Photochem. Photobiol. B Biol. 2018, 189, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.P.; Al Mamun, A.; Kozlowski, P.M. How does the mutation in the cap domain of methylcobalamin-dependent methionine synthase influence the photoactivation of the Co–C bond? Phys. Chem. Chem. Phys. 2019, 21, 20628–20640. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Jaworska, M.; Lodowski, P.; Kumar, M.; Kozlowski, P. Electronic structure of cofactor-substrate reactant complex involved in the methyl transfer reaction catalyzed by cobalamin-dependent methionine synthase. J. Phys. Chem. B 2011, 115, 6722–6731. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Alfonso-Prieto, M.; Rovira, C.; Lodowski, P.; Jaworska, M.; Kozlowski, P.M. Role of the axial base in the modulation of the cob(I)alamin electronic properties: Insight from QM/MM, DFT, and CASSCF calculations. J. Chem. Theory Comput. 2011, 7, 1541–1551. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Kozlowski, P.M. Mechanistic insights for formation of an organometallic Co–C bond in the methyl transfer reaction catalyzed by methionine synthase. J. Phys. Chem. B 2013, 117, 16044–16057. [Google Scholar] [CrossRef] [PubMed]
- Spataru, T.; Birke, R.L. Carbon-cobalt bond distance and bond cleavage in one-electron reduced methylcobalamin: A failure of the conventional DFT method. J. Phys. Chem. A 2006, 110, 8599–8604. [Google Scholar] [CrossRef] [PubMed]
- Spataru, T.; Fernandez, F. The nature of the Co-C bond cleavage processes in the methylcob(II)alamin and adenosylcob(III)alamin. Chem. J. Mold. 2016, 11, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.P.; Ryde, U. Conversion of homocysteine to methionine by methionine synthase: A density functional study. J. Am. Chem. Soc. 2003, 125, 13970–13971. [Google Scholar] [CrossRef]
- Alfonso-Prieto, M.; Biarnés, X.; Kumar, M.; Rovira, C.; Kozlowski, P.M. Reductive cleavage mechanism of Co−C bond in cobalamin-dependent methionine synthase. J. Phys. Chem. B 2010, 114, 12965–12971. [Google Scholar] [CrossRef]
- Brooks, A.J.; Vlasie, M.; Banerjee, R.; Brunold, T.C. Spectroscopic and computational studies on the adenosylcobalamin-dependent methylmalonyl-CoA mutase: Evaluation of enzymatic contributions to Co-C bond activation in the Co3+ ground state. J. Am. Chem. Soc. 2004, 126, 8167–8180. [Google Scholar] [CrossRef]
- Abdel-Azeim, S.; Li, X.; Chung, L.W.; Morokuma, K. Zinc–Homocysteine binding in cobalamin-dependent methionine synthase and its role in the substrate activation: DFT, ONIOM, and QM/MM molecular dynamics studies. Comput. Chem. 2011, 32, 3154–3167. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.P.; Ryde, U. How the Co-C bond is cleaved in coenzyme B12 enzymes: A theoretical study. J. Am. Chem. Soc. 2005, 127, 9117–9128. [Google Scholar] [CrossRef] [PubMed]
- Kwiecien, R.A.; Khavrutskii, I.V.; Musaev, D.G.; Morokuma, K.; Banerjee, R.; Paneth, P. Computational insights into the mechanism of radical generation in B12-dependent methylmalonyl-CoA mutase. J. Am. Chem. Soc. 2006, 128, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chung, L.W.; Paneth, P.; Morokuma, K. DFT and ONIOM (DFT:MM) studies on Co−C bond cleavage and hydrogen transfer in B12-dependent methylmalonyl-CoA mutase. stepwise or concerted mechanism? J. Am. Chem. Soc. 2009, 131, 5115–5125. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Bucher, D.; Kozlowski, P.M. Mechanistic implications of reductive Co−C bond cleavage in B12-dependent methylmalonyl CoA mutase. J. Phys. Chem. B 2019, 123, 2210–2216. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Liu, S.; Kozlowski, P.M. Charge separation propensity of the coenzyme B12–tyrosine complex in adenosylcobalamin-dependent methylmalonyl–CoA mutase enzyme. J. Phys. Chem. Lett. 2012, 3, 1035–1038. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Kozlowski, P.M. Role of tyrosine residue in the activation of Co−C bond in coenzyme B12-dependent enzymes: Another case of proton-coupled electron transfer? J. Phys. Chem. B 2009, 113, 9050–9054. [Google Scholar] [CrossRef]
- Pawel, M.K.; Kamachi, T.; Kumar, M.; Yoshizawa, K. Initial step of B12-dependent enzymatic catalysis: Energetic implications regarding involvement of the one-electron-reduced form of adenosylcobalamin. J. Biol. Inorg. Chem. 2012, 17, 293–300. [Google Scholar]
- Conrad, K.S.; Jordan, C.D.; Brown, K.L.; Brunold, T.C. Spectroscopic and computational studies of cobalamin species with variable lower axial ligation: Implications for the mechanism of Co–C bond activation by class I cobalamin-dependent isomerases. Inorg. Chem. 2015, 54, 3736–3747. [Google Scholar] [CrossRef]
- Brooks, A.J.; Fox, C.C.; Marsh, E.N.G.; Vlasie, M.; Banerjee, R.; Brunold, T.C. Electronic structure studies of the adenosylcobalamin cofactor in glutamate mutase. Biochemistry 2005, 44, 15167–15181. [Google Scholar] [CrossRef]
- Spataru, T. The complete electronic structure and mechanism of the methionine synthase process as determined by the MCSCF method. J. Organomet. Chem. 2021, 942, 181211–181221. [Google Scholar] [CrossRef]
- Spataru, T. The electronic structure and mechanism of the adenosylcobalamin-dependent bio-processes as determined by the MCSCF method. J. Med. Chem. 2021, 11, 595–606. [Google Scholar]
- Spataru, T. The First Step and the Cob(II)alamin Cofactor Inactive Particles Reactivation in the Updated Mechanism of the Methionine Synthase Process. Reactions 2023, 4, 274–285. [Google Scholar] [CrossRef]
- Spataru, T. The Co-N bond cleavage in the adenosylcobalamin cofactor in advance to Glutamete Mutase and Methylmalonyl-Co-A Mutase processes. Chem. J. Mold. 2023, 18, 96–104. [Google Scholar] [CrossRef]
- Li, Y.N.; Gulati, S.; Baker, P.J.; Brody, L.C.; Banerjee, R.; Kruger, W.D. Cloning, mapping and RNA analysis of the human methionine synthase gene. Hum. Mol. Genet. 1996, 5, 1851–1858. [Google Scholar] [CrossRef]
- Marmion, C.J. Interrelations Between Essential Metal Ions and Human Diseases. In Metal Ions in Life Sciences; Sigel, A., Sigel, H., Sigel, R.K.O., Eds.; Springer: Dordrecht, The Netherland, 2013; Volume 13, pp. 295–320. [Google Scholar]
- Wald, I.; Członkowska, A.; Dowżenko, A. Clinical Neurology; National Institute of Medical Publications: Warsaw, Poland, 1987; p. 451. (in Polish) [Google Scholar]
- Morris, M.C.; Evans, D.A.; Schneider, J.A.; Tangney, C.C.; Bienias, J.L.; Aggarwal, N.T. Dietary folate and vitamins B12 and B6 not associated with incident alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 435–443. [Google Scholar] [CrossRef]
- Siuda, J.; Gorzkowska, A.; Patalong-Ogiewa, M.; Krzystanek, E.; Czech, E.; Wiechuła, B.; Garczorz, W.; Danch, A.; Jasińska-Myga, B.; Opala, G. From mild cognitive impairment to alzheimer’s disease—Influence of homocysteine, vitamin B12 and folate on cognition over time: Results from one-year follow-up. Pol. J. Neurol. Neurosurg. 2009, 43, 321–329. (In Polish) [Google Scholar]
- Kivipelto, M.; Annerbo, S.; Hultdin, J.; Bäckman, L.; Viitanen, M.; Fratiglioni, L.; Lökk, J. Homocysteine and holo-transcobalamin and the risk of dementia and alzheimer’s disease: A prospective study. Eur. J. Neurol. 2009, 16, 808–813. [Google Scholar] [CrossRef]
- Kageyama, M.; Hiraoka, M.; Kagawa, Y. Relationship between genetic polymorphism, serum folate and homocysteine in Alzheimer’s disease. Asia-Pac. J. Public Health 2008, 20, 111–117. [Google Scholar]
- Prodan, C.I.; Cowan, L.D.; Stoner, J.A.; Ross, E.D. Cumulative incidence of vitamin B12 deficiency in patients with Alzheimer’s disease. J. Neurol. Sci. 2009, 284, 144–148. [Google Scholar] [CrossRef]
- Neil, E.; Marsh, G.; Patterson, D.P.; Lei, L. Adenosyl radical: Reagent and catalyst in enzyme reactions. ChemBioChem 2010, 11, 604–621. [Google Scholar]
- Martin, B.D.; Finke, R.G. Methylcobalamin’s full- vs. half-strength cobalt-carbon sigma bonds and bond dissociation enthalpies: A>1015 Co-CH3 homolysis rate enhancement following one-antibonding-electron reduction of methylcobalamin. J. Am. Chem. Soc. 1992, 114, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, H.; Vogler, A. Photolysis of methylcobalamin. nature of the reactive excited state. J. Organomet. Chem. 1993, 453, 269–272. [Google Scholar] [CrossRef]
- Walker LA, I.I.; Jarrett, J.T.; Anderson, N.A.; Pullen, S.H.; Matthews, R.G.; Sension, R.J. Time-resolved spectroscopic studies of B12 coenzymes: The identification of a metastable cob(III)alamin photoproduct in the photolysis of methylcobalamin. J. Am. Chem. Soc. 1998, 120, 3597–3603. [Google Scholar] [CrossRef]
- Luo, L.B.; Chen, H.L.; Fu, S.W.; Zhang, S.Y. Laser-induced photoacoustic calorimetric determination of enthalpy and volume changes in the photolysis of 5′-deoxyadenosylcobalamin and methylcobalamin. J. Chem. Soc. Dalton Trans. 1998, 12, 2103–2107. [Google Scholar] [CrossRef]
- Shiang, J.J.; Walker, L.A.; Anderson, N.A.; Cole, A.G.; Sension, R.J. The time-resolved spectroscopic studies of B12 coenzymes: The photolysis of methylcobalamin is wavelength dependent. J. Phys. Chem. B 1999, 103, 10532–10539. [Google Scholar] [CrossRef]
- Cole, A.G.; Yoder, L.M.; Shiang, J.J.; Andeson, N.A.; Walker, L.A.; Banaszak Holl, M.M.; Sension, R.J. Time-resolved spectroscopic studies of B12 coenzymes: A comparison of the primary photolysis mechanism in methyl-, ethyl-, n-propyl-, and 5′-deoxyadenosylcobalamin. J. Am. Chem. Soc. 2002, 124, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Orio, M.; Pantazis, A.D.; Neese, F. Density functional theory. Photosynth. Res. 2009, 102, 443–453. [Google Scholar] [CrossRef]
- Warshel, A.; Levitt, M. Theoretical studies of enzymatic reactions: Dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol. 1976, 103, 227–249. [Google Scholar] [CrossRef]
- Kozlowski, P.M.; Kuta, J.; Galezowski, W. Reductive cleavage mechanism of methylcobalamin: Elementary steps of Co-C bond breaking. J. Phys. Chem. B 2007, 111, 7638–7645. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, I.B. Limitations of density functional theory in application to degenerate states. J. Comput. Chem. 1997, 2, 260–267. [Google Scholar] [CrossRef]
- Öpik, U.; Pryce, M.H.L. Studies of the jahn-teller effect. I. a survey of the static problem. Proc. R. Soc. London. Ser. A Math. Phys. Sci. R. Soc. 1957, 238, 425–447. [Google Scholar]
- Bader, R.F.W. An interpretation of potential interaction constants in terms of low-lying excited states. Mol. Phys. 1960, 3, 137–151. [Google Scholar] [CrossRef]
- Fulton, R.L.; Gouterman, M. Vibronic coupling. I. mathematical treatment for two electronic states. J. Chem. Phys. 1961, 35, 1059–1071. [Google Scholar] [CrossRef]
- Bersuker, I.B. On the origin of ferroelectricity in perovskite-type crystals. Phys. Lett. 1966, 20, 589–590. [Google Scholar] [CrossRef]
- Bersuker, I.B.; Stavrov, S.S. Structure and properties of metalloporphyrins and hemoproteins: The vibronic approach. Coord. Chem. Rev. 1988, 88, 1–68. [Google Scholar] [CrossRef]
- Bersuker, I.B.; Polinger, V.Z. Vibronic Interactions in Molecules and Crystals; Springer Series in Chemical Physics; Springer: Berlin/Heidelberg, Germany, 1989; p. 49. [Google Scholar]
- Bersuker Issak, B. The Jahn–Teller Effect; Cambridge University Press: Cambridge, UK, 2006. [Google Scholar]
- Bearpark, M.J.; Blancafort, L.; Robb, M.A. The pseudo-jahn—Teller effect: A CASSCF diagnostic. Mol. Phys. 2002, 100, 1735–1739. [Google Scholar] [CrossRef]
- Ouyang, L.; Rulis, P.; Ching, W.Y.; Nardin, G.; Randaccio, L. Accurate redetermination of the X-ray structure and electronic bonding in adenosylcobalamin. Inorg. Chem. 2004, 43, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Gerzberg, G.; Monfils, A. The dissociation energies of the H2, HD, and D2 molecules. J. Mol. Spectrosc. 1961, 5, 482–498. [Google Scholar] [CrossRef]
- Randaccio, L.; Furlan, M.; Geremia, S.; Šlouf, M.; Srnova, I.; Toffoli, D. Similarities and differences between cobalamins and cobaloximes. accurate structural determination of methylcobalamin and LiCl- and KCl-containing cyanocobalamins by synchrotron radiation. Inorg. Chem. 2000, 39, 3403–3413. [Google Scholar] [CrossRef]
- Shaik, S.; Cohen, S.; Wang, Y.; Chen, H.; Kumar, D.; Thiel, W. P450 enzymes: Their structure, reactivity, and selectivity modeled by QM/MM calculations. Chem. Rev. 2010, 110, 949–1017. [Google Scholar] [CrossRef] [PubMed]
- Norton, J.R.; Spataru, T.; Camaioni, D.M.; Lee, S.-J.; Li, G.; Choi, J.; Franz, J.A. Kinetics and mechanism of the hydrogenation of CpCr(CO)3•/[CpCr(CO)3]2 equilibrium to CpCr(CO)3H. Organometallics 2014, 33, 2496–2502. [Google Scholar] [CrossRef]
- Han, A.; Spataru, T.; Hartung, J.; Li, G.; Norton, J.R. Effect of double-bond substituents on the rate of cyclization of α-carbomethoxyhex-5-enyl, radicals. J. Org. Chem. 2014, 79, 1938–1946. [Google Scholar] [CrossRef] [PubMed]
- Kohen, A.; Pklinman, J. Hydrogen tunneling in biology. Chem. Biol. 1999, 6, R191–R198. [Google Scholar] [CrossRef] [PubMed]
- Lerner, T. Encyclopedia of Physics, 2nd ed.; VCH: New York, NY, USA, 1991; pp. 1308–1408. [Google Scholar]
- Gorinchoy, N.; Balan, I.; Polinger, V.; Bersuker, I. Pseudo jahn-teller origin of the proton-transfer energy barrier in the hydrogen-bonded [FHF]-system. Chem. J. Moldova. Gen. Ind. Ecol. Chem. 2021, 16, 115–120. [Google Scholar] [CrossRef]
- Shibata, N.; Tamagaki, H.; Hieda, N.; Akita, K.; Komori, H.; Shomura, Y.; Terawaki, S.; Mori, K.; Noritake Yasuoka, N.; Higuchi, Y.; et al. Crystal structures of ethanolamine ammonia-lyase complexed with coenzyme B12 analogs and substrates. J. Biol. Chem. 2010, 285, 26484–26493. [Google Scholar] [CrossRef]
- Dobrzyńska, E.; Pośniak, M.; Szewczyńska, M.; Buszewski, B. Chlorinated Volatile Organic Compounds Old, However, Actual Analytical and Toxicological Problem. Crit. Rev. Anal. Chem. 2010, 40, 41–57. [Google Scholar] [CrossRef]
- Lee, M.; Wells, E.; Wong, Y.K.; Koenig, J.; Adrian, L.; Richnow, H.H.; Manefield, M. Relative Contributions of Dehalobacter and Zerovalent Iron in the Degradation of Chlorinated Methanes. Environ. Sci. Technol. 2015, 49, 4481–4489. [Google Scholar] [CrossRef]
- Torrentó, C.; Palau, J.; Rodríguez-Fernández, D.; Heckel, B.; Meyer, A.; Domènech, C.; Rosell, M.; Soler, A.; Elsner, M.; Hunkeler, D. Carbon and Chlorine Isotope Fractionation Patterns Associated with Different Engineered Chloroform Transformation Reactions. Environ. Sci. Technol. 2017, 51, 6174–6184. [Google Scholar] [CrossRef]
- Palau, J.; Shouakar-Stash, O.; Hatijah Mortan, S.; Yu, R.; Rosell, M.; Marco-Urrea, E.; Freedman, D.L.; Aravena, R.; Soler, A.; Hunkeler, D. Hydrogen Isotope Fractionation during the Biodegradation of 1,2-Dichloroethane: Potential for Pathway Identification Using a Multielement (C, Cl, and H) Isotope Approach. Environ. Sci. Technol. 2017, 51, 10526–10535. [Google Scholar] [CrossRef]
- Carter, J.M.; Lapham, W.W.; Zogorski, J.S. Occurrence of Volatile Organic Compounds in Aquifers of the United States1. J. Am. Water Resour. Assoc. 2008, 44, 399–416. [Google Scholar] [CrossRef]
- Thakur, M.; Pathania, D. Chapter 12—Environmental fate of organic pollutants and effect on human health. In Abatement of Environmental Pollutants Trends and Strategies; Elsveier: Amsterdam, The Netherlands, 2020; pp. 245–262. [Google Scholar]
- Lacrampe-Couloume, G.; Edwards, E.A.; Sherwood Lollar, B. Large Carbon Isotope Fractionation during Biodegradation of Chloroform by Dehalobacter Cultures. Environ. Sci. Technol. 2012, 46, 10154–10160. [Google Scholar]
- Palau, J.; Shouakar-Stash, O.; Hunkeler, D. Carbon and Chlorine Isotope Analysis to Identify Abiotic Degradation Pathways of 1,1,1-Trichloroethane. Environ. Sci. Technol. 2014, 48, 14400–14408. [Google Scholar] [CrossRef]
- Hartenbach, A.; Hofstetter, T.B.; Berg, M.; Bolotin, J.; Schwarzenbach, R.P. Using Nitrogen Isotope Fractionation To Assess Abiotic Reduction of Nitroaromatic Compounds. Environ. Sci. Technol. 2006, 40, 7710–7716. [Google Scholar] [CrossRef]
- Nijenhuis, I.; Richnow, H.H. Stable isotope fractionation concepts for characterizing biotransformation of organohalides. Curr.Opin. Biotechnol. 2016, 41, 108–113. [Google Scholar] [CrossRef]
- Chen, G.; Shouakar-Stash, O.; Phillips, E.; Justicia-Leon, S.D.; Gilevska, T.; Sherwood Lollar, B.; Mack, E.E.; Seger, E.S.; Löffler, F.E. Dual Carbon-Chlorine Isotope Analysis Indicates Distinct Anaerobic Dichloromethane Degradation Pathways in Two Members of Peptococcaceae. Environ. Sci. Technol. 2018, 52, 8607–8616. [Google Scholar] [CrossRef]
- Zwank, L.; Elsner, M.; Aeberhard, A.; Schwarzenbach, R.P.; Haderlein, S.B. Carbon isotope fractionation in the reductive dehalogenation of carbon tetrachloride at iron (hydro)oxide and iron sulfide minerals. Environ. Sci. Technol. 2005, 39, 5634–5641. [Google Scholar] [CrossRef]
- Rodríguez-Fernández, D.; Torrentó, C.; Guivernau, M.; Viñas, M.; Hunkeler, D.; Soler, A.; Domènech, C.; Rosell, M. Vitamin B12 effects on chlorinated methanes-degrading microcosms: Dual isotope and metabolically active microbial populations assessment. Sci. Total Environ. 2018, 621, 1615–1625. [Google Scholar] [CrossRef]
- Spataru, T.; Fernandez, F.; Sista, J.; Spataru, P.; Povar, I. Disposal of Poisonous Organic Halides by Using the Electrochemical Method: DFT Simulation. Chem. J. Mold. 2016, 11, 93–98. [Google Scholar] [CrossRef]
- Lapeyrouse, N.; Liu, M.; Zou, S.; Booth, G.; Yestrebsky, C.L. Remediation of Chlorinated Alkanes by Vitamin B12 and Zero-Valent Iron. J. Chem. 2019, 2019, 7565464. [Google Scholar] [CrossRef]
- Heckel, B.; Elsner, M. Exploring Mechanisms of Biotic Chlorinated Alkane Reduction: Evidence of Nucleophilic Substitution (SN2) with Vitamin B12. Environ. Sci. Technol. 2022, 56, 6325–6336. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Lorenz, A.; Schüürmann, G. Interaction Mode and Regioselectivity in Vitamin B12-Dependent Dehalogenation of Aryl Halides by Dehalococcoides mccartyi Strain CBDB1. Environ. Sci. Technol. 2018, 52, 1834–1843. [Google Scholar] [CrossRef] [PubMed]
- Heckel, B.; McNeill, K.; Elsner, M. Chlorinated Ethene Reactivity with Vitamin B12 Is Governed by Cobalamin Chloroethylcarbanions as Crossroads of Competing Pathways. ACS Catal. 2018, 8, 3054–3066. [Google Scholar] [CrossRef]
- Payne, K.P.A.P.; Quezada, C.P.; Fisher, K.; Dunstan, M.S.; Collins, F.A.; Sjuts, H. Reductive dehalogenase structure suggests a mechanism for B12-dependent dehalogenation. Nature 2015, 517, 513–516. [Google Scholar] [CrossRef] [PubMed]
- Löffler, F.E.; Ritalahti, K.M.; Zinder, S.H. Dehalococcoides and reductive dechlorination of chlorinated solvents. In Bioaugmentation for Groundwater Remediation; Stroo, H.F., Leeson, A., Ward, C.H., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 39–88. [Google Scholar]
- Hendrickson, E.R.; Payne, J.A.; Young, R.M.; Starr, M.G.; Perry, M.P.; Fahnestock, S.; Ellis, D.E.; Ebersole, R.C. Molecular analysis of Dehalococcoides 16S ribosomal DNA from chloroethene-contaminated sites throughout North America and Europe. Appl. Environ. Microbiol. 2002, 68, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.A.; Rosner, B.M.; Von Abendroth, G.; Meshulam-Simon, G.; McCarty, P.L.; Spormann, A.M. Molecular identification of the catabolic vinyl chloride reductase from Dehalococcoides sp. strain VSand its environmental distribution. Appl. Environ. Microbiol. 2004, 70, 4880–4888. [Google Scholar] [CrossRef] [PubMed]
- Villemur, R. The pentachlorophenol-dehalogenating Desulf itobacterium haf niense strain PCP-1. Philos. Trans. R. Soc. B 2013, 368, 20120319. [Google Scholar] [CrossRef]
- Leys, D.; Adrian, L.; Smidt, H. Organohalide respiration: Microbes breathing chlorinated molecules. Philos. Trans. R. Soc. B 2013, 368, 20120316. [Google Scholar] [CrossRef]
- Hug, L.A.; Maphosa, F.; Leys, D.; Löffler, F.E.; Smidt, H.; Edwards, E.A.; Adrian, L. Overview of organohalide-respiring bacteria and a proposal for a classification system for reductive dehalogenases. Philos. Trans. R. Soc. B 2013, 368, 20120322. [Google Scholar] [CrossRef]
- Schipp, C.J.; Marco-Urrea, E.; Kublik, A.; Seifert, J.; Adrian, L. Organic cofactors in the metabolism of Dehalococcoides mccartyi strains. Philos. Trans. R. Soc. B 2013, 368, 20120321. [Google Scholar] [CrossRef]
- Cooper, M.; Wagner, A.; Wondrousch, D.; Sonntag, F.; Sonnabend, A.; Brehm, M.; Schüürmann, G.; Adrian, L. Anaerobic microbial transformation of halogenated aromatics and fate prediction using electron density modeling. Environ. Sci. Technol. 2015, 49, 6018–6028. [Google Scholar] [CrossRef]
- Zhang, S.; Wondrousch, D.; Cooper, M.; Zinder, S.H.; Schüürmann, G.; Adrian, L. Anaerobic Dehalogenation of Chloroanilines by Dehalococcoides mccartyi Strain CBDB1 and Dehalobacter Strain 14DCB1 via Different Pathways as Related to Molecular Electronic Structure. Environ. Sci. Technol. 2017, 51, 3714–3724. [Google Scholar] [CrossRef]
- Spataru, T.; Dumitrescu, T.; Moraru, M.; Fernandez, F.; Spataru, P. The Mechanism of the Toxic Organic Halides Disposal under the Catalytic Influence of the Vitamin B12. J. Toxicol. Risk Assess. 2023, 9, 1–9. [Google Scholar]








































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spataru, T. The Miracle of Vitamin B12 Biochemistry. Reactions 2024, 5, 20-76. https://doi.org/10.3390/reactions5010002
Spataru T. The Miracle of Vitamin B12 Biochemistry. Reactions. 2024; 5(1):20-76. https://doi.org/10.3390/reactions5010002
Chicago/Turabian StyleSpataru, Tudor. 2024. "The Miracle of Vitamin B12 Biochemistry" Reactions 5, no. 1: 20-76. https://doi.org/10.3390/reactions5010002
APA StyleSpataru, T. (2024). The Miracle of Vitamin B12 Biochemistry. Reactions, 5(1), 20-76. https://doi.org/10.3390/reactions5010002