CCR6–CCL20 Axis in IBD: What Have We Learnt in the Last 20 Years?


{kind=link}
Abstract
1. Introduction
1.1. Inflammatory Bowel Disease (IBD)
1.2. Chemokines—Chemokine Receptor 6 (CCR6) and CC—Chemokine Ligand 20 (CCL20)
1.3. Immunological Pathogenesis in IBD
2. CCR6 and Its Role in Potentiating Pro-Inflammatory Properties in IBD
2.1. Effect of Chemokines (CCR6 and CCL20) on T Cells in IBD
2.2. The Influence of TH17 Cells in IBD
2.3. The Influence of Gamma Delta (γδ) T Cells in IBD
2.4. The Influence of CCR6+ Innate Lymphoid Cells (ILC) in IBD
2.5. The Effect of CCR6+ Intestinal Epithelial Cells on IBD
2.6. The Impact of CCR6 Expression on Colonic Histology
2.7. CCR6 Association with Colitis and Colorectal Cancer
2.8. The Impact of CCR6+ T Cells in the Gut on Human Immunodeficiency Virus (HIV)
2.9. The Effect of CCR6+ Cells in the Gut on Obesity
3. CCR6 and Its Role in Potentiating Anti-Inflammatory Properties
3.1. The Effect of Regulatory Treg Cells in IBD
3.2. The Influence of TH17/Treg Balance in IBD
3.3. The Effect of CCR6 on Dendritic Cells (DC) in IBD
3.4. The Influence of Natural Killer T (NKT) Cells
4. Future Directions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Graham, D.B.; Xavier, R.J. From genetics of inflammatory bowel disease towards mechanistic insights. Trends Immunol. 2013, 34, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Shouval, D.S.; Rufo, P.A. The role of environmental factors in the pathogenesis of inflammatory bowel diseases: A review. JAMA Pediatr. 2017, 171, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Rosen, M.J.; Dhawan, A.; Saeed, S.A. Inflammatory bowel disease in children and adolescents. JAMA Pediatr. 2015, 169, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Zeissig, S.; Blumberg, R.S. Inflammatory Bowel Disease. Annu. Rev. Immunol. 2010, 28, 573–621. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.J. Inflammatory bowel disease goes global. JAMA 2018, 319, 648. [Google Scholar] [CrossRef] [PubMed]
- Niewiadomski, O.; Studd, C.; Hair, C.; Wilson, J.; Ding, N.S.; Heerasing, N.; Ting, A.; McNeill, J.; Knight, R.; Santamaria, J.; et al. Prospective population-based cohort of inflammatory bowel disease in the biologics era: Disease course and predictors of severity. J. Gastroenterol. Hepatol. 2015, 30, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Crohn’s and Colitis Foundation Australia Website. 2013. Available online: http://www.crohnsandcolitis.com.au (accessed on 21 July 2018).
- Niewiadomski, O.; Studd, C.; Wilson, J.; Williams, J.; Hair, C.; Knight, R.; Prewett, E.; Dabkowski, P.; Alexander, S.; Allen, B.; et al. Influence of food and lifestyle on the risk of developing inflammatory bowel disease. J. Intern. Med. 2016, 46, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Ranasinghe, R.; Eri, R. Pleiotropic immune functions of chemokine receptor 6 in health and disease. Medicines 2018, 5, 69. [Google Scholar] [CrossRef] [PubMed]
- Basheer, W.; Kunde, D.; Eri, R. Role of chemokine ligand CCL20 and its receptor CCR6 in intestinal inflammation. Immunol. Infect. Dis. 2013, 1, 30–37. [Google Scholar]
- Lee, A.Y.S.; Eri, R.; Lyons, A.B.; Grimm, M.C.; Korner, H. CC chemokine ligand CCL20 and its cognate receptor CCR6 in mucosal T cell immunology and inflammatory bowel disease: Odd couple or axis of evil? Front. Immunol. 2013, 4, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.S.; Phan, T.K.; Hulett, M.D.; Korner, H. The relationship between CCR6 and its binding partners: Does the CCR6-CCL20 axis have to be extended? Cytokine 2015, 72, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.Y.; Lu, S.S.; Chang, S.L.; Liao, F. The phosphorylation of CCR6 on distinct Ser/Thr residues in the carboxyl terminus differentially regulates biological function. Front. Immunol. 2018, 9, 415. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kwon, J.; Cho, M.L. Immunological pathogenesis of inflammatory bowel disease. Intest. Res. 2018, 16, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Guan, O.; Zhang, J. Recent advances: The imbalance of cytokines in the pathogenesis of inflammatory bowel disease. Mediat. Inflamm. 2017. [Google Scholar] [CrossRef] [PubMed]
- Strober, W.; Fuss, I.J. Pro-inflammatory cytokines in the pathogenesis of IBD. Gastroenterology 2011, 14, 1756–1767. [Google Scholar] [CrossRef] [PubMed]
- Soufli, I.; Toumi, R.; Rafa, H.; Touil-Boukoffa, C. Overview of cytokines and nitric oxide involvement in immune-pathogenesis of inflammatory bowel diseases. World J. Gastrointest. Pharmacol. Ther. 2016, 7, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Rafa, H.; Saoula, H.; Belkhelfa, M.; Medjeber, O.; Soufli, I.; Toumi, R.; de Launoit, Y.; Morales, O.; Nakmouche, M.; Delhem, N.; et al. IL-23/IL-17A axis correlates with the nitric oxide pathway in inflammatory bowel disease: Immunomodulatory effect of retinoic acid. J. Interferon Cytokine Res. 2013, 33, 353. [Google Scholar] [CrossRef] [PubMed]
- Scheerens, H.; Hessel, E.; deWaal-Malefyt, R.; Leach, M.W.; Rennick, D. Characterization of chemokines and chemokine receptors in two murine models of inflammatory bowel disease: IL-10-/- mice and Rag-2-/- mice reconstituted with CD4+CD45RBhigh T cells. Eur. J. Immunol. 2001, 31, 1465–1474. [Google Scholar] [CrossRef]
- Varona, R.; Cadenas, V.; Flores, J.; Martinez, A.C.; Marquez, G. CCR6 has a non-redundant role in the development of inflammatory bowel disease. Eur. J. Immunol. 2003, 33, 2937–2946. [Google Scholar] [CrossRef] [PubMed]
- Teramoto, K.; Miura, S.; Tsuzuki, Y.; Hokari, R.; Watanabe, C.; Inamura, T.; Ogawa, T.; Hosoe, N.; Nagata, H.; Ishii, H.; et al. Increased lymphocyte trafficking to colonic microvessels is dependent on MAdCAM-1 and C-C chemokine mLARC/CCL20 in DSS—Induced mice colitis. Clin. Exp. Immunol. 2005, 139, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Katchar, K.; Kelly, C.P.; Keates, S.; O’brien, M.J.; Keates, A.C. MIP-3alpha neutralizing monoclonal antibody protects against TNBS-induced colonic injury and inflammation in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1263–G1271. [Google Scholar] [CrossRef] [PubMed]
- Eijkelkamp, N.; Heijnen, C.J.; Lucas, A.; Premont, R.T.; Elsenbruch, S.; Schedlowski, M.; Kavelaars, A. G protein-coupled receptor kinase 6 controls chronicity and severity of dextran sodium sulphate-induced colitis in mice. Gut 2007, 56, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Blazquez, A.B.; Knight, A.K.; Getachew, H.; Bromberg, J.S.; Lira, S.A.; Mayer, L.; Berin, M.C. A functional role for CCR6 on pro-allergic T cells in the gastrointestinal tract. Gastroenterology 2010, 138, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Atreya, R.; Neurath, M.F. Chemokines in inflammatory bowel diseases. Dig. Dis. 2010, 28, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K.; Koike, Y.; Hashimoto, K.; Saigusa, S.; Inoue, M.; Otake, K.; Tanaka, K.; Matsushita, K.; Okita, Y.; Fujikawa, H.; et al. The increased expression of CCL20 and CCR6 in rectal mucosa correlated to severe inflammation in pediatric ulcerative colitis. Gastroenterol. Res. Pract. 2015. [Google Scholar] [CrossRef] [PubMed]
- Pezoldt, J.; Huehn, J. Tissue specific induction of CCR6 and Nrp1 during early CD4+ T cell differentiation. Eur. J. Microbiol. Immunol. 2016, 6, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Wang, D.; Li, Y.; Sang, L.; Zhu, J.; Wang, J.; Wei, B.; Lu, C.; Sun, X. Th1/Th2 balance and Th17/Treg –mediated immunity in relation to murine resistance to dextran sulfate-induced colitis. J. Immunol. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.W.; Lee, J.; Hillsamer, P.; Kim, C.H. Human Th17 cells share major trafficking receptors with both polarized effector T cells and FOXP3+ regulatory T cells. J. Immunol. 2008, 180, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Kang, S.G.; Lee, J.; Sun, Z.; Kim, C.H. The roles of CCR6 in migration of Th17 cells and regulation of effector T cell balance in the gut. Mucosal Immunol. 2009, 2, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Kleinschek, M.A.; Boniface, K.; Sadekova, S.; Grein, J.; Murphy, E.E.; Turner, S.P.; Raskin, L.; Desai, B.; Faubion, W.A.; de Waal Malefyt, R.; et al. Circulating and gut-resident human Th17 cells express CD161 and promote intestinal inflammation. J. Exp. Med. 2009, 206, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Ahern, P.P.; Schiering, C.; Buonocore, S.; McGeachy, M.J.; Cua, D.J.; Maloy, K.J.; Powrie, F. Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity 2010, 33, 279–288. [Google Scholar] [CrossRef] [PubMed]
- El-Bassat, H.; Ali, L.A.; El Yamany, S.; Al Shenawy, H.; Al Din, R.A.; Taha, A. Interleukin-23p 19 expression in patients with ulcerative colitis and its relation to disease severity. Adv. Dig. Med. 2016, 3, 88–94. [Google Scholar] [CrossRef]
- Esplugues, E.; Huber, S.; Gagliani, N.; Hauser, A.E.; Town, T.; Wan, Y.Y.; O’Connor, W., Jr.; Rongvaux, A.; Van Rooijen, N.; Haberman, A.M.; et al. Control of Th17 cells occurs in the small intestine. Nature 2011, 475, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ferguson, J.; Ng, S.M.; Hui, K.; Goh, G.; Lin, A.; Esplugues, E.; Flavell, R.A.; Abraham, C.; Zhao, H.; et al. Effector CD4+ T cell expression signatures and immune-mediated disease associated genes. PLoS ONE 2012, 7, e38510. [Google Scholar] [CrossRef] [PubMed]
- Duhen, T.; Campbell, D.J. IL-1β promotes the differentiation of polyfunctional human CCR6+CXCR3+ Th1/17 cells that are specific for pathogenic and commensal microbes. J. Immunol. 2014, 193, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Huang, H. The Role of CD39 in Modulating Effector Immune Responses in Inflammatory Bowel Disease. Ph.D. Thesis, Harvard Medical School, Boston, MA, USA, 2015. [Google Scholar]
- Longhi, M.S.; Vuerich, M.; Kalbasi, A.; Kenison, J.E.; Yeste, A.; Csizmadia, E.; Vaughn, B.; Feldbrugge, L.; Mitsuhashi, S.; Wegiel, B.; et al. Bilirubin suppresses Th17 immunity in colitis by upregulating CD39. JCI Insight 2017, 2, e92791. [Google Scholar] [CrossRef] [PubMed]
- Hildner, K.; Punkenburg, E.; Abendroth, B.; Neurath, M.F. Immunopathogenesis of IBD: Batf as a key driver of disease activity. Dig. Dis. 2016, 34 (Suppl. S1), 40–47. [Google Scholar] [CrossRef] [PubMed]
- Haas, J.D.; Gonzalez, F.H.; Schmitz, S.; Chennupati, V.; Fohse, L.; Kremmer, E.; Forster, R.; Prinz, I. CCR6 and NK1.1 distinguish between IL-17A and IFN- gamma-producing gammadelta effector T cells. Eur. J. Immunol. 2009, 39, 3488–3497. [Google Scholar] [CrossRef] [PubMed]
- Do, J.; Visperas, A.; Dong, C.; Baldwin, W.M., III; Min, B. Generation of colitogenic Th17 CD4 T cells is enhanced by IL-17+γδ T cells. J. Immunol. 2011, 186, 4546–4550. [Google Scholar] [CrossRef] [PubMed]
- Geremia, A.; Arancibia-Carcamo, C.V.; Fleming, M.P.; Rust, N.; Singh, B.; Mortensen, N.J.; Travis, S.P.; Powrie, F. IL-23—responsive innate lymphoid cells are increased in inflammatory bowel disease. J. Exp. Med. 2011, 208, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Hepworth, M.R.; Fung, T.C.; Masur, S.H.; Kelsen, J.R.; McConnell, F.M.; Dubrot, J.; Withers, D.R.; Hugues, S.; Farrar, M.A.; Reith, W.; et al. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4+ T cells. Science 2015, 348, 1031–1035. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.H.; Keates, S.; Bassani, L.; Mayer, L.F.; Keates, A.C. Colonic epithelial cells are a major site of macrophage inflammatory protein 3 alpha (MIP-3alpha) production in normal colon and inflammatory bowel disease. Gut 2002, 51, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Choi, S.C.; Lee, M.H.; Oh, H.M.; Choi, E.Y.; Choi, E.J.; Yun, K.J.; Seo, G.S.; Kim, S.W.; Lee, J.G.; et al. Increased expression of MIP-3alpha/CCL20 in peripheral blood mononuclear cells from patients with ulcerative colitis and its down-regulation by sulfasalazine and glucocorticoid treatment. Inflamm. Bowel Dis. 2005, 11, 1070–1079. [Google Scholar] [CrossRef] [PubMed]
- Puleston, J.; Cooper, M.; Murch, S.; Bid, K.; Makh, S.; Ashwood, P.; Bingham, A.H.; Green, H.; Moss, P.; Dhillon, A.; et al. A distinct subset of chemokines dominates the mucosal chemokine response in inflammatory bowel disease. Aliment. Pharmacol. Ther. 2005, 21, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Macho-Fernandez, E.; Koroleva, E.P.; Spencer, C.M.; Tighe, M.; Torrado, E.; Cooper, A.M.; Fu, Y.X.; Tumanov, A.V. Lymphotoxin beta receptor signalling limits mucosal damage through driving IL-23 production by epithelial cells. Mucosal Immunol. 2015, 8, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Marafini, I.; Monteleone, I.; Dinallo, V.; Di Fusco, D.; De Simone, V.; Laudisi, F.; Fantini, M.C.; Di Sabatino, A.; Pallone, F.; Monteleone, G. CCL20 is negatively regulated by TGF-β1 in intestinal epithelial cells and reduced in Crohn’s disease patients with a successful response to Mongersen, a smad7 antisense oligonucleotide. J. Crohns Colitis 2017, 11, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.L.; Ip, P.P.; Liao, F. CCR6 deficiency impairs IgA production and dysregulates antimicrobial peptide production, altering the intestinal flora. Front. Immunol. 2017, 8, 805. [Google Scholar] [CrossRef] [PubMed]
- Lugering, A.; Floer, M.; Westphal, S.; Maaser, C.; Spahn, T.W.; Schmidt, M.A.; Domschke, W.; Williams, I.R.; Kucharzik, T. Absence of CCR6 inhibits CD4+ regulatory T cell development and M cell formation inside peyer’s patches. Am. J. Pathol. 2005, 166, 1647–1654. [Google Scholar] [CrossRef]
- Williams, I.R. CCR6 and CCL20 partners in intestinal immunity and lymphorganogenesis. Ann. N. Y. Acad. Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Westphal, S.; Lugering, A.; von Wedel, J.; von Eiff, C.; Maaser, C.; Spahn, T.; Heusipp, G.; Schmidt, M.A.; Herbst, H.; Williams, I.R.; et al. Resistance of chemokine receptor 6-deficient mice to Yersinia Enterocolitica infection: Evidence of defective M-cell formation in vivo. Am. J. Pathol. 2008, 172, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Saigusa, S.; Araki, T.; Tanaka, K.; Okita, Y.; Fujikawa, H.; Kawamura, M.; Okugawa, Y.; Toiyama, Y.; Inoue, Y.; et al. Correlation of CCL20 expression in rectal mucosa with the development of ulcerative colitis-associated neoplasia. Oncol. Lett. 2013, 6, 1271–1276. [Google Scholar] [CrossRef] [PubMed]
- Brand, S.; Olszak, T.; Beigel, F.; Diebold, J.; Otte, J.M.; Eichhorst, S.T.; Goke, B.; Dambacher, J. Cell differentiation dependent expressed CCR6 mediates ERK-1/2, SAPK/JNK and Akt signalling resulting in proliferation and migration of colorectal cancer cells. J. Cell Biochem. 2006, 97, 709–723. [Google Scholar] [CrossRef] [PubMed]
- Frick, V.O.; Rubie, C.; Kolsch, K.; Wagner, M.; Ghadjar, P.; Graeber, S.; Glanemann, M. CCR6/CCL20 chemokine expression profile in distinct colorectal malignancies. Scand. J. Immunol. 2013, 78, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Nandi, B.; Shapiro, M.; Samur, M.K.; Pai, C.; Frank, N.Y.; Yoon, C.; Prabhala, R.H.; Munshi, N.C.; Gold, J.S. Stromal CCR6 drives tumor growth in a murine transplantable colon cancer through recruitment of tumor-promoting macrophages. Oncoimmunology 2016, 5, e1189052. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, P.; Gosselin, A.; Wacleche, V.S.; El-Far, M.; Said, E.A.; Kared, H.; Grandvaux, N.; Boulassel, M.R.; Routy, J.P.; Ancuta, P. Memory CCR6+CD4+ T cells are preferential targets for productive HIV type 1 infection regardless of their expression of integrin β7. J. Immunol. 2011, 186, 4618–4630. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, C.M.; Ackermann, P.J.; Ostermann, A.L.; Adams-Quack, P.; Vogt, C.; Tran, M.L.; Nikolaiev, A.; Waisman, A.; Garbers, C.; Theurich, S.; et al. Obesity exacerbates colitis-associated cancer via IL-6-regulated macrophage polarisation and CCL-20/CCR-6-mediated lymphocyte recruitment. Nat. Commun. 2018, 9, 1646. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Farber, J.M.; Kelsall, B.L. CCR6 marks regulatory T cells as a colon-tropic, IL-10-producing phenotype. J. Immunol. 2010, 185, 3295–3304. [Google Scholar] [CrossRef] [PubMed]
- Rivino, L.; Gruarin, P.; Haringer, B.; Steinfelder, S.; Lozza, L.; Steckel, B.; Weick, A.; Sugliano, E.; Jarrossay, D.; Kuhl, A.A.; et al. CCR6 is expressed on an IL-10-producing, autoreactive memory T cell population with context-dependent regulatory function. J. Exp. Med. 2010, 207, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Wu, K.; Zhao, E.; Wei, S.; Vatan, L.; Szeliga, W.; Huang, E.; Greenson, J.; Chang, A.; Rolinski, J.; et al. IL-17+ regulatory T cells in the microenvironments of chronic inflammation and cancer. J. Immunol. 2011, 186, 4388–4395. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Wang, L.; Wu, K.; Li, W.; Zhao, E.; Cui, T.; Wei, S.; Liu, Y.; Wang, Y.; Vatan, L.; et al. Inflammatory regulatory T cells in the microenvironments of ulcerative colitis and colon carcinoma. Oncoimmunology 2016, 5, e1105430. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, N.; Meitei, H.T.; Sonar, S.A.; Sharma, P.K.; Mujeeb, V.R.; Srivastava, S.; Boppana, R.; Lal, G. CCR6 signaling inhibits suppressor function of induced-Treg during gut inflammation. J. Autoimmun. 2018, 88, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Godefroy, E.; Alameddine, J.; Montassier, E.; Mathé, J.; Desfrançois-Noël, J.; Marec, N.; Bossard, C.; Jarry, A.; Bridonneau, C.; Le Roy, A.; et al. Expression of CCR6 and CXCR6 by gut-derived CD4+/CD8alpha+ T-regulatory cells which are decreased in blood samples from patients with inflammatory bowel diseases. Gastroenterology 2018. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Yang, X.O.; Chung, Y.; Fukunaga, A.; Nurieva, R.; Pappu, B.; Martin-Orozco, N.; Kang, H.S.; Ma, L.; Panopoulos, A.D.; et al. CCR6 regulates the migration of inflammatory and regulatory T cells. J. Immunol. 2008, 181, 8391–8401. [Google Scholar] [CrossRef] [PubMed]
- Lochner, M.; Peduto, L.; Cherrier, M.; Sawa, S.; Langa, F.; Varona, R.; Riethmacher, D.; Si-Tahar, M.; Di Santo, J.P.; Eberl, G. In vivo equilibrium of pro-inflammatory IL-17+ and regulatory IL-10+ Foxp3+ RORγt+ T cells. J. Exp. Med. 2008, 205, 1381–1393. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, A.; Rudra, D.; Treuting, P.; Samstein, R.M.; Liang, Y.; Kas, A.; Rudensky, A.Y. CD4+ regulatory T cells control Th17 responses in a Stat3-dependent manner. Science 2009, 326, 986–991. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Ludwiczek, T.; Holzmann, S.; Moschen, A.R.; Weiss, G.; Enrich, B.; Graziadei, I.; Dunzendorfer, S.; Wiedermann, C.J.; Murzl, E.; et al. Increased expression of CCL20 in human inflammatory bowel disease. J. Clin. Immunol. 2004, 24, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Dieu-Nosjean, M.; Massacrier, C.; Vanbervliet, B.; Fridman, W.; Caux, C. IL-10 induces CCR6 expression during Langerhans cell development while IL-4 and IFN-γ suppress it. J. Immunol. 2001, 167, 5594–5602. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Gonzalez, R.M.; Niess, J.H.; Zammit, D.J.; Ravindran, R.; Srinivasan, A.; Maxwell, J.R.; Stoklasek, T.; Yadav, R.; Williams, I.R.; Gu, X.; et al. CCR6-mediated dendritic cell activation of pathogen-specific T cells in Peyer’s patches. Immunity 2006, 24, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Yamakawa, M.; Hiroaki, T.; Kawata, S.; Kimura, O. Correlation of dendritic cell infiltration with active crypt inflammation in ulcerative colitis. Clin. Immunol. 2007, 122, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, K.R.R.; Laffont, S.; Powrie, F. E-Cadherin marks a subset of inflammatory dendritic cells that promote T cell mediated-colitis. Immunity 2010, 32, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Hornung, M.; Werner, J.M.; Farkas, S.; Schlitt, H.J.; Geissler, E.K. Migration and chemokine receptor pattern of colitis-preventing DX5+NKT cells. Int. J. Colorectal Dis. 2011, 26, 1423–1433. [Google Scholar] [CrossRef] [PubMed]
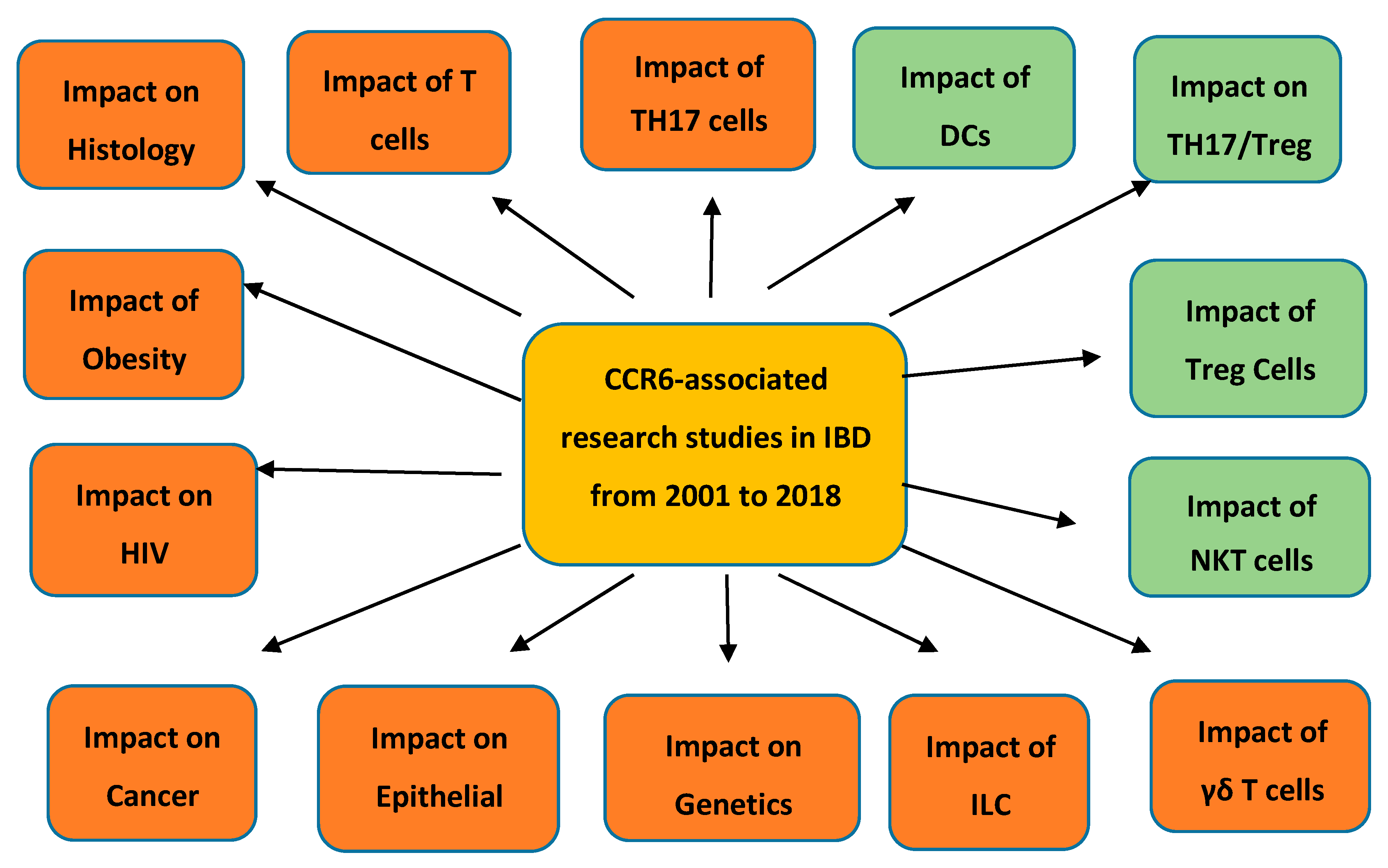
 Pro-inflammatory analyses;
Pro-inflammatory analyses;  Anti-inflammatory analyses. This figure describes both the pro-inflammatory and anti-inflammatory analyses involving the chemokines CCR6 and CCL20 in inflammatory bowel disease (IBD) published by research workers during the time of 2001 to 2018.
Pro-inflammatory analyses; Anti-inflammatory analyses. This figure describes both the pro-inflammatory and anti-inflammatory analyses involving the chemokines CCR6 and CCL20 in inflammatory bowel disease (IBD) published by research workers during the time of 2001 to 2018.
Anti-inflammatory analyses. This figure describes both the pro-inflammatory and anti-inflammatory analyses involving the chemokines CCR6 and CCL20 in inflammatory bowel disease (IBD) published by research workers during the time of 2001 to 2018.
Pro-inflammatory analyses; Anti-inflammatory analyses. This figure describes both the pro-inflammatory and anti-inflammatory analyses involving the chemokines CCR6 and CCL20 in inflammatory bowel disease (IBD) published by research workers during the time of 2001 to 2018.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranasinghe, R.; Eri, R. CCR6–CCL20 Axis in IBD: What Have We Learnt in the Last 20 Years? Gastrointest. Disord. 2019, 1, 57-74. https://doi.org/10.3390/gidisord1010006
Ranasinghe R, Eri R. CCR6–CCL20 Axis in IBD: What Have We Learnt in the Last 20 Years? Gastrointestinal Disorders. 2019; 1(1):57-74. https://doi.org/10.3390/gidisord1010006
Chicago/Turabian StyleRanasinghe, Ranmali, and Rajaraman Eri. 2019. "CCR6–CCL20 Axis in IBD: What Have We Learnt in the Last 20 Years?" Gastrointestinal Disorders 1, no. 1: 57-74. https://doi.org/10.3390/gidisord1010006
APA StyleRanasinghe, R., & Eri, R. (2019). CCR6–CCL20 Axis in IBD: What Have We Learnt in the Last 20 Years? Gastrointestinal Disorders, 1(1), 57-74. https://doi.org/10.3390/gidisord1010006