

The First Step and the Cob(II)alamin Cofactor Inactive Particles Reactivation in the Updated Mechanism of the Methionine Synthase Process

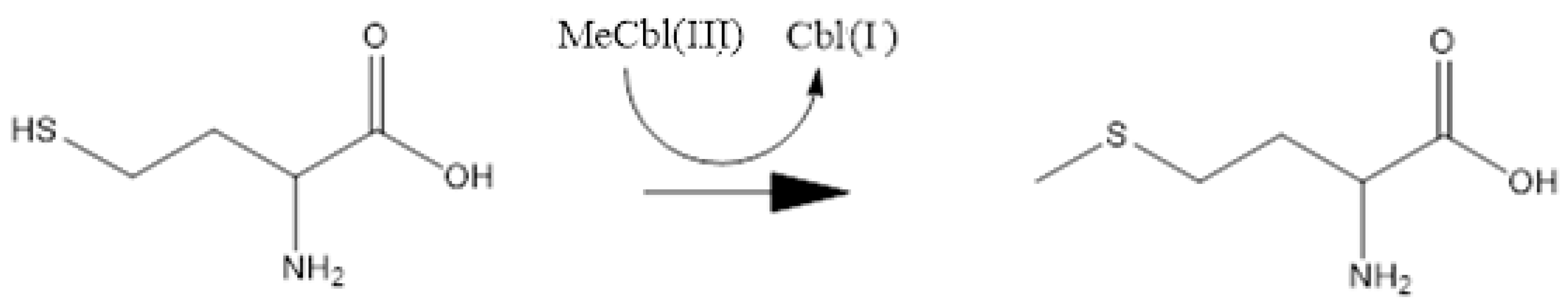{kind=link}
{kind=link}
{kind=link}
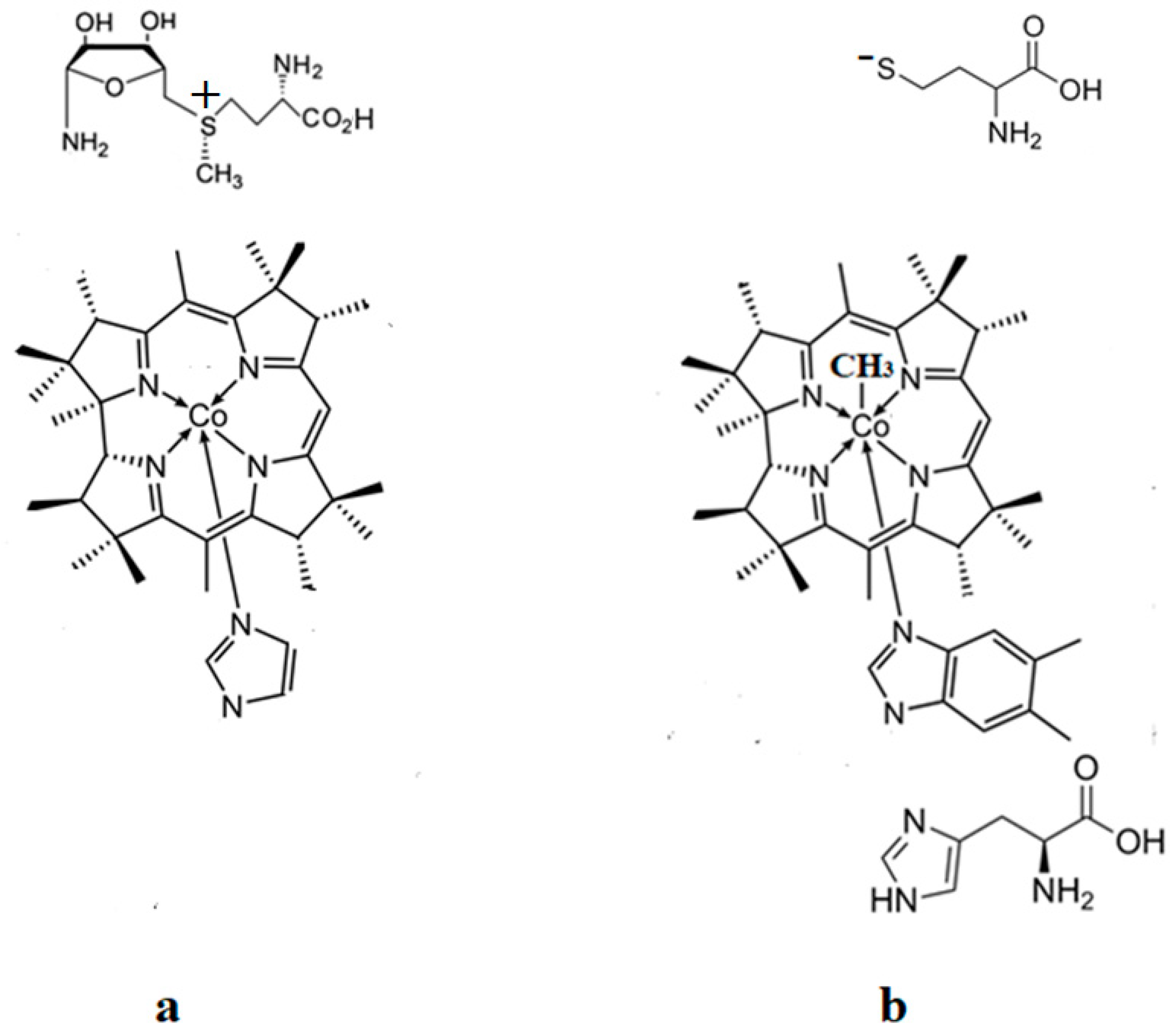{kind=link}
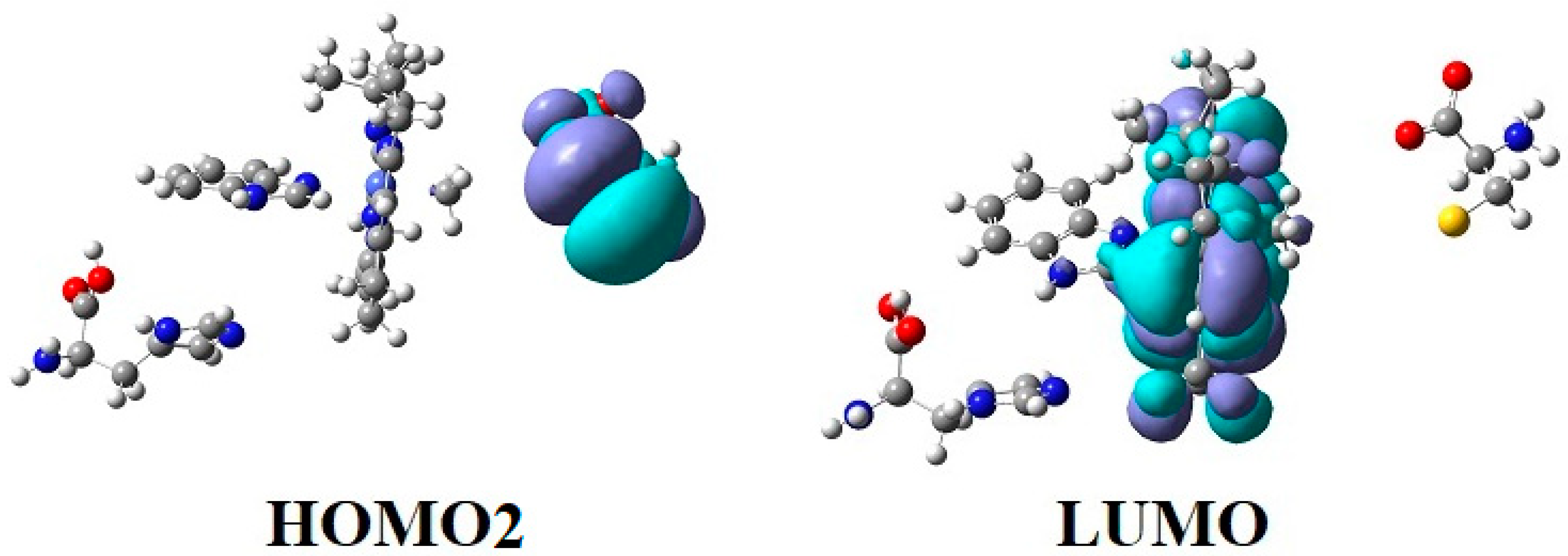{kind=link}
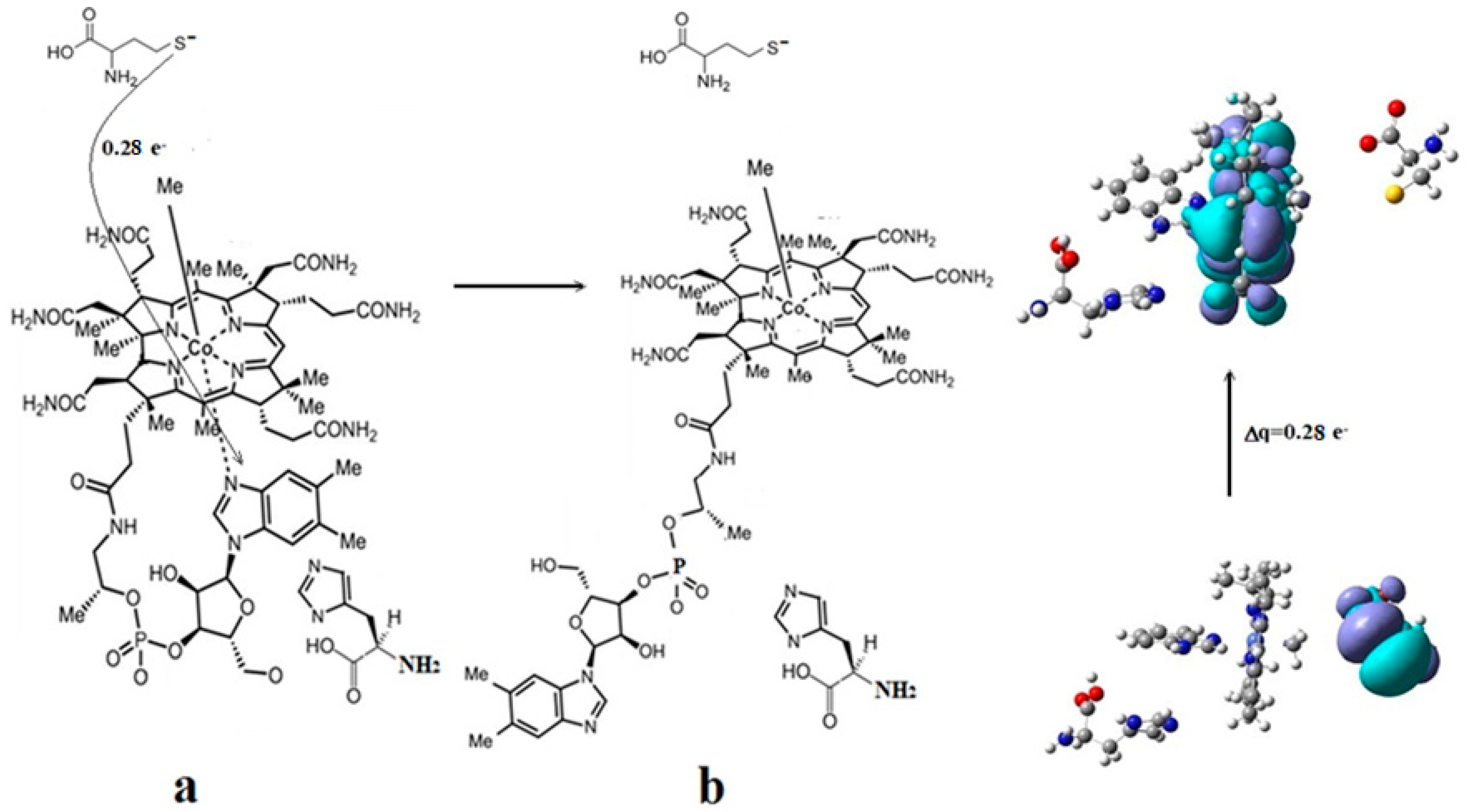{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Computational Details
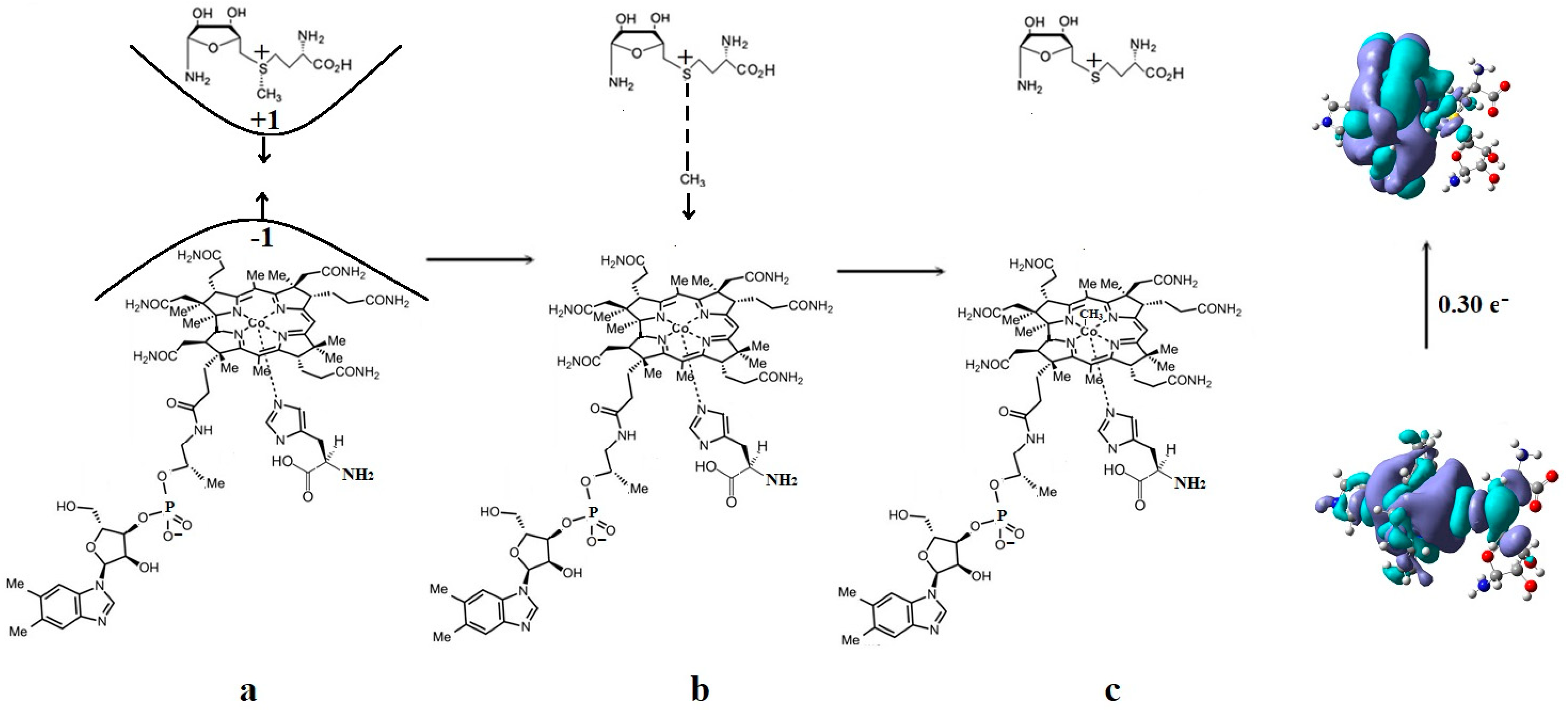
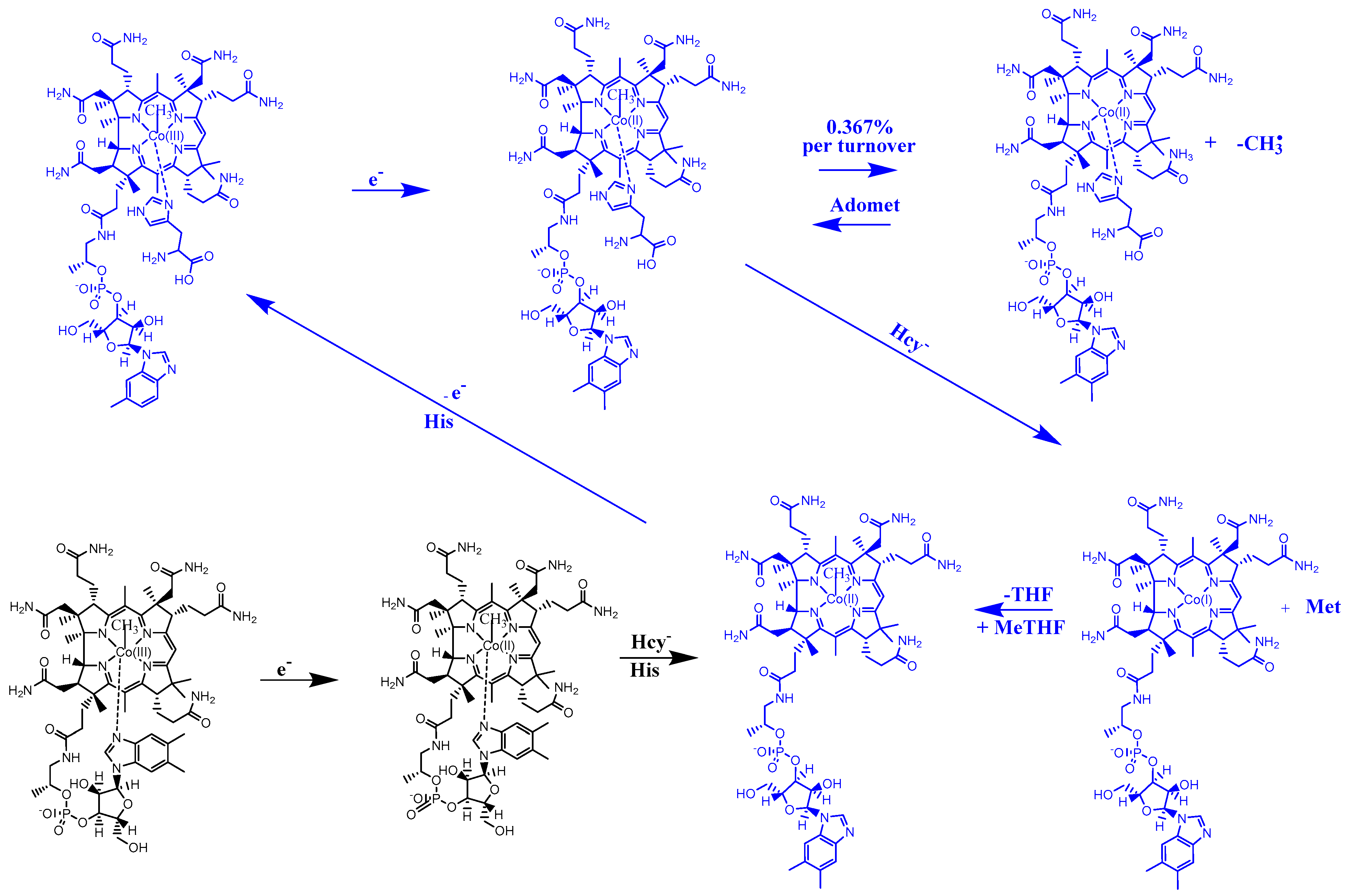
3. Results and Discussion
4. Conclusions
Supplementary Materials
Funding
Data Availability Statement
Conflicts of Interest
References
- Pratt, J.M. The roles of Co, corrin, and protein. I. Co-ligand bonding and the trans effect. In Chemistry and Biochemistry of B12; Banerjee, R., Ed.; John Wiley & Sons: New York, NY, USA, 1999; pp. 73–112. [Google Scholar]
- Matthews, R.G. Cobalamin-dependent methionine synthase. In Chemistry and Biochemistry of B12; Banerjee, R., Ed.; John Wiley & Sons: New York, NY, USA, 1999; pp. 681–706. [Google Scholar]
- Banerjee, R.V.; Matthews, R.G. Cobalamin-dependent methionine synthase. FASEB J. 1990, 4, 1450–1459. [Google Scholar] [CrossRef]
- Matthews, R.G. Cobalamin-Dependent Methyltransferases. Acc. Chem. Res. 2001, 34, 681–689. [Google Scholar] [CrossRef]
- Matthews, R.G.; Koutmos, M.; Datta, S. Cobalamin-dependent and cobamidedependent methyltransferases. Curr. Opin. Struct. Biol. 2008, 18, 658–666. [Google Scholar] [CrossRef]
- Drennan, C.L.; Huang, S.; Drummond, J.T.; Matthews, R.G.; Ludwig, M.L. How a protein binds B12: A 3.0 Å x-ray structure of B12-binding domains of methionine synthase. Science 1994, 266, 1669–1674. [Google Scholar] [CrossRef]
- Mancia, F.; Keep, N.M.; Nakagawa, A.; Leadlay, P.F.; McSweeney, S.; Rasmussen, B.; Bosecke, P.; Diat, O.; Evans, P.F. How coenzyme B12 radicals are generated: The crystal structure of methylmalonyl-coenzyme A mutase at 2 Å resolution. Structure 1996, 4, 339–350. [Google Scholar] [CrossRef]
- Koutmos, M.; Datta, S.; Pattridge, K.A.; Smith, J.L.; Matthews, R.G. Insights into the reactivation of cobalamin-dependent methionine synthase. Proc. Natl. Acad. Sci. USA 2009, 106, 18527–18532. [Google Scholar] [CrossRef]
- Hagemeier, C.H.; Kruer, M.; Rudolf, K.; Thauer, R.K.; Eberhard, W.; Ermler, U. Insight into the mechanism of biological methanol activation based on the crystal structure of the methanol-cobalamin methyltransferase complex. Proc. Natl. Acad. Sci. USA 2006, 103, 18917–18922. [Google Scholar] [CrossRef]
- Reitzer, R.; Gruber, K.; Jogl, G.; Wagner, U.G.; Bothe, H.; Buckel, W.; Kratky, C. Glutamate mutase from Clostridium cochlearium: The structure of a coenzyme B12-dependent enzyme provides new mechanistic insights. Structure 1999, 7, 891–902. [Google Scholar] [CrossRef]
- Jensen, K.P.; Ryde, U. Conversion of Homocysteine to Methionine by Methionine Synthase: A Density Functional Study. J. Am. Chem. Soc. 2003, 125, 13970–13971. [Google Scholar] [CrossRef]
- Kozlowski, P.M.; Kuta, J.; Galezowski, W. Reductive Cleavage Mechanism of Methylcobalamin: Elementary Steps of Co-C Bond Breaking. J. Phys. Chem. B 2007, 111, 7638–7645. [Google Scholar] [CrossRef]
- Kozlowski, P.M.; Kamachi, T. Reductive elimination pathway for homocysteine to methionine conversion in cobalamin-dependent methionine synthase. J. Biol. Inorg. Chem. 2012, 17, 611–619. [Google Scholar] [CrossRef]
- Alfonso-Prieto, M.; Biarnes, X.; Kumar, M.; Rovira, C.; Kozlowski, P.M. Reductive Cleavage Mechanism of Co-C Bond in Cobalamin-Dependent Methionine Synthase. J. Phys. Chem. B 2010, 114, 12965–12971. [Google Scholar] [CrossRef]
- Spataru, T.; Birke, R.L. Carbon-Cobalt Bond Distance and Bond Cleavage in OneElectron Reduced Methylcobalamin: A Failure of the Conventional DFT Method. J. Phys. Chem. A 2006, 110, 8599–8604. [Google Scholar] [CrossRef]
- Spataru, T.; Fernandez, F. The nature of the Co-C bond cleavage processes in the methylcob(II)alamin and adenosylcob(III)alamin. Chem. J. Mold. 2016, 11, 10–20. [Google Scholar] [CrossRef]
- Birke, R.L.; Huang, Q.; Spataru, T.; Gosser, D.K., Jr. Electroreduction of an of Alkylcobalamins: Mechanism of Stepwise Reductive Cleavage of the Co-C Bond. J. Am. Chem. Soc. 2006, 128, 1922–1936. [Google Scholar] [CrossRef]
- Spataru, T.; Birke, R.L. The effect of solvent on the electrode process of methylcobalamin as studied by cyclic voltammetry. J. Electroanal. Chem. 2006, 593, 74–86. [Google Scholar] [CrossRef]
- Lexa, D.; Savéant, J.-M. Electrochemistry of vitamin B12. 3. One-electron intermediates in the reduction of methylcobalamin and methylcobinamide. J. Am. Chem. Soc. 1978, 100, 3220–3222. [Google Scholar] [CrossRef]
- Bersuker, I.B. Limitations of Density Functional Theory in Application to the Degenerate States. J. Comp. Chem. 1997, 2, 260–267. [Google Scholar] [CrossRef]
- Chen, S.-L.; Blomberg, M.R.A.; Siegbahn, P.E.M. How Is a Co-Methyl Intermediate Formed in the Reaction of Cobalamin-Dependent Methionine Synthase? Theoretical Evidence for a Two-Step Methyl Cation Transfer Mechanism. J. Phys. Chem. B 2011, 115, 4066–4077. [Google Scholar] [CrossRef]
- Spataru, T. The complete electronic structure and mechanism of the methionine synthase process as determined by the MCSCF method. J. Organomet. Chem. 2021, 942, 121811. [Google Scholar] [CrossRef]
- James, T.; Drummond, J.T.; Sha, H.; Blumenthal, R.M.; Matthews, R.G. Assignment of Enzymatic Function to Specific Protein Regions of Cobalamin-Dependent Methionine Synthase from Escherichia coli. Biochemistry 1993, 32, 9290–9295. [Google Scholar]
- Spataru, T. The Electronic Structure and Mechanism of the AdenosylcobalaminDependent Bio-processes as Determined by the MCSCF Method. J. Med. Chem. 2021, 11, 595. [Google Scholar]
- Valiev, M.; Bylaska, E.J.; Govind, N.; Kowalski, K.; Straatsma, T.P.; van Dam, H.J.J.; Wang, D.; Nieplocha, D.; Apra, E.; Windus, T.L.; et al. “NwChem”: A comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 2010, 181, 1477. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spataru, T. The First Step and the Cob(II)alamin Cofactor Inactive Particles Reactivation in the Updated Mechanism of the Methionine Synthase Process. Reactions 2023, 4, 274-285. https://doi.org/10.3390/reactions4020016
Spataru T. The First Step and the Cob(II)alamin Cofactor Inactive Particles Reactivation in the Updated Mechanism of the Methionine Synthase Process. Reactions. 2023; 4(2):274-285. https://doi.org/10.3390/reactions4020016
Chicago/Turabian StyleSpataru, Tudor. 2023. "The First Step and the Cob(II)alamin Cofactor Inactive Particles Reactivation in the Updated Mechanism of the Methionine Synthase Process" Reactions 4, no. 2: 274-285. https://doi.org/10.3390/reactions4020016
APA StyleSpataru, T. (2023). The First Step and the Cob(II)alamin Cofactor Inactive Particles Reactivation in the Updated Mechanism of the Methionine Synthase Process. Reactions, 4(2), 274-285. https://doi.org/10.3390/reactions4020016