
The Role of TGF-β, Activin and Follistatin in Inflammatory Bowel Disease


Abstract
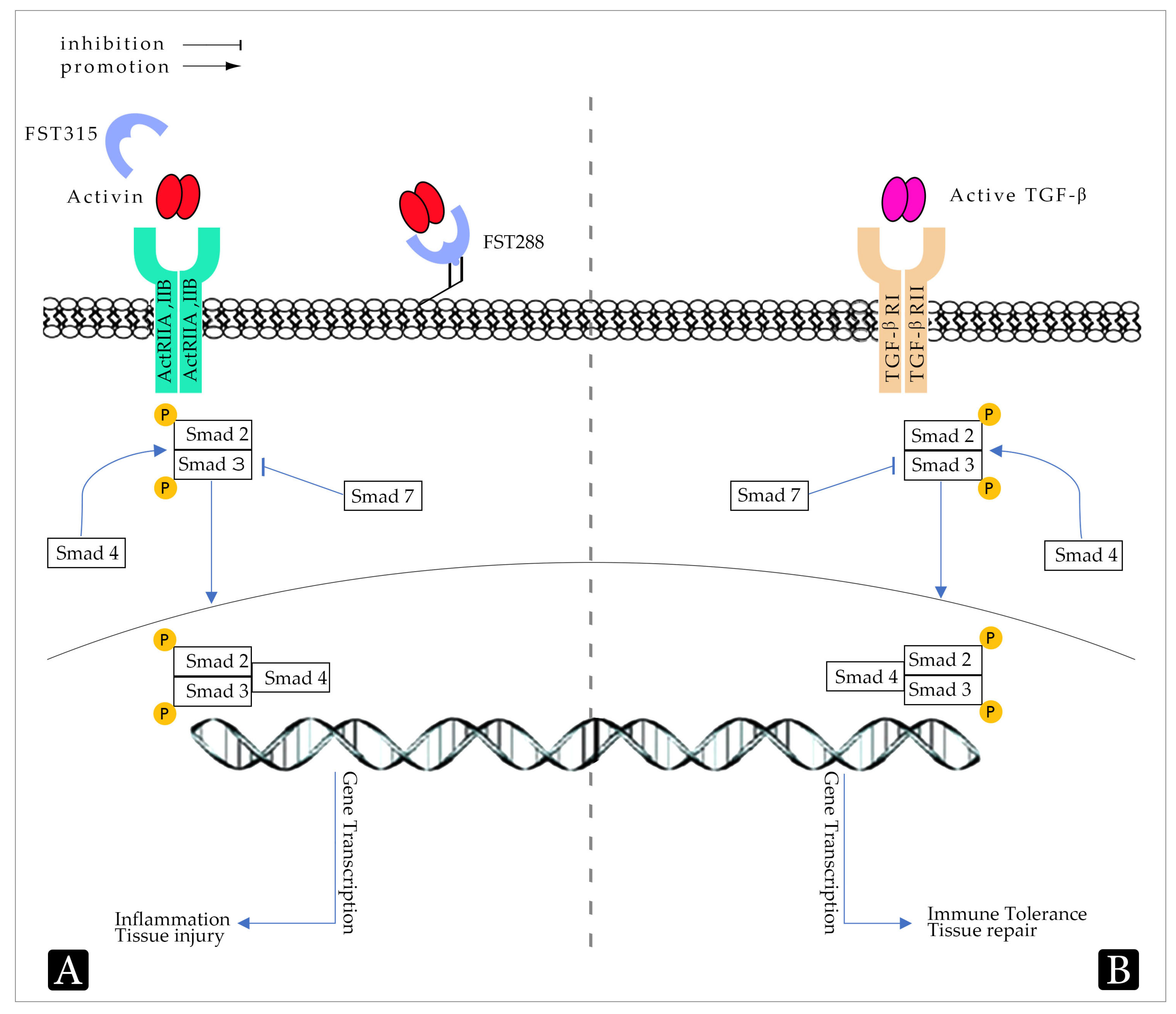
1. Introduction
2. TGF-β
2.1. General Aspects of TGF-β
2.2. Biological Roles of TGF-β
2.3. Role of Transforming Growth Factor-β in IBD
2.4. Therapeutic Approaches Related to TGF-β

{kind=link}
{kind=link}
| Product | Route of Administration | Effect | Model | Ref. |
|---|---|---|---|---|
| TGF-β/AΤRA-loaded microspheres | Gavage | Dose-dependent effects. Encapsulated TGF-β prevented weight loss, reduced average disease score, SAA levels, colon weight-to-length ratio and histological score. Encapsulated TGF-β/ATRA reduced gut inflammation, slowed disease progression and prolonged survival with no increased lung and intestinal fibrosis. Significant expression of Foxp3 in colonic lamina propria CD4+ CD25+ T-cells. | Mouse CD4 + CD25- T cell transfer | [56] |
| TreXTAM® (a combination of microencapsulated (ATRA), and encapsulated TGF-β (TPX6001) | Gavage | Significant reduction in serum and tissue TGF-β levels. TPX6001 only reduced weight loss transiently. TreXTAM or TPX6001 was safe and well tolerated at the highest doses. | Rat, Mouse CD4 + CD25-T cell transfer | [57] |
| TGF-β2 | enteral supplementation | Protection against NEC-like intestinal injury | transgenic mice with defect in TGF-β signalling platelet-activating factor and LPS induced | [58] |
| TGF-β1 | Gavage | Activation of Smad2 in intestinal epithelium—decrease in NEC severity and incidence. Inhibition of NF-kB activation, maintainance of Ikβα expression, reduction of proinflammatory cytokine production (IL-6 and IFN-γ) | NEC Neonatal Rat | [59] |
| TGF-β2 | Supplemented in diet | Reversed intestinal damage, stimulated intestinal recovery, enhanced cell proliferation and inhibition of apoptosis (up-regulation of BCL-2 and downregulation of BAx expression). Significant increase in bowel and mucosal weight, DNA and protein content, pERK, IL-1β and b-catenin protein levels in intestinal mucosa. | Rat methotrexate-induced | [60] |
| Modulen | Enteral diet | More weight gain, no diarrhoea or prolapse, lower pathological scores, SSA and TNFα; higher hematocrit. | Mouse IL-10-/- | [4] |
| Modulen | Supplemented in diet | Protection against histologic damage, weight loss, hypoalbuminemia, acidosis and GI damage. | Rat methotrexate-induced | [61] |
| Casein based diet | Supplemented in diet | Lower inflammatory score, mucosal thickness, IFN-γ mRNA expression, leukocytosis and acute phase response. Improved diarrhoea, increased fecal dry matter, MUC2 protein expression and the muscle catabolic response. Restoration of immune homeostasis. | Rat HLA-B27 | [70] |
| Modulen IBD | Supplemented in diet | Lower clinical and inflammatory score. Lower myeloperoxidase, inhibited intestinal angiogenesis. Significant decrease in the numbers of T cells, natural killer cells, dendritic cells and significant decrease in cytokine expression. | Mouse DSS-induced | [71] |
| Product | Route of Administration | Effect | Subjects | Ref. |
|---|---|---|---|---|
| CT3211 | Polymeric formula as EEN | Mucosal healing and down regulation of mucosal pro-inflammatory cytokine mRNA. Increase in ileal TGF-β mRNA. Improved clinical remission. Decreased PCDAI and CRP. Increased weight standard deviation score. | Active CD (29 children) | [68] |
| AL110 | Polymeric formula as EEN | Increased serum albumin and weight gain. Improvement in endoscopic appearance and mucosal healing. Reduction in mucosal inflammation, Lloyd Still index, ESR and CRP. | Active CD (7 children) | [69] |
| AL004 | Polymeric formula as EEN | Clinical remission and improvement in quality-of-life scores. | Active CD (26 children) | [72] |
| Modulen | Polymeric formula as EEN | Clinical remission, inflammatory remission, decrease in endoscopic and histological scores. | Active CD (32 children) | [73] |
| Modulen IBD | Enteral nutrition as EEN | Improvement in remission of CD, height and weight z scores. Decreased inflammatory parameters (62%) of patients with UC had improved clinical remission and laboratory values | Active UC and CD (73 children) | [74] |
| Modulen IBD | Supplementary formula | Decreased ESR, CRP and PCDAI normalisation of weight and height z scores, continuing remission | Active CD (28 children) | [75] |
| Modulen IBD | Polymeric formula as EEN | Improved ESR, CRP, albumin and platelet count; improved growth; induction of remission | Active CD (27 children) | [76] |
| Modulen IBD | Polymeric formula as EEN | Improved ESR, CRP and albumin; decreased PCDAI; increased weight gain induction and maintenance of remission | Active and recently diagnosed CD (19 children) | [77] |
| Modulen IBD | Polymeric formula as EEN | Induction clinical remission; normalisation of inflammatory markers, improvements in weight/BMI Z-scores. | Active CD (114 children) | [78] |
| Modulen IBD | Systematic review of EEN | Improvements in growth and development, CRP, ESR and albumin | Active CD (147 children) | [79] |
| Modulen IBD | Polymeric formula as EEN | Improved ESR, CRP, hemoglobin, platelets and albumin; decreased PCDAI, induction and maintenance of remission, increased weight and height. | CD (106 children) | [80] |
| Modulen IBD | Polymeric formula as EEN | Clinical improvement in 69% patients. Reduced CRP and ESR. Improved nutritional parameters, anthropometric parameters, weight, and general wellbeing | Active CD (29 adults) | [14] |
| Modulen IBD | Nutritional supplement | Improved histologic parameters and significant reduction in CRP. | Active CD (38 adults) | [62] |
| Modulen IBD | Polymeric formula as EEN | Decreased postoperative morbidity. Discontinuation of steroids, decreased postoperative complications in patients at low or high risk. | preoperative CD (35 adults) | [63] |
| whey protein with TGF-β | Nutritional supplement | Increased lean body mass. | Active CD (22 adults) | [64] |
| Modulen IBD | Nutritional supplement | Improvement in all anthropometric and many nutritional and biochemical parameters. Improvement in HDL and reduction in LDL serum level. Maintenance of remission in quiescent CD. | mild to moderately active CD (32 adults) | [65] |
| Modulen IBD | Polymeric formula as EEN | Remission of severe scleritis and psoriasis in one patient | Active CD (one adult) | [67] |
3. Activins
3.1. General Aspects of Activins
3.2. Biological Activity of Activins
3.3. Inflammatory and Physiological Roles of Activins
3.4. Activins and T Helper 17 Cells
3.5. Roles of Activin in Inflammatory Bowel Disease
3.6. Activin Signalling as an Emerging Target for Therapeutic Interventions
4. Follistatin
4.1. General Aspects of Follistatin
4.2. The Role of Follistatin in Inflammation and Tissue Repair
4.3. The Role of Follistatin in Intestinal Inflammation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McQuaid, K.R. Inflammatory Bowel Disease. In Current Medical Diagnosis & Treatment; Papadakis, M.A., McPhee, S.J., Rabow, M.W., McQuaid, K.R., Eds.; McGraw-Hill Education: New York, NY, USA, 2022. [Google Scholar]
- Ledder, O.; Sonnino, M.; Birimberg-Schwartz, L.; Escher, J.C.; Russell, R.K.; Orlanski-Meyer, E.; Matar, M.; Assa, A.; Tzion, R.L.; Shteyer, E.; et al. Appraisal of the PIBD-classes Criteria: A Multicentre Validation. J. Crohn’s Colitis 2020, 14, 1672–1679. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, B.; Moraes, L.; Magnusson, M.K.; Öhman, L. Immunopathogenesis of inflammatory bowel disease and mechanisms of biological therapies. Scand. J. Gastroenterol. 2018, 53, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Oz, H.S.; Ray, M.; Chen, T.S.; McClain, C.J. Efficacy of a transforming growth factor β2 containing nutritional support formula in a murine model of inflammatory bowel disease. J. Am. Coll. Nutr. 2004, 23, 220–226. [Google Scholar] [CrossRef]
- Poniatowski, Ł.A.; Wojdasiewicz, P.; Gasik, R.; Szukiewicz, D. Transforming growth factor Beta family: Insight into the role of growth factors in regulation of fracture healing biology and potential clinical applications. Mediat. Inflamm. 2015, 2015, 137823. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β family: Context-dependent roles in cell and tissue physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef]
- de Kretser, D.M.; O’Hehir, R.E.; Hardy, C.L.; Hedger, M.P. The roles of activin A and its binding protein, follistatin, in inflammation and tissue repair. Mol. Cell. Endocrinol. 2012, 359, 101–106. [Google Scholar] [CrossRef]
- Phillips, D.J.; Jones, K.L.; Scheerlinck, J.Y.; Hedger, M.P.; de Kretser, D.M. Evidence for activin A and follistatin involvement in the systemic inflammatory response. Mol. Cell. Endocrinol. 2001, 180, 155–162. [Google Scholar] [CrossRef]
- Hedger, M.P.; de Kretser, D.M. The activins and their binding protein, follistatin—Diagnostic and therapeutic targets in inflammatory disease and fibrosis. Cytokine Growth Factor Rev. 2013, 24, 285–295. [Google Scholar] [CrossRef]
- Chang, H.; Brown, C.W.; Matzuk, M.M. Genetic analysis of the mammalian transforming growth factor-β superfamily. Endocr. Rev. 2002, 23, 787–823. [Google Scholar] [CrossRef]
- Govinden, R.; Bhoola, K. Genealogy, expression, and cellular function of transforming growth factor-β. Pharmacol. Ther. 2003, 98, 257–265. [Google Scholar] [CrossRef]
- Massagué, J.; Blain, S.W.; Lo, R.S. TGFβ signaling in growth control, cancer, and heritable disorders. Cell 2000, 103, 295–309. [Google Scholar] [CrossRef]
- Letterio, J.J.; Roberts, A.B. Regulation of immune responses by TGF-beta. Annu. Rev. Immunol. 1998, 16, 137. [Google Scholar] [CrossRef] [PubMed]
- Triantafillidis, J.K.; Stamataki, A.; Gikas, A.; Sklavaina, M.; Mylonaki, M.; Georgopoulos, F.; Mastragelis, A.; Cheracakis, P. Beneficial effect of a polymeric feed, rich in TGF-b, on adult patients with active Crohn’s disease: A pilot study. Ann. Gastroenterol. 2006, 19, 66–71. [Google Scholar]
- Zhang, Y.-Z.; Li, Y.-Y. Inflammatory bowel disease: Pathogenesis. World J. Gastroenterol. 2014, 20, 91. [Google Scholar] [CrossRef]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.; Flavell, R.A. Transforming growth factor-beta regulation of immune responses. Annu. Rev. Immunol. 2006, 24, 99. [Google Scholar] [CrossRef]
- Kubiczkova, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. TGF-β–an excellent servant but a bad master. J. Transl. Med. 2012, 10, 1–24. [Google Scholar] [CrossRef]
- Smythies, L.E.; Sellers, M.; Clements, R.H.; Mosteller-Barnum, M.; Meng, G.; Benjamin, W.H.; Orenstein, J.M.; Smith, P.D. Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J. Clin. Investig. 2005, 115, 66–75. [Google Scholar] [CrossRef]
- Shen, Y.; Zhang, C.; Chen, Y. TGF-β in inflammatory bowel diseases: A tale of the Janus-like cytokine. Crit. Rev. Eukaryot. Gene Expr. 2015, 25, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Huse, M.; Muir, T.W.; Xu, L.; Chen, Y.-G.; Kuriyan, J.; Massagué, J. The TGFβ receptor activation process: An inhibitor-to substrate-binding switch. Mol. Cell 2001, 8, 671–682. [Google Scholar] [CrossRef]
- Li, M.O.; Flavell, R.A. TGF-β: A master of all T cell trades. Cell 2008, 134, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.; Ju, W.; Heyer, J.; Wittek, B.; Haneke, T.; Knaus, P.; Kucherlapati, R.; Böttinger, E.P.; Nitschke, L.; Kneitz, B. B cell-specific deficiency for Smad2 in vivo leads to defects in TGF-β-directed IgA switching and changes in B cell fate. J. Immunol. 2006, 176, 2389–2396. [Google Scholar] [CrossRef]
- Verstockt, B.; Ferrante, M.; Vermeire, S.; Van Assche, G. New treatment options for inflammatory bowel diseases. J. Gastroenterol. 2018, 53, 585–590. [Google Scholar] [CrossRef]
- Monteleone, G.; Neurath, M.F.; Ardizzone, S.; Di Sabatino, A.; Fantini, M.C.; Castiglione, F.; Scribano, M.L.; Armuzzi, A.; Caprioli, F.; Sturniolo, G.C. Mongersen, an oral SMAD7 antisense oligonucleotide, and Crohn’s disease. N. Engl. J. Med. 2015, 372, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, T.; Wakabayashi, Y.; Sekiya, T.; Inoue, N.; Morita, R.; Ichiyama, K.; Takahashi, R.; Asakawa, M.; Muto, G.; Mori, T. Smad2 and Smad3 are redundantly essential for the TGF-β–mediated regulation of regulatory T plasticity and Th1 development. J. Immunol. 2010, 185, 842–855. [Google Scholar] [CrossRef]
- McKarns, S.C.; Schwartz, R.H.; Kaminski, N.E. Smad3 is essential for TGF-β1 to suppress IL-2 production and TCR-induced proliferation, but not IL-2-induced proliferation. J. Immunol. 2004, 172, 4275–4284. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.-G.; Li, C.; Qiao, W.; Mamura, M.; Kasperczak, B.; Anver, M.; Wolfraim, L.; Hong, S.; Mushinski, E.; Potter, M. Smad4 signalling in T cells is required for suppression of gastrointestinal cancer. Nature 2006, 441, 1015–1019. [Google Scholar] [CrossRef]
- Monteleone, G.; Kumberova, A.; Croft, N.M.; McKenzie, C.; Steer, H.W.; MacDonald, T.T. Blocking Smad7 restores TGF-β1 signaling in chronic inflammatory bowel disease. J. Clin. Investig. 2001, 108, 601–609. [Google Scholar] [CrossRef]
- Monteleone, G.; Blanco, G.D.V.; Monteleone, I.; Fina, D.; Caruso, R.; Gioia, V.; Ballerini, S.; Federici, G.; Bernardini, S.; Pallone, F. Post-transcriptional regulation of Smad7 in the gut of patients with inflammatory bowel disease. Gastroenterology 2005, 129, 1420–1429. [Google Scholar] [CrossRef]
- MacDonald, T.T.; DiSabatino, A.; Gordon, J.N. Immunopathogenesis of Crohn’s disease. J. Parenter. Enter. Nutr. 2005, 29, S118–S125. [Google Scholar] [CrossRef]
- Yoshimura, A.; Wakabayashi, Y.; Mori, T. Cellular and molecular basis for the regulation of inflammation by TGF-β. J. Biochem. 2010, 147, 781–792. [Google Scholar] [CrossRef]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.; Li, L.; Marinos, N. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, e86. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Lopes, J.E.; Chong, M.M.; Ivanov, I.I.; Min, R.; Victora, G.D.; Shen, Y.; Du, J.; Rubtsov, Y.P.; Rudensky, A.Y. TGF-β-induced Foxp3 inhibits TH17 cell differentiation by antagonizing RORγt function. Nature 2008, 453, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, M.; Almeida, C.F.; Caridade, M.; Ribot, J.C.; Duarte, J.; Agua-Doce, A.; Wollenberg, I.; Silva-Santos, B.; Graca, L. Identification of regulatory Foxp3+ invariant NKT cells induced by TGF-β. J. Immunol. 2010, 185, 2157–2163. [Google Scholar] [CrossRef]
- Kullberg, M.C.; Hay, V.; Cheever, A.W.; Mamura, M.; Sher, A.; Letterio, J.J.; Shevach, E.M.; Piccirillo, C.A. TGF-β1 production by CD4+ CD25+ regulatory T cells is not essential for suppression of intestinal inflammation. Eur. J. Immunol. 2005, 35, 2886–2895. [Google Scholar] [CrossRef]
- Sakaguchi, S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu. Rev. Immunol. 2004, 22, 531. [Google Scholar] [CrossRef]
- Goto, Y.; Panea, C.; Nakato, G.; Cebula, A.; Lee, C.; Diez, M.G.; Laufer, T.M.; Ignatowicz, L.; Ivanov, I.I. Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity 2014, 40, 594–607. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef]
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Konkel, J.E.; Maruyama, T.; Carpenter, A.C.; Xiong, Y.; Zamarron, B.F.; Hall, B.E.; Kulkarni, A.B.; Zhang, P.; Bosselut, R.; Chen, W. Control of the development of CD8αα+ intestinal intraepithelial lymphocytes by TGF-β. Nat. Immunol. 2011, 12, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Biancheri, P.; Di Sabatino, A.; Corazza, G.R.; MacDonald, T.T. Proteases and the gut barrier. Cell Tissue Res. 2013, 351, 269–280. [Google Scholar] [CrossRef]
- Ruiz, P.A.; Shkoda, A.; Kim, S.C.; Sartor, R.B.; Haller, D. IL-10 gene-deficient mice lack TGF-beta/Smad-mediated TLR2 degradation and fail to inhibit proinflammatory gene expression in intestinal epithelial cells under conditions of chronic inflammation. Ann. N. Y. Acad. Sci. 2006, 1072, 389–394. [Google Scholar] [CrossRef]
- Ding, A.; Nathan, C.; Graycar, J.; Derynck, R.; Stuehr, D.; Srimal, S. Macrophage deactivating factor and transforming growth factors-beta 1-beta 2 and-beta 3 inhibit induction of macrophage nitrogen oxide synthesis by IFN-gamma. J. Immunol. 1990, 145, 940–944. [Google Scholar] [CrossRef]
- Nelson, B.; Ralph, P.; Green, S.; Nacy, C. Differential susceptibility of activated macrophage cytotoxic effector reactions to the suppressive effects of transforming growth factor-beta 1. J. Immunol. 1991, 146, 1849–1857. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Kato, J.; Solomon, M.J.; Sherr, C.J.; Massague, J.; Roberts, J.M.; Koff, A. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 1994, 8, 9–22. [Google Scholar] [CrossRef]
- Jung, H.C.; Eckmann, L.; Yang, S.; Panja, A.; Fierer, J.; Morzycka-Wroblewska, E.; Kagnoff, M. A distinct array of proinflammatory cytokines is expressed in human colon epithelial cells in response to bacterial invasion. J. Clin. Investig. 1995, 95, 55–65. [Google Scholar] [CrossRef]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef]
- Mladenova, D.; Kohonen-Corish, M.R. Mouse models of inflammatory bowel disease-insights into the mechanisms of inflammation-associated colorectal cancer. In Vivo 2012, 26, 627–646. [Google Scholar]
- Li, M.O.; Flavell, R.A. Contextual regulation of inflammation: A duet by transforming growth factor-β and interleukin-10. Immunity 2008, 28, 468–476. [Google Scholar] [CrossRef]
- Ihara, S.; Hirata, Y.; Koike, K. TGF-β in inflammatory bowel disease: A key regulator of immune cells, epithelium, and the intestinal microbiota. J. Gastroenterol. 2017, 52, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Feagins, L.A. Role of transforming growth factor-β in inflammatory bowel disease and colitis-associated colon cancer. Inflamm. Bowel Dis. 2010, 16, 1963–1968. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, G.; Mann, J.; Monteleone, I.; Vavassori, P.; Bremner, R.; Fantini, M.; Blanco, G.D.V.; Tersigni, R.; Alessandroni, L.; Mann, D. A failure of transforming growth factor-β1 negative regulation maintains sustained NF-κB activation in gut inflammation. J. Biol. Chem. 2004, 279, 3925–3932. [Google Scholar] [CrossRef]
- Mangan, P.R.; Harrington, L.E.; O’Quinn, D.B.; Helms, W.S.; Bullard, D.C.; Elson, C.O.; Hatton, R.D.; Wahl, S.M.; Schoeb, T.R.; Weaver, C.T. Transforming growth factor-β induces development of the TH17 lineage. Nature 2006, 441, 231–234. [Google Scholar] [CrossRef]
- Di Sabatino, A.; Pickard, K.M.; Rampton, D.; Kruidenier, L.; Rovedatti, L.; Leakey, N.A.; Corazza, G.R.; Monteleone, G.; MacDonald, T.T. Blockade of transforming growth factor β upregulates T-box transcription factor T-bet, and increases T helper cell type 1 cytokine and matrix metalloproteinase-3 production in the human gut mucosa. Gut 2008, 57, 605–612. [Google Scholar] [CrossRef]
- Conway, T.F.; Hammer, L.; Furtado, S.; Mathiowitz, E.; Nicoletti, F.; Mangano, K.; Egilmez, N.K.; Auci, D.L. Oral delivery of particulate transforming growth factor beta 1 and all-trans retinoic acid reduces gut inflammation in murine models of inflammatory bowel disease. J. Crohn’s Colitis 2015, 9, 647–658. [Google Scholar] [CrossRef]
- Hammer, L.; Furtado, S.; Mathiowitz, E.; Auci, D.L. Oral encapsulated transforming growth factor β1 reduces endogenous levels: Effect on inflammatory bowel disease. World J. Gastrointest. Pharmacol. Ther. 2020, 11, 79. [Google Scholar] [CrossRef]
- Maheshwari, A.; Kelly, D.R.; Nicola, T.; Ambalavanan, N.; Jain, S.K.; Murphy–Ullrich, J.; Athar, M.; Shimamura, M.; Bhandari, V.; Aprahamian, C. TGF-β2 suppresses macrophage cytokine production and mucosal inflammatory responses in the developing intestine. Gastroenterology 2011, 140, 242–253. [Google Scholar] [CrossRef]
- Shiou, S.-R.; Yu, Y.; Guo, Y.; Westerhoff, M.; Lu, L.; Petrof, E.O.; Sun, J.; Claud, E.C. Oral administration of transforming growth factor-β1 (TGF-β1) protects the immature gut from injury via Smad protein-dependent suppression of epithelial nuclear factor κB (NF-κB) signaling and proinflammatory cytokine production. J. Biol. Chem. 2013, 288, 34757–34766. [Google Scholar] [CrossRef] [PubMed]
- Ben-Lulu, S.; Pollak, Y.; Mogilner, J.; Bejar, J.; Coran, A.G.; Sukhotnik, I. Dietary transforming growth factor-beta 2 (TGF-β2) supplementation reduces methotrexate-induced intestinal mucosal injury in a rat. PLoS ONE 2012, 7, e45221. [Google Scholar] [CrossRef] [PubMed]
- Harsha, W.T.; Kalandarova, E.; McNutt, P.; Irwin, R.; Noel, J. Nutritional supplementation with transforming growth factor-β, glutamine, and short chain fatty acids minimizes methotrexate-induced injury. J. Pediatr. Gastroenterol. Nutr. 2006, 42, 53–58. [Google Scholar] [CrossRef]
- Ferreira, T.M.R.; Albuquerque, A.; Cancela Penna, F.G.; Macedo Rosa, R.; Correia, M.I.T.D.; Barbosa, A.J.A.; Salles Cunha, A.; Ferrari, M.d.L.A. Effect of Oral nutrition supplements and TGF-β2 on nutrition and inflammatory patterns in patients with active Crohn’s disease. Nutr. Clin. Pract. 2020, 35, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Beaupel, N.; Brouquet, A.; Abdalla, S.; Carbonnel, F.; Penna, C.; Benoist, S. Preoperative oral polymeric diet enriched with transforming growth factor-beta 2 (Modulen) could decrease postoperative morbidity after surgery for complicated ileocolonic Crohn’s disease. Scand. J. Gastroenterol. 2017, 52, 5–10. [Google Scholar] [CrossRef]
- Davanço, T.; Leal, R.F.; Oya, V.; Lomazi, E.; dos Santos Vilela, M.M.; Coy, S.R.; Sgarbieri, V.; Ayrizono, M.d.L.S. Nutritional supplementation assessment with whey proteins and TGF-β in patients with Crohn’s disease. Nutr. Hosp. 2012, 27, 1286–1292. [Google Scholar] [CrossRef]
- Triantafillidis, J.; Stamataki, A.; Gikas, A.; Malgarinos, G. Maintenance treatment of Crohn’s disease with a polymeric feed rich in TGF-β. Ann. Gastroenterol. 2010, 23, 113–118. [Google Scholar]
- Tabet, F.; Rye, K.-A. High-density lipoproteins, inflammation and oxidative stress. Clin. Sci. 2009, 116, 87–98. [Google Scholar] [CrossRef]
- Triantafillidis, J.K.; Mantzaris, G.; Stamataki, A.; Asvestis, K.; Malgarinos, G.; Gikas, A. Complete remission of severe scleritis and psoriasis in a patient with active Crohn’s disease using Modulen IBD as an exclusive immunomodulating diet. J. Clin. Gastroenterol. 2008, 42, 550–551. [Google Scholar] [CrossRef]
- Fell, J.; Paintin, M.; Arnaud-Battandier, F.; Beattie, R.; Hollis, A.; Kitching, P.; Donnet-Hughes, A.; MacDonald, T.; Walker-Smith, J. Mucosal healing and a fall in mucosal pro-inflammatory cytokine mRNA induced by a specific oral polymeric diet in paediatric Crohn’s disease. Aliment. Pharmacol. Ther. 2000, 14, 281–290. [Google Scholar] [CrossRef]
- Beattie, R.; Schiffrin, E.; Donnet-Hughes, A.; Huggett, A.; Domizio, P.; MacDonald, T.; Walker-Smith, J. Polymeric nutrition as the primary therapy in children with small bowel Crohn’s disease. Aliment. Pharmacol. Ther. 1994, 8, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Schiffrin, E.J.; Yousfi, M.E.; Faure, M.; Combaret, L.; Donnet, A.; Blum, S.; Obled, C.; Breuillé, D. Milk casein-based diet containing TGF-β controls the inflammatory reaction in the HLA-B27 transgenic rat model. J. Parenter. Enter. Nutr. 2005, 29, S141–S150. [Google Scholar] [CrossRef]
- Kanwar, J.; Kanwar, R.; Stathopoulos, S.; Haggarty, N.; MacGibbon, A.; Palmano, K.; Roy, K.; Rowan, A.; Krissansen, G. Comparative activities of milk components in reversing chronic colitis. J. Dairy Sci. 2016, 99, 2488–2501. [Google Scholar] [CrossRef]
- Afzal, N.; Van der Zaag-Loonen, H.; Arnaud-Battandier, F.; Davies, S.; Murch, S.; Derkx, B.; Heuschkel, R.; Fell, J. Improvement in quality of life of children with acute Crohn’s disease does not parallel mucosal healing after treatment with exclusive enteral nutrition. Aliment. Pharmacol. Ther. 2004, 20, 167–172. [Google Scholar] [CrossRef]
- Bascietto, C.; Borrelli, O.; Di Nardo, G.; Ambrosini, A.; Cirulli, M.; Bosco, S.; Cucchiara, S. P0129 pp nutritional therapy alone with a polymeric diet (Modulen) is more effective than corticosteroids in inducing healing of intestinal mucosal lesions in active Crohn’s disease. J. Pediatr. Gastroenterol. Nutr. 2004, 39, S106–S107. [Google Scholar] [CrossRef]
- Agin, M.; Yucel, A.; Gumus, M.; Yuksekkaya, H.A.; Tumgor, G. The effect of enteral nutrition support rich in TGF-β in the treatment of inflammatory bowel disease in childhood. Medicina 2019, 55, 620. [Google Scholar] [CrossRef]
- Hartman, C.; Berkowitz, D.; Weiss, B.; Shaoul, R.; Levine, A.; Eshach Adiv, O.; Shapira, R.; Fradkin, A.; Wilschanski, M.; Tamir, A. Nutritional supplementation with polymeric diet enriched with transforming growth factor-beta 2 for children with Crohn’s disease. Isr. Med. Assoc. J. 2008, 10, 503. [Google Scholar]
- Day, A.S.; Whitten, K.E.; Lemberg, D.A.; Clarkson, C.; Vitug-Sales, M.; Jackson, R.; Bohane, T.D. Exclusive enteral feeding as primary therapy for Crohn’s disease in Australian children and adolescents: A feasible and effective approach. J. Gastroenterol. Hepatol. 2006, 21, 1609–1614. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, O.; Cordischi, L.; Cirulli, M.; Paganelli, M.; Labalestra, V.; Uccini, S.; Russo, P.M.; Cucchiara, S. Polymeric diet alone versus corticosteroids in the treatment of active pediatric Crohn’s disease: A randomized controlled open-label trial. Clin. Gastroenterol. Hepatol. 2006, 4, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, E.; Gaunt, W.; Cardigan, T.; Garrick, V.; McGrogan, P.; Russell, R. The use of exclusive enteral nutrition for induction of remission in children with Crohn’s disease demonstrates that disease phenotype does not influence clinical remission. Aliment. Pharmacol. Ther. 2009, 30, 501–507. [Google Scholar] [CrossRef]
- Heuschkel, R.B.; Menache, C.C.; Megerian, T.J.; Baird, A.E. Enteral nutrition and corticosteroids in the treatment of acute Crohn’s disease in children. J. Pediatr. Gastroenterol. Nutr. 2000, 31, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Rubio, A.; Pigneur, B.; Garnier-Lengliné, H.; Talbotec, C.; Schmitz, J.; Canioni, D.; Goulet, O.; Ruemmele, F. The efficacy of exclusive nutritional therapy in paediatric Crohn’s disease, comparing fractionated oral vs. continuous enteral feeding. Aliment. Pharmacol. Ther. 2011, 33, 1332–1339. [Google Scholar] [CrossRef]
- Kingsley, D. The TGF-beta superfamily: New members, new receptors, and new genetic tests of function in different organisms. Genes Dev. 1994, 8, 133–146. [Google Scholar] [CrossRef]
- Massague, J. The transforming growth factor-beta family. Annu. Rev. Cell Biol. 1990, 6, 597–641. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-G.; Lui, H.M.; Lin, S.-L.; Lee, J.M.; Ying, S.-Y. Regulation of cell proliferation, apoptosis, and carcinogenesis by activin. Exp. Biol. Med. 2002, 227, 75–87. [Google Scholar] [CrossRef]
- Tsuchida, K.; Nakatani, M.; Hitachi, K.; Uezumi, A.; Sunada, Y.; Ageta, H.; Inokuchi, K. Activin signaling as an emerging target for therapeutic interventions. Cell Commun. Signal. 2009, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Mather, J.P.; Moore, A.; Li, R.-H. Activins, inhibins, and follistatins: Further thoughts on a growing family of regulators. Proc. Soc. Exp. Biol. Med. 1997, 215, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.-Y.; Zhang, Z.; Furst, B.; Batres, Y.; Huang, G.; Li, G. Activins and activin receptors in cell growth. Proc. Soc. Exp. Biol. Med. 1997, 214, 114–122. [Google Scholar] [CrossRef]
- Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; Benhadji, K.A. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Devel. Ther. 2015, 9, 4479. [Google Scholar] [CrossRef]
- Anderton, M.J.; Mellor, H.R.; Bell, A.; Sadler, C.; Pass, M.; Powell, S.; Steele, S.J.; Roberts, R.R.; Heier, A. Induction of heart valve lesions by small-molecule ALK5 inhibitors. Toxicol. Pathol. 2011, 39, 916–924. [Google Scholar] [CrossRef]
- Pearsall, R.S.; Canalis, E.; Cornwall-Brady, M.; Underwood, K.W.; Haigis, B.; Ucran, J.; Kumar, R.; Pobre, E.; Grinberg, A.; Werner, E.D. A soluble activin type IIA receptor induces bone formation and improves skeletal integrity. Proc. Natl. Acad. Sci. USA 2008, 105, 7082–7087. [Google Scholar] [CrossRef]
- Massagué, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810. [Google Scholar] [CrossRef]
- De Caestecker, M. The transforming growth factor-β superfamily of receptors. Cytokine Growth Factor Rev. 2004, 15, 1–11. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Landström, M.; Moustakas, A. Mechanism of TGF-β signaling to growth arrest, apoptosis, and epithelial–mesenchymal transition. Curr. Opin. Cell Biol. 2009, 21, 166–176. [Google Scholar] [CrossRef]
- Zhang, L.; Deng, M.; Parthasarathy, R.; Wang, L.; Mongan, M.; Molkentin, J.D.; Zheng, Y.; Xia, Y. MEKK1 transduces activin signals in keratinocytes to induce actin stress fiber formation and migration. Mol. Cell. Biol. 2005, 25, 60–65. [Google Scholar] [CrossRef]
- Chen, N.; Laadem, A.; Wilson, D.M.; Zhang, X.; Sherman, M.L.; Ritland, S.; Attie, K.M. Pharmacokinetics and exposure-response of luspatercept in patients with beta-thalassemia: Preliminary results from phase 2 studies. Blood 2016, 128, 2463. [Google Scholar] [CrossRef]
- Jones, K.L.; Brauman, J.N.; Groome, N.P.; de Kretser, D.M.; Phillips, D.J. Activin A release into the circulation is an early event in systemic inflammation and precedes the release of follistatin. J. Endocrinol. 2000, 141, 1905–1908. [Google Scholar] [CrossRef]
- Wu, H.; Chen, Y.; Winnall, W.R.; Phillips, D.J.; Hedger, M.P. Acute regulation of activin A and its binding protein, follistatin, in serum and tissues following lipopolysaccharide treatment of adult male mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R665–R675. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.; Dolter, K.; Shao, L.; Yu, J. Suppression of IL-6 biological activities by activin A and implications for inflammatory arthropathies. J. Clin. Exp. Immunol. 1998, 112, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Steel, D.M.; Whitehead, A.S. The major acute phase reactants: C-reactive protein, serum amyloid P component and serum amyloid A protein. Immunol. Today 1994, 15, 81–88. [Google Scholar] [CrossRef]
- Abe, M.; Shintani, Y.; Eto, Y.; Harada, K.; Kosaka, M.; Matsumoto, T. Potent induction of activin A secretion from monocytes and bone marrow stromal fibroblasts by cognate interaction with activated T cells. J. Leukoc. Biol. 2002, 72, 347–352. [Google Scholar] [CrossRef]
- Karagiannidis, C.; Hense, G.; Martin, C.; Epstein, M.; Rückert, B.; Mantel, P.-Y.; Menz, G.; Uhlig, S.; Blaser, K.; Schmidt-Weber, C.B. Activin A is an acute allergen-responsive cytokine and provides a link to TGF-β–mediated airway remodeling in asthma. J. Allergy Clin. Immunol. 2006, 117, 111–118. [Google Scholar] [CrossRef]
- Iemura, S.-i.; Yamamoto, T.S.; Takagi, C.; Uchiyama, H.; Natsume, T.; Shimasaki, S.; Sugino, H.; Ueno, N. Direct binding of follistatin to a complex of bone-morphogenetic protein and its receptor inhibits ventral and epidermal cell fates in early Xenopus embryo. Proc. Natl. Acad. Sci. USA 1998, 95, 9337–9342. [Google Scholar] [CrossRef] [PubMed]
- Sugino, K.; Kurosawa, N.; Nakamura, T.; Takio, K.; Shimasaki, S.; Ling, N.; Titani, K.; Sugino, H. Molecular heterogeneity of follistatin, an activin-binding protein. Higher affinity of the carboxyl-terminal truncated forms for heparan sulfate proteoglycans on the ovarian granulosa cell. J. Biol. Chem. 1993, 268, 15579–15587. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.P.; Gregory, L.G.; Causton, B.; Campbell, G.A.; Lloyd, C.M. Activin A and TGF-β promote TH9 cell–mediated pulmonary allergic pathology. J. Allergy Clin. Immunol. 2012, 129, 1000–1010. [Google Scholar] [CrossRef] [PubMed]
- Locci, M.; Wu, J.E.; Arumemi, F.; Mikulski, Z.; Dahlberg, C.; Miller, A.T.; Crotty, S. Activin A programs the differentiation of human TFH cells. Nat. Immunol. 2016, 17, 976–984. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Funaba, M.; Chen, Y.; Tsujimoto, M. Activin A functions as a Th2 cytokine in the promotion of the alternative activation of macrophages. J. Immunol. 2006, 177, 6787–6794. [Google Scholar] [CrossRef]
- Harrison, C.A.; Gray, P.C.; Vale, W.W.; Robertson, D.M. Antagonists of activin signaling: Mechanisms and potential biological applications. Trends Endocrinol. Metab. 2005, 16, 73–78. [Google Scholar] [CrossRef]
- Westall, G.P.; Snell, G.I.; Loskot, M.; Levvey, B.; O’Hehir, R.; Hedger, M.P.; de Kretser, D.M. Activin biology after lung transplantation. Transpl. Direct 2017, 3, e159. [Google Scholar] [CrossRef]
- Hedger, M.P.; Winnall, W.R.; Phillips, D.J.; de Kretser, D.M. The regulation and functions of activin and follistatin in inflammation and immunity. Vitam. Horm. 2011, 85, 255–297. [Google Scholar] [CrossRef]
- Forrester, H.B.; De Kretser, D.M.; Leong, T.; Hagekyriakou, J.; Sprung, C.N. Follistatin attenuates radiation-induced fibrosis in a murine model. PLoS ONE 2017, 12, e0173788. [Google Scholar] [CrossRef]
- Yndestad, A.; Larsen, K.-O.; Øie, E.; Ueland, T.; Smith, C.; Halvorsen, B.; Sjaastad, I.; Skjønsberg, O.H.; Pedersen, T.M.; Anfinsen, O.-G. Elevated levels of activin A in clinical and experimental pulmonary hypertension. J. Appl. Physiol. 2009, 106, 1356–1364. [Google Scholar] [CrossRef]
- Phillips, D.J.; de Kretser, D.M.; Hedger, M.P. Activin and related proteins in inflammation: Not just interested bystanders. Cytokine Growth Factor Rev. 2009, 20, 153–164. [Google Scholar] [CrossRef]
- Michel, U.; Ebert, S.; Phillips, D.; Nau, R. Serum concentrations of activin and follistatin are elevated and run in parallel in patients with septicemia. Eur. J. Endocrinol. 2003, 148, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Aoki, F.; Kurabayashi, M.; Hasegawa, Y.; Kojima, I. Attenuation of bleomycin-induced pulmonary fibrosis by follistatin. Am. J. Respir. Crit. Care Med. 2005, 172, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Gaublomme, J.T.; Yosef, N.; Lee, Y.; Gertner, R.S.; Yang, L.V.; Wu, C.; Pandolfi, P.P.; Mak, T.; Satija, R.; Shalek, A.K. Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell 2015, 163, 1400–1412. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Awasthi, A.; Yosef, N.; Quintana, F.J.; Xiao, S.; Peters, A.; Wu, C.; Kleinewietfeld, M.; Kunder, S.; Hafler, D.A. Induction and molecular signature of pathogenic TH17 cells. Nat. Immunol. 2012, 13, 991–999. [Google Scholar] [CrossRef]
- McGeachy, M.J.; Bak-Jensen, K.S.; Chen, Y.; Tato, C.M.; Blumenschein, W.; McClanahan, T.; Cua, D.J. TGF-β and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain TH-17 cell–mediated pathology. Nat. Immunol. 2007, 8, 1390–1397. [Google Scholar] [CrossRef]
- Omenetti, S.; Bussi, C.; Metidji, A.; Iseppon, A.; Lee, S.; Tolaini, M.; Li, Y.; Kelly, G.; Chakravarty, P.; Shoaie, S. The intestine harbors functionally distinct homeostatic tissue-resident and inflammatory Th17 cells. Immunity 2019, 51, 77–89. [Google Scholar] [CrossRef]
- Wang, C.; Yosef, N.; Gaublomme, J.; Wu, C.; Lee, Y.; Clish, C.B.; Kaminski, J.; Xiao, S.; Zu Horste, G.M.; Pawlak, M. CD5L/AIM regulates lipid biosynthesis and restrains Th17 cell pathogenicity. Cell 2015, 163, 1413–1427. [Google Scholar] [CrossRef]
- Wu, B.; Zhang, S.; Guo, Z.; Bi, Y.; Zhou, M.; Li, P.; Seyedsadr, M.; Xu, X.; Li, J.-l.; Markovic-Plese, S. The TGF-β superfamily cytokine Activin-A is induced during autoimmune neuroinflammation and drives pathogenic Th17 cell differentiation. Immunity 2021, 54, 308–323. [Google Scholar] [CrossRef]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 Cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef]
- Sideras, P.; Apostolou, E.; Stavropoulos, A.; Sountoulidis, A.; Gavriil, A.; Apostolidou, A.; Andreakos, E. Activin, neutrophils, and inflammation: Just coincidence? Semin. Immunopathol. 2013, 35, 481–499. [Google Scholar] [CrossRef]
- Werner, S.; Alzheimer, C. Roles of activin in tissue repair, fibrosis, and inflammatory disease. Cytokine Growth Factor Rev. 2006, 17, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Sonoyama, K.; Rutatip, S.; Kasai, T. Gene expression of activin, activin receptors, and follistatin in intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 278, G89–G97. [Google Scholar] [CrossRef]
- Dignass, A.; Jung, S.; Harder-d’Heureuse, J.; Wiedenmann, B. Functional relevance of activin A in the intestinal epithelium. Scand. J. Gastroenterol. 2002, 37, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Hübner, G.; Brauchle, M.; Gregor, M.; Werner, S. Activin A: A novel player and inflammatory marker in inflammatory bowel disease? Lab. Investig. 1997, 77, 311–318. [Google Scholar] [CrossRef]
- Lodberg, A. Principles of the activin receptor signaling pathway and its inhibition. Cytokine Growth Factor Rev. 2021, 60, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Dohi, T.; Ejima, C.; Kato, R.; Kawamura, Y.I.; Kawashima, R.; Mizutani, N.; Tabuchi, Y.; Kojima, I. Therapeutic potential of follistatin for colonic inflammation in mice. Gastroenterology 2005, 128, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Souza, T.A.; Chen, X.; Guo, Y.; Sava, P.; Zhang, J.; Hill, J.J.; Yaworsky, P.J.; Qiu, Y. Proteomic identification and functional validation of activins and bone morphogenetic protein 11 as candidate novel muscle mass regulators. Mol. Endocrinol. 2008, 22, 2689–2702. [Google Scholar] [CrossRef]
- McPherron, A.C.; Huynh, T.V.; Lee, S.-J. Redundancy of myostatin and growth/differentiation factor 11 function. BMC Dev. Biol. 2009, 9, 24. [Google Scholar] [CrossRef]
- Sinha, M.; Jang, Y.C.; Oh, J.; Khong, D.; Wu, E.Y.; Manohar, R.; Miller, C.; Regalado, S.G.; Loffredo, F.S.; Pancoast, J.R. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science 2014, 344, 649–652. [Google Scholar] [CrossRef]
- Egerman, M.A.; Cadena, S.M.; Gilbert, J.A.; Meyer, A.; Nelson, H.N.; Swalley, S.E.; Mallozzi, C.; Jacobi, C.; Jennings, L.L.; Clay, I. GDF11 increases with age and inhibits skeletal muscle regeneration. Cell Metab. 2015, 22, 164–174. [Google Scholar] [CrossRef]
- Hinken, A.C.; Powers, J.M.; Luo, G.; Holt, J.A.; Billin, A.N.; Russell, A.J. Lack of evidence for GDF 11 as a rejuvenator of aged skeletal muscle satellite cells. Aging Cell 2016, 15, 582–584. [Google Scholar] [CrossRef] [PubMed]
- Latres, E.; Pangilinan, J.; Miloscio, L.; Bauerlein, R.; Na, E.; Potocky, T.B.; Huang, Y.; Eckersdorff, M.; Rafique, A.; Mastaitis, J. Myostatin blockade with a fully human monoclonal antibody induces muscle hypertrophy and reverses muscle atrophy in young and aged mice. Skelet. Muscle 2015, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Lord, S.R.; Studenski, S.A.; Warden, S.J.; Fielding, R.A.; Recknor, C.P.; Hochberg, M.C.; Ferrari, S.L.; Blain, H.; Binder, E.F. Myostatin antibody (LY2495655) in older weak fallers: A proof-of-concept, randomised, phase 2 trial. Lancet Diabetes Endocrinol. 2015, 3, 948–957. [Google Scholar] [CrossRef] [PubMed]
- Bogdanovich, S.; Perkins, K.J.; Krag, T.O.; Whittemore, L.-A.; Khurana, T.S. Myostatin propeptide-mediated amelioration of dystrophic pathophysiology. FASEB J. 2005, 19, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, M.D.; Pistilli, E.E.; Balduzzi, A.; Birnbaum, M.J.; Lachey, J.; Khurana, T.S.; Ahima, R.S. Akt deficiency attenuates muscle size and function but not the response to ActRIIB inhibition. PLoS ONE 2010, 5, e12707. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, J.L.; Lu, J.; Song, Y.; Kwak, K.S.; Jiao, Q.; Rosenfeld, R.; Chen, Q.; Boone, T.; Simonet, W.S. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010, 142, 531–543. [Google Scholar] [CrossRef]
- Pearsall, R.; Davies, M.; Cannell, M.; Li, J.; Widrick, J.; Mulivor, A.; Wallner, S.; Troy, M.; Spaits, M.; Liharska, K. Follistatin-based ligand trap ACE-083 induces localized hypertrophy of skeletal muscle with functional improvement in models of neuromuscular disease. Sci. Rep. 2019, 9, 11392. [Google Scholar] [CrossRef]
- Pistilli, E.E.; Bogdanovich, S.; Goncalves, M.D.; Ahima, R.S.; Lachey, J.; Seehra, J.; Khurana, T. Targeting the activin type IIB receptor to improve muscle mass and function in the mdx mouse model of Duchenne muscular dystrophy. Am. J. Pathol. 2011, 178, 1287–1297. [Google Scholar] [CrossRef]
- Sepulveda, P.V.; Lamon, S.; Hagg, A.; Thomson, R.E.; Winbanks, C.E.; Qian, H.; Bruce, C.R.; Russell, A.P.; Gregorevic, P. Evaluation of follistatin as a therapeutic in models of skeletal muscle atrophy associated with denervation and tenotomy. Sci. Rep. 2015, 5, 17535. [Google Scholar] [CrossRef]
- Meier, D.; Lodberg, A.; Gvozdenovic, A.; Pellegrini, G.; Neklyudova, O.; Born, W.; Fuchs, B.; Eijken, M.; Botter, S.M. Inhibition of the activin receptor signaling pathway: A novel intervention against osteosarcoma. Cancer Med. 2021, 10, 286–296. [Google Scholar] [CrossRef]
- Dankbar, B.; Fennen, M.; Brunert, D.; Hayer, S.; Frank, S.; Wehmeyer, C.; Beckmann, D.; Paruzel, P.; Bertrand, J.; Redlich, K. Myostatin is a direct regulator of osteoclast differentiation and its inhibition reduces inflammatory joint destruction in mice. Nat. Med. 2015, 21, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Rajan, V.; Lin, E.; Hu, Z.; Han, H.; Zhou, X.; Song, Y.; Min, H.; Wang, X.; Du, J. Pharmacological inhibition of myostatin suppresses systemic inflammation and muscle atrophy in mice with chronic kidney disease. FASEB J. 2011, 25, 1653–1663. [Google Scholar] [CrossRef]
- Zeng, R.; Glaubitz, S.; Schmidt, J. Antibody therapies in autoimmune inflammatory myopathies: Promising treatment options. Neurotherapeutics 2022, 19, 911–921. [Google Scholar] [CrossRef]
- Joshi, S.R.; Liu, J.; Bloom, T.; Karaca Atabay, E.; Kuo, T.-H.; Lee, M.; Belcheva, E.; Spaits, M.; Grenha, R.; Maguire, M.C. Sotatercept analog suppresses inflammation to reverse experimental pulmonary arterial hypertension. Sci. Rep. 2022, 12, 1–18. [Google Scholar] [CrossRef]
- Joshi, S.; Liu, J.; Pearsall, R.; Andre, P.; Li, G.; Kumar, R. Activin receptor type IIA-Fc (Sotatercept) suppresses inflammation to alleviate pulmonary arterial hypertension in preclinical models. Am. J. Respir. Crit. Care Med. 2020, 201, A3835. [Google Scholar]
- Raje, N.; Vallet, S. Sotatercept, a soluble activin receptor type 2A IgG-Fc fusion protein for the treatment of anemia and bone loss. Curr. Opin. Mol. Ther. 2010, 12, 586–597. [Google Scholar] [PubMed]
- Abdulkadyrov, K.M.; Salogub, G.N.; Khuazheva, N.K.; Sherman, M.L.; Laadem, A.; Barger, R.; Knight, R.; Srinivasan, S.; Terpos, E. Sotatercept in patients with osteolytic lesions of multiple myeloma. Br. J. Haematol. 2014, 165, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.L.; Borgstein, N.G.; Mook, L.; Wilson, D.; Yang, Y.; Chen, N.; Kumar, R.; Kim, K.; Laadem, A. Multiple-dose, safety, pharmacokinetic, and pharmacodynamic study of sotatercept (ActRIIA-IgG1), a novel erythropoietic agent, in healthy postmenopausal women. J. Clin. Pharmacol. 2013, 53, 1121–1130. [Google Scholar] [CrossRef] [PubMed]
- Raftopoulos, H.; Laadem, A.; Hesketh, P.J.; Goldschmidt, J.; Gabrail, N.; Osborne, C.; Ali, M.; Sherman, M.L.; Wang, D.; Glaspy, J.A. Sotatercept (ACE-011) for the treatment of chemotherapy-induced anemia in patients with metastatic breast cancer or advanced or metastatic solid tumors treated with platinum-based chemotherapeutic regimens: Results from two phase 2 studies. Support. Care Cancer 2016, 24, 1517–1525. [Google Scholar] [CrossRef]
- Humbert, M.; McLaughlin, V.; Gibbs, J.S.R.; Gomberg-Maitland, M.; Hoeper, M.M.; Preston, I.R.; Souza, R.; Waxman, A.B.; Ghofrani, H.-A.; Subias, P.E. Sotatercept for the treatment of pulmonary arterial hypertension: PULSAR open-label extension. Eur. Respir. J. 2023, 61, 2201347. [Google Scholar] [CrossRef]
- Bose, P.; Pemmaraju, N.; Masarova, L.; Bledsoe, S.D.; Daver, N.; Jabbour, E.; Kadia, T.M.; Estrov, Z.E.; Kornblau, S.M.; Andreeff, M. Sotatercept (ACE-011) for anemia of myelofibrosis: A phase 2 study. Blood 2020, 136, 10–11. [Google Scholar] [CrossRef]
- Fenaux, P.; Kiladjian, J.J.; Platzbecker, U. Luspatercept for the treatment of anemia in myelodysplastic syndromes and primary myelofibrosis. Blood 2019, 133, 790–794. [Google Scholar] [CrossRef] [PubMed]
- Jelkmann, W. Activin receptor ligand traps in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2018, 27, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Platzbecker, U.; Mufti, G.J.; Garcia-Manero, G.; Buckstein, R.; Santini, V.; Díez-Campelo, M.; Finelli, C.; Cazzola, M.; Ilhan, O. Luspatercept in patients with lower-risk myelodysplastic syndromes. N. Engl. J. Med. 2020, 382, 140–151. [Google Scholar] [CrossRef]
- Amato, A.A.; Sivakumar, K.; Goyal, N.; David, W.S.; Salajegheh, M.; Praestgaard, J.; Lach-Trifilieff, E.; Trendelenburg, A.-U.; Laurent, D.; Glass, D.J. Treatment of sporadic inclusion body myositis with bimagrumab. Neurology 2014, 83, 2239–2246. [Google Scholar] [CrossRef]
- Hanna, M.G.; Badrising, U.A.; Benveniste, O.; Lloyd, T.E.; Needham, M.; Chinoy, H.; Aoki, M.; Machado, P.M.; Liang, C.; Reardon, K.A. Safety and efficacy of intravenous bimagrumab in inclusion body myositis (RESILIENT): A randomised, double-blind, placebo-controlled phase 2b trial. Lancet Neurol. 2019, 18, 834–844. [Google Scholar] [CrossRef]
- Rooks, D.S.; Laurent, D.; Praestgaard, J.; Rasmussen, S.; Bartlett, M.; Tankó, L.B. Effect of bimagrumab on thigh muscle volume and composition in men with casting-induced atrophy. J. Cachexia Sarcopenia Muscle 2017, 8, 727–734. [Google Scholar] [CrossRef]
- Sivakumar, K.; Cochrane, T.I.; Sloth, B.; Ashar, H.; Laurent, D.; Tankó, L.B.; Amato, A.A. Long-term safety and tolerability of bimagrumab (BYM338) in sporadic inclusion body myositis. Neurology 2020, 95, e1971–e1978. [Google Scholar] [CrossRef]
- Jagtap, K.; Hoff, L.S.; Conticini, E.; Naveen, R.; Gupta, L. Inflammaging in muscle the missing link between sarcopenia and idiopathic inflammatory myopathies. J. Anti. Aging Med. 2022, 1, 63–72. [Google Scholar] [CrossRef]
- Glasser, C.E.; Gartner, M.R.; Wilson, D.; Miller, B.; Sherman, M.L.; Attie, K.M. Locally acting ACE-083 increases muscle volume in healthy volunteers. Muscle Nerve 2018, 57, 921–926. [Google Scholar] [CrossRef]
- Statland, J.M.; Campbell, C.; Desai, U.; Karam, C.; Díaz-Manera, J.; Guptill, J.T.; Korngut, L.; Genge, A.; Tawil, R.N.; Elman, L. Randomized phase 2 study of ACE-083, a muscle-promoting agent, in facioscapulohumeral muscular dystrophy. Muscle Nerve 2022, 66, 50–62. [Google Scholar] [CrossRef]
- Shy, M.; Herrmann, D.; Thomas, F.; Quinn, C.; Statland, J.; Walk, D.; Johnson, N.; Subramony, S.; Karam, C.; Mozaffar, T. CMT and neurogenic disease: P. 339Preliminary phase 2 results for ACE-083, local muscle therapeutic, in patients with CMT1 and CMTX. Neuromuscul. Disord. 2018, 28, S132–S133. [Google Scholar] [CrossRef]
- Thomas, F.P.; Brannagan, T.H.; Butterfield, R.J.; Desai, U.; Habib, A.A.; Herrmann, D.N.; Eichinger, K.J.; Johnson, N.E.; Karam, C.; Pestronk, A. Randomized phase 2 study of ACE-083 in patients with charcot-marie-tooth disease. Neurology 2022, 98, e2356–e2367. [Google Scholar] [CrossRef]
- Statland, J.; Bravver, E.; Karam, C.; Elman, L.; Johnson, N.; Joyce, N. Results for a dose-escalation phase 2 study to evaluate ACE-083, a local muscle therapeutic, in patients with facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 2018, 28, S140. [Google Scholar] [CrossRef]
- Thomas, F.P.; Shy, M.; Quinn, C.; Desai, U.; Herrmann, D.; Statland, J.; Subramony, S.; Brannagan, T.; Habib, A.A.; Karam, C. Results of a phase 2 double-blind placebo-controlled study of a local muscle therapeutic, ACE-083, in subjects with charcot-marie-tooth (CMT) disease (1514). Neurology 2020, 14, 1514. [Google Scholar]
- Statland, J.; Amato, A.; Bravver, E.; Campbell, C.; Elman, L.; Johnson, N.; Joyce, N.; Karam, C.; Kissel, J.; Korngut, L. Preliminary results from a phase 2 study to evaluate ACE-083, a local muscle therapeutic, in patients with facioscapulohumeral muscular dystrophy (S38. 001). Neurology 2018, 90, S38.001. [Google Scholar]
- Ghasemi, M.; Emerson Jr, C.P.; Hayward, L.J. Outcome measures in facioscapulohumeral muscular dystrophy clinical trials. Cells 2022, 11, 687. [Google Scholar] [CrossRef]
- Lodberg, A.; van der Eerden, B.C.; Boers-Sijmons, B.; Thomsen, J.S.; Brüel, A.; van Leeuwen, J.P.; Eijken, M. A follistatin-based molecule increases muscle and bone mass without affecting the red blood cell count in mice. FASEB J. 2019, 33, 6001–6010. [Google Scholar] [CrossRef]
- Latres, E.; Mastaitis, J.; Fury, W.; Miloscio, L.; Trejos, J.; Pangilinan, J.; Okamoto, H.; Cavino, K.; Na, E.; Papatheodorou, A. Activin A more prominently regulates muscle mass in primates than does GDF8. Nat. Commun. 2017, 8, 151153. [Google Scholar] [CrossRef]
- Gregory, S.J.; Kaiser, U.B. Regulation of gonadotropins by inhibin and activin. Semin. Reprod. Med. 2004, 22, 253–267. [Google Scholar] [CrossRef]
- Garito, T.; Zakaria, M.; Papanicolaou, D.A.; Li, Y.; Pinot, P.; Petricoul, O.; Laurent, D.; Rooks, D.; Rondon, J.C.; Roubenoff, R. Effects of bimagrumab, an activin receptor type II inhibitor, on pituitary neurohormonal axes. Clin. Endocrinol. 2018, 88, 908–919. [Google Scholar] [CrossRef]
- Pabla, N.; Dong, Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008, 73, 994–1007. [Google Scholar] [CrossRef]
- Castonguay, R.; Lachey, J.; Wallner, S.; Strand, J.; Liharska, K.; Watanabe, A.E.; Cannell, M.; Davies, M.V.; Sako, D.; Troy, M.E. Follistatin-288-Fc fusion protein promotes localized growth of skeletal muscle. J. Pharmacol. Exp. Ther. 2019, 368, 435–445. [Google Scholar] [CrossRef]
- Cash, J.N.; Rejon, C.A.; McPherron, A.C.; Bernard, D.J.; Thompson, T.B. The structure of myostatin: Follistatin 288: Insights into receptor utilization and heparin binding. EMBO J. 2009, 28, 2662–2676. [Google Scholar] [CrossRef]
- Lerch, T.F.; Shimasaki, S.; Woodruff, T.K.; Jardetzky, T.S. Structural and biophysical coupling of heparin and activin binding to follistatin isoform functions. J. Biol. Chem. 2007, 282, 15930–15939. [Google Scholar] [CrossRef]
- Wang, E.Y.; Draper, L.B.; Lee, E.; Polak, A.; Sluss, P.; Weiss, J.; Woodruff, T.K. Identification of naturally occurring follistatin complexes in human biological fluids. Biol. Reprod. 1999, 60, 8–13. [Google Scholar] [CrossRef]
- Schneyer, A.L.; Wang, Q.; Sidis, Y.; Sluss, P.M. Differential distribution of follistatin isoforms: Application of a new FS315-specific immunoassay. J. Clin. Endocrinol. Metab. 2004, 89, 5067–5075. [Google Scholar] [CrossRef]
- Thompson, T.B.; Lerch, T.F.; Cook, R.W.; Woodruff, T.K.; Jardetzky, T.S. The structure of the follistatin: Activin complex reveals antagonism of both type I and type II receptor binding. Dev. Cell 2005, 9, 535–543. [Google Scholar] [CrossRef]
- Sidis, Y.; Mukherjee, A.; Keutmann, H.; Delbaere, A.; Sadatsuki, M.; Schneyer, A. Biological activity of follistatin isoforms and follistatin-like-3 is dependent on differential cell surface binding and specificity for activin, myostatin, and bone morphogenetic proteins. J. Endocrinol. 2006, 147, 3586–3597. [Google Scholar] [CrossRef]
- Hashimoto, O.; Nakamura, T.; Shoji, H.; Shimasaki, S.; Hayashi, Y.; Sugino, H. A novel role of follistatin, an activin-binding protein, in the inhibition of activin action in rat pituitary cells: Endocytotic degradation of activin and its acceleration by follistatin associated with cell-surface heparan sulfate. J. Biol. Chem. 1997, 272, 13835–13842. [Google Scholar] [CrossRef]
- Jones, K.L.; Mansell, A.; Patella, S.; Scott, B.J.; Hedger, M.P.; de Kretser, D.M.; Phillips, D.J. Activin A is a critical component of the inflammatory response, and its binding protein, follistatin, reduces mortality in endotoxemia. Proc. Natl. Acad. Sci. USA 2007, 104, 16239–16244. [Google Scholar] [CrossRef]
- Hidalgo, I.J.; Raub, T.J.; Borchardt, R.T. Characterization of the human colon carcinoma cell line (Caco-2) as a model system for intestinal epithelial permeability. Gastroenterology 1989, 96, 736–749. [Google Scholar] [CrossRef]
- Moustakas, A.; Kardassis, D. Regulation of the human p21/WAF1/Cip1 promoter in hepatic cells by functional interactions between Sp1 and Smad family members. Proc. Natl. Acad. Sci. USA 1998, 95, 6733–6738. [Google Scholar] [CrossRef]
- Oda, S.; Nishimatsu, S.-I.; Murakami, K.; Ueno, N. Molecular cloning and functional analysis of a new activin β subunit: A dorsal mesoderm-inducing activity in Xenopus. Biochem. Biophys. Res. Commun. 1995, 210, 581–588. [Google Scholar] [CrossRef]
- Parker, S.B.; Eichele, G.; Zhang, P.; Rawls, A.; Sands, A.T.; Bradley, A.; Olson, E.N.; Harper, J.W.; Elledge, S.J. p53-independent expression of p21Cip1 in muscle and other terminally differentiating cells. Science 1995, 267, 1024–1027. [Google Scholar] [CrossRef]
- Evers, B.M.; Ko, T.C.; Li, J.; Thompson, E.A. Cell cycle protein suppression and p21 induction in differentiating Caco-2 cells. Am. J. Physiol. Gastrointest. Liver Physiol. 1996, 271, G722–G727. [Google Scholar] [CrossRef]
- Kogure, K.; Omata, W.; Kanzaki, M.; Zhang, Y.-Q.; Yasuda, H.; Mine, T.; Kojima, I. A single intraportal administration of follistatin accelerates liver regeneration in partially hepatectomized rats. Gastroenterology 1995, 108, 1136–1142. [Google Scholar] [CrossRef]
- Ishiki, Y.; Ohnishi, H.; Muto, Y.; Matsumoto, K.; Nakamura, T. Direct evidence that hepatocyte growth factor is a hepatotrophic factor for liver regeneration and has a potent antihepatitis effect in vivo. Hepatology 1992, 16, 1227–1235. [Google Scholar] [CrossRef]
- Staudacher, J.J.; Bauer, J.; Jana, A.; Tian, J.; Carroll, T.; Mancinelli, G.; Özden, Ö.; Krett, N.; Guzman, G.; Kerr, D. Activin signaling is an essential component of the TGF-β induced pro-metastatic phenotype in colorectal cancer. Sci. Rep. 2017, 7, 5569. [Google Scholar] [CrossRef]
- Zhang, Y.-Q.; Resta, S.; Jung, B.; Barrett, K.E.; Sarvetnick, N. Upregulation of activin signaling in experimental colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G768–G780. [Google Scholar] [CrossRef]
- Stoitzner, P.; Stössel, H.; Wankell, M.; Hofer, S.; Heufler, C.; Werner, S.; Romani, N. Langerhans cells are strongly reduced in the skin of transgenic mice overexpressing follistatin in the epidermis. Eur. J. Cell Biol. 2005, 84, 733–741. [Google Scholar] [CrossRef]
- Yaden, B.C.; Croy, J.E.; Wang, Y.; Wilson, J.M.; Datta-Mannan, A.; Shetler, P.; Milner, A.; Bryant, H.U.; Andrews, J.; Dai, G. Follistatin: A novel therapeutic for the improvement of muscle regeneration. J. Pharmacol. Exp. Ther. 2014, 349, 355–371. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hatamzade Esfahani, N.; Day, A.S. The Role of TGF-β, Activin and Follistatin in Inflammatory Bowel Disease. Gastrointest. Disord. 2023, 5, 167-186. https://doi.org/10.3390/gidisord5020015
Hatamzade Esfahani N, Day AS. The Role of TGF-β, Activin and Follistatin in Inflammatory Bowel Disease. Gastrointestinal Disorders. 2023; 5(2):167-186. https://doi.org/10.3390/gidisord5020015
Chicago/Turabian StyleHatamzade Esfahani, Nasim, and Andrew S. Day. 2023. "The Role of TGF-β, Activin and Follistatin in Inflammatory Bowel Disease" Gastrointestinal Disorders 5, no. 2: 167-186. https://doi.org/10.3390/gidisord5020015
APA StyleHatamzade Esfahani, N., & Day, A. S. (2023). The Role of TGF-β, Activin and Follistatin in Inflammatory Bowel Disease. Gastrointestinal Disorders, 5(2), 167-186. https://doi.org/10.3390/gidisord5020015