The Effect of Mn Doping and Ti3+ Defects at TiO2 Surfaces in NO and SO2 Gas Capture Investigated Using Near-Ambient Pressure X-ray Photoelectron Spectroscopy



Abstract

1. Introduction
2. Experimental Section
2.1. Samples
2.2. XPS Measurements
3. Results and Discussion
3.1. Single Crystal TiO2(110) Surfaces
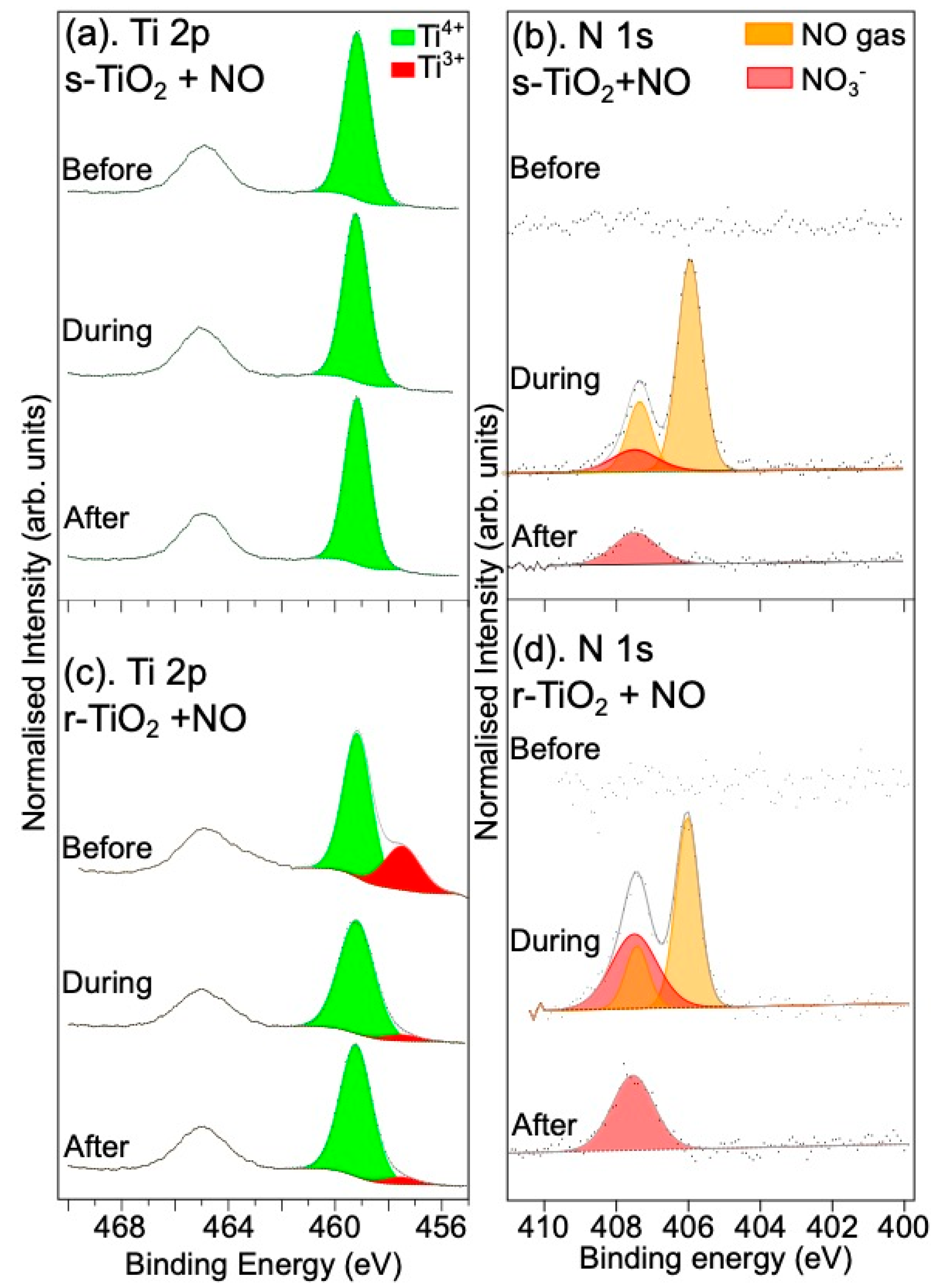
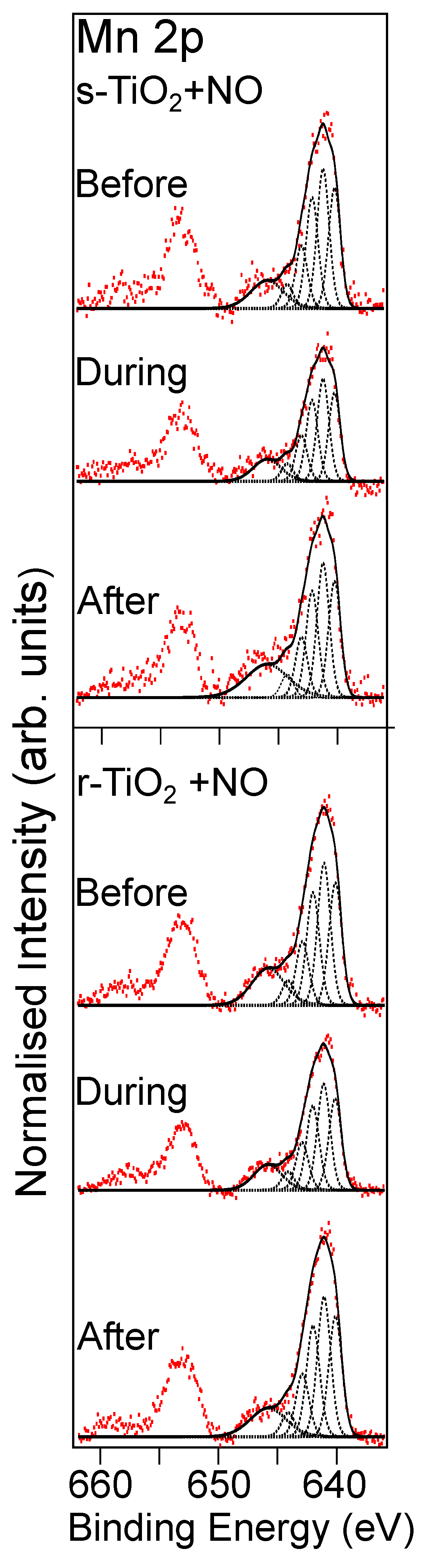
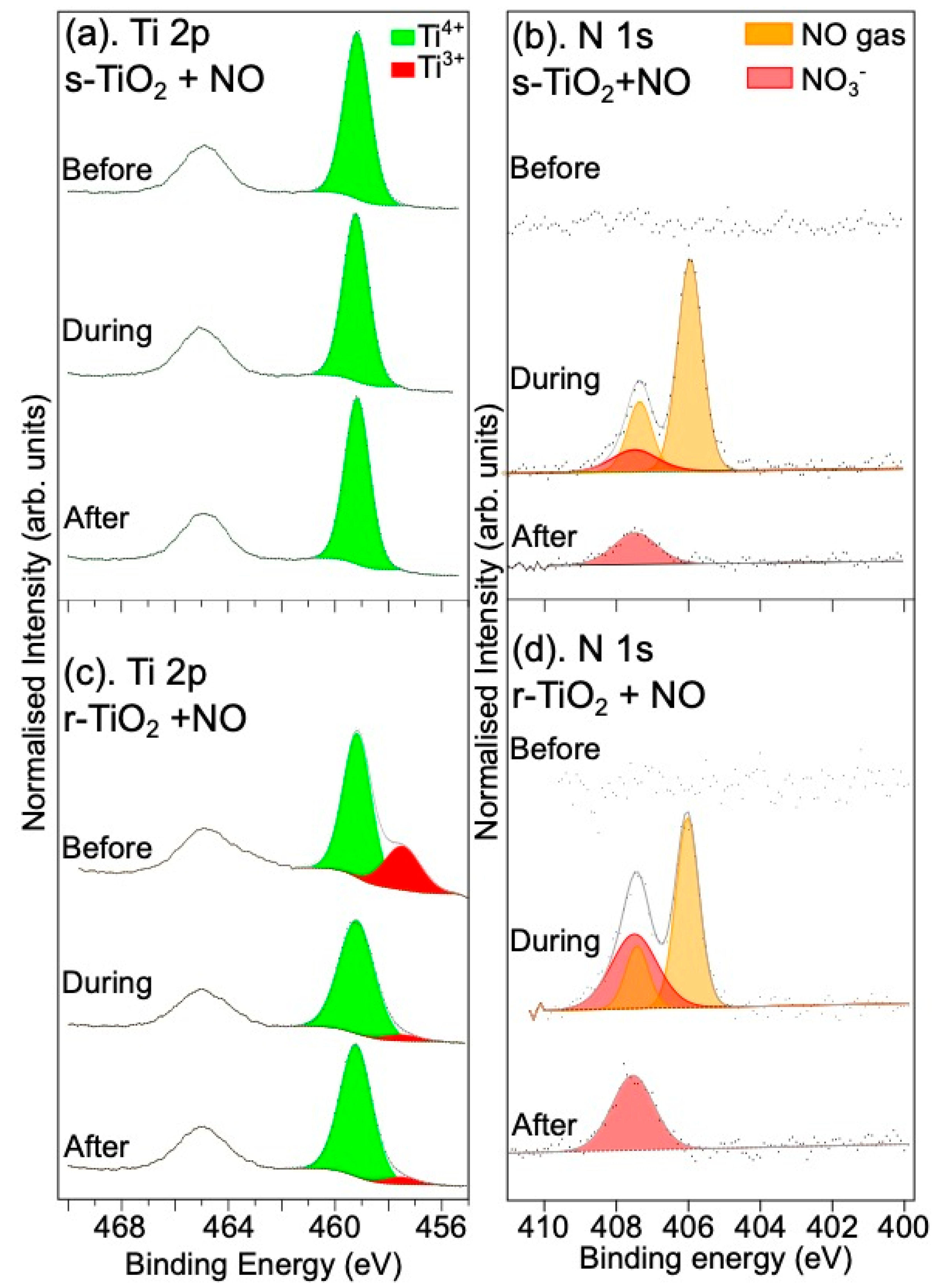
3.1.1. Reaction with NO Gas
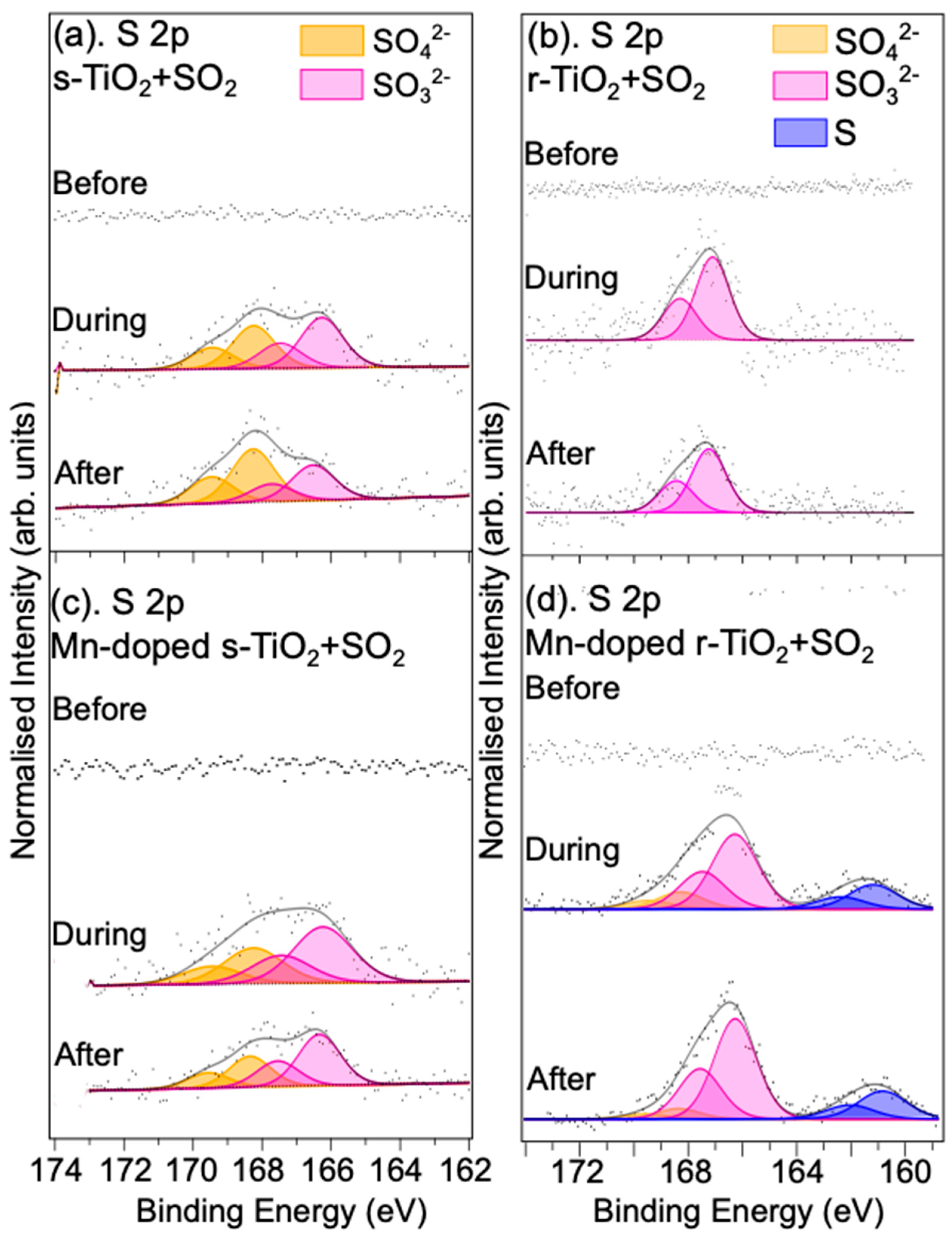
3.1.2. Reaction with SO2 Gas
3.2. TiO2 Nanopowders
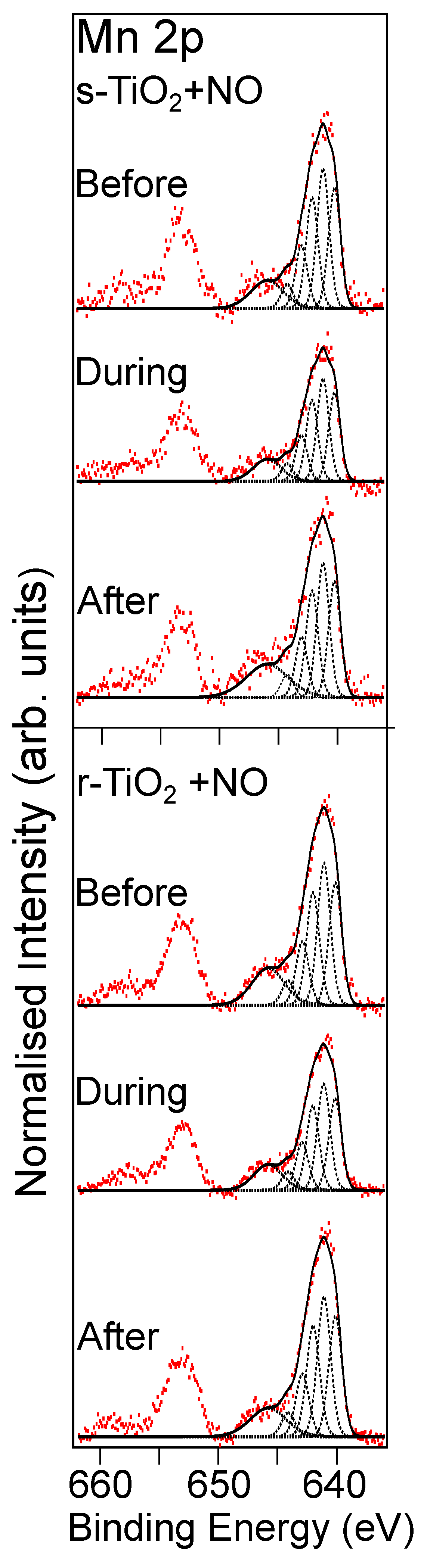
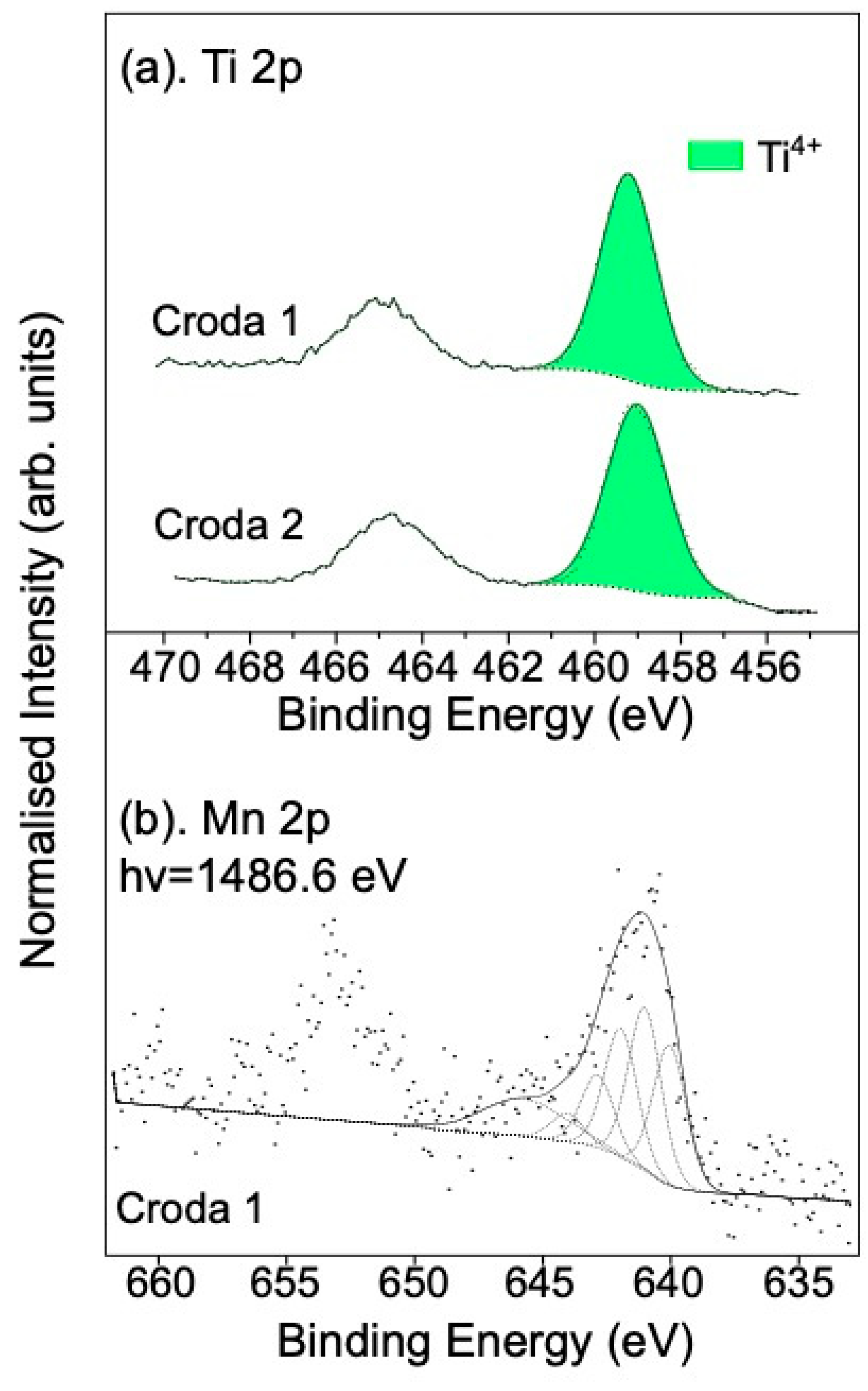
3.2.1. Reaction with NO Gas
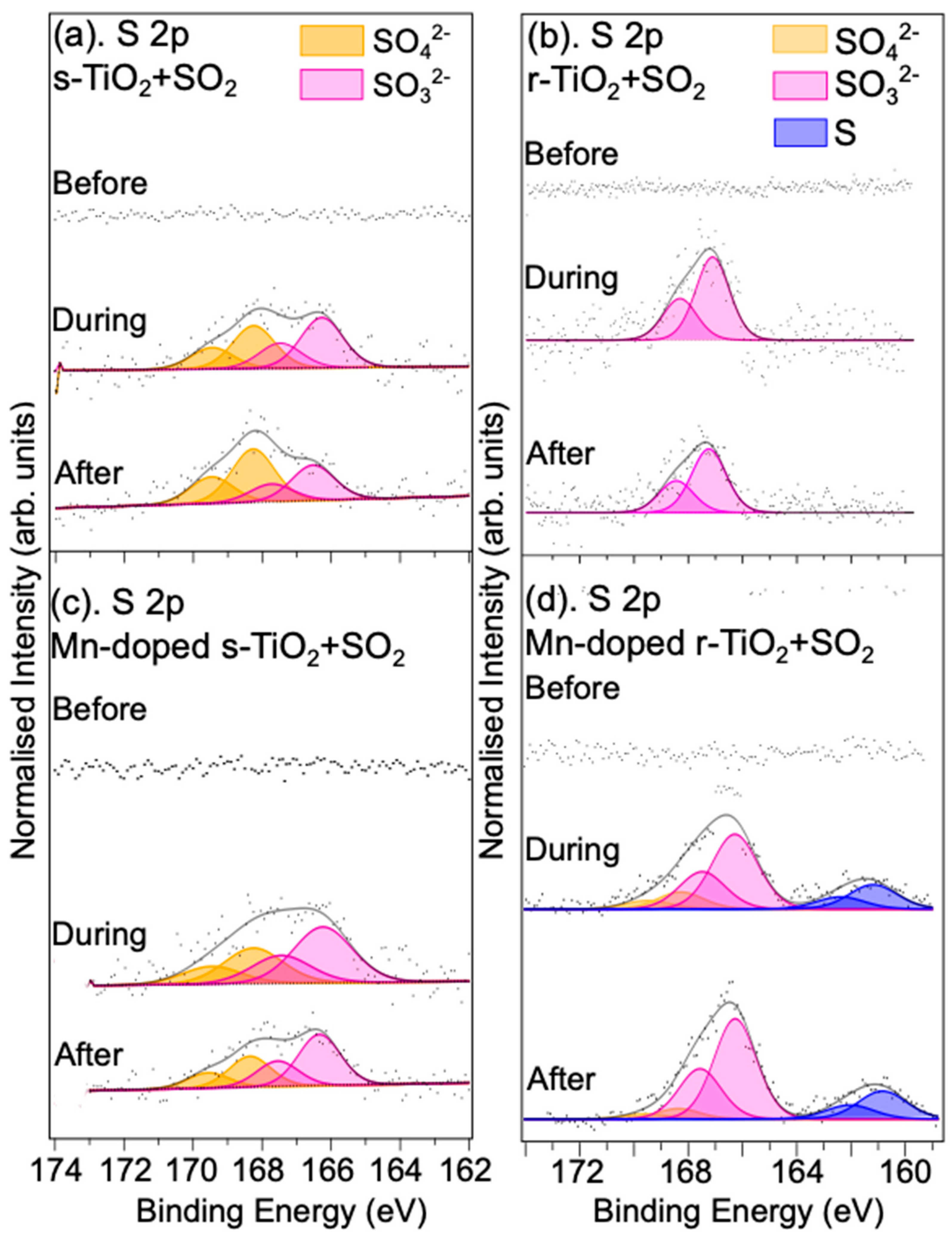
3.2.2. Reaction with SO2/Ar Gas
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jaroenworaluck, A.; Sunsaneeyametha, W.; Kosachan, N.; Stevens, R. Characteristics of Silica-Coated TiO2 and Its UV Absorption for Sunscreen Cosmetic Applications. Surf. Interface Anal. 2006, 38, 473–477. [Google Scholar] [CrossRef]
- Zinelis, S.; Silikas, N.; Thomas, A.; Syres, K.; Eliades, G. Surface Characterization of SLActive Dental Implants. Eur. J. Esthet. Dent. Off. J. Eur. Acad. Esthet. Dent. 2012, 7, 72–92. [Google Scholar]
- Thomas, A.; Syres, K. Adsorption of Organic Molecules on Rutile TiO2 and Anatase TiO2 Single Crystal Surfaces. Chem. Soc. Rev. 2012, 41, 4207–4217. [Google Scholar] [CrossRef]
- Morris, D.; Egdell, R.G. Application of V-Doped TiO2 as a Sensor for Detection of SO2. J. Mater. Chem. 2001, 11, 3207–3210. [Google Scholar] [CrossRef]
- Dalton, J.S.; Janes, P.A.; Jones, N.G.; Nicholson, J.A.; Hallam, K.R.; Allen, G.C. Photocatalytic Oxidation of NOx Gases Using TiO2: A Surface Spectroscopic Approach. Environ. Pollut. 2002, 120, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Kampa, M.; Castanas, E. Human Health Effects of Air Pollution. Environ. Pollut. 2008, 151, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Mancebo, S.E.; Wang, S.Q. Recognizing the Impact of Ambient Air Pollution on Skin Health. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 2326–2332. [Google Scholar] [CrossRef]
- Cerza, F.; Renzi, M.; Gariazzo, C.; Davoli, M.; Michelozzi, P.; Forastiere, F.; Cesaroni, G. Long-Term Exposure to Air Pollution and Hospitalization for Dementia in the Rome Longitudinal Study. Environ. Health 2019, 18, 72. [Google Scholar] [CrossRef]
- Rosseler, O.; Sleiman, M.; Montesinos, V.N.; Shavorskiy, A.; Keller, V.; Keller, N.; Litter, M.I.; Bluhm, H.; Salmeron, M.; Destaillats, H. Chemistry of NOx on TiO2 Surfaces Studied by Ambient Pressure XPS: Products, Effect of UV Irradiation, Water, and Coadsorbed K+. J. Phys. Chem. Lett. 2013, 4, 536–541. [Google Scholar] [CrossRef]
- Ettireddy, P.R.; Ettireddy, N.; Mamedov, S.; Boolchand, P.; Smirniotis, P.G. Surface Characterization Studies of TiO2 Supported Manganese Oxide Catalysts for Low Temperature SCR of NO with NH3. Appl. Catal. B Environ. 2007, 76, 123–134. [Google Scholar] [CrossRef]
- Khlyustova, A.; Sirotkin, N.; Kusova, T.; Kraev, A.; Titov, V.; Agafonov, A. Doped TiO2: The Effect of Doping Elements on Photocatalytic Activity. Mater. Adv. 2020, 1, 1193–1201. [Google Scholar] [CrossRef]
- Janczarek, M.; Kowalska, E. Defective Dopant-Free TiO2 as an Efficient Visible Light-Active Photocatalyst. Catalysts 2021, 11, 978. [Google Scholar] [CrossRef]
- Fan, L.; Ichikuni, N.; Shimazu, S.; Uematsu, T. Preparation of Au/TiO2 Catalysts by Suspension Spray Reaction Method and Their Catalytic Property for CO Oxidation. Appl. Catal. A Gen. 2003, 246, 87–95. [Google Scholar] [CrossRef]
- Binas, V.D.; Sambani, K.; Maggos, T.; Katsanaki, A.; Kiriakidis, G. Synthesis and Photocatalytic Activity of Mn-Doped TiO2 Nanostructured Powders under UV and Visible Light. Appl. Catal. B Environ. 2012, 113–114, 79–86. [Google Scholar] [CrossRef]
- Nam, K.B.; Kwon, D.W.; Hong, S.C. DRIFT Study on Promotion Effects of Tungsten-Modified Mn/Ce/Ti Catalysts for the SCR Reaction at Low-Temperature. Appl. Catal. A Gen. 2017, 542, 55–62. [Google Scholar] [CrossRef]
- Jia, B.; Guo, J.; Luo, H.; Shu, S.; Fang, N.; Li, J. Study of NO Removal and Resistance to SO2 and H2O of MnOx/TiO2, MnOx/ZrO2 and MnOx/ZrO2–TiO2. Appl. Catal. A Gen. 2018, 553, 82–90. [Google Scholar] [CrossRef]
- Shayegan, Z.; Lee, C.-S.; Haghighat, F. TiO2 Photocatalyst for Removal of Volatile Organic Compounds in Gas Phase—A Review. Chem. Eng. J. 2018, 334, 2408–2439. [Google Scholar] [CrossRef]
- Venezia, A.M. X-Ray Photoelectron Spectroscopy (XPS) for Catalysts Characterization. Catal. Today 2003, 77, 359–370. [Google Scholar] [CrossRef]
- Zhong, L.; Chen, D.; Zafeiratos, S. A Mini Review of in Situ Near-Ambient Pressure XPS Studies on Non-Noble, Late Transition Metal Catalysts. Catal. Sci. Technol. 2019, 9, 3851–3867. [Google Scholar] [CrossRef]
- Wagstaffe, M.; Hussain, H.; Acres, M.J.; Jones, R.; Syres, K.L.; Thomas, A.G. Structure and Reactivity of a Model Oxide Supported Silver Nanocluster Catalyst Studied by Near Ambient Pressure X-ray Photoelectron Spectroscopy. J. Phys. Chem. C 2017, 121, 21383–21389. [Google Scholar] [CrossRef]
- Herranz, T.; Deng, X.; Cabot, A.; Alivisatos, P.; Liu, Z.; Soler-Illia, G.; Salmeron, M. Reactivity of Au Nanoparticles Supported over SiO2 and TiO2 Studied by Ambient Pressure Photoelectron Spectroscopy. Catal. Today 2009, 143, 158–166. [Google Scholar] [CrossRef]
- Jackman, M.J.; Thomas, A.G.; Muryn, C. Photoelectron Spectroscopy Study of Stoichiometric and Reduced Anatase TiO2(101) Surfaces: The Effect of Subsurface Defects on Water Adsorption at Near-Ambient Pressures. J. Phys. Chem. C 2015, 119, 13682–13690. [Google Scholar] [CrossRef]
- Chun-Ren Ke, J.; Walton, A.S.; Lewis, D.J.; Tedstone, A.; O’Brien, P.; Thomas, A.G.; Flavell, W.R. In Situ Investigation of Degradation at Organometal Halide Perovskite Surfaces by X-ray Photoelectron Spectroscopy at Realistic Water Vapour Pressure. Chem. Commun. 2017, 53, 5231–5234. [Google Scholar] [CrossRef] [PubMed]
- Diebold, U. The Surface Science of Titanium Dioxide. Surf. Sci. Rep. 2003, 48, 53–229. [Google Scholar] [CrossRef]
- Wendt, S.; Schaub, R.; Matthiesen, J.; Vestergaard, E.K.; Wahlstrom, E.; Rasmussen, M.D.; Thostrup, P.; Molina, L.M.; Laegsgaard, E.; Stensgaard, I.; et al. Oxygen Vacancies on TiO2(110) and Their Interaction with H2O and O2: A Combined High-Resolution STM and DFT Study. Surf. Sci. 2005, 598, 226–245. [Google Scholar] [CrossRef]
- Byrne, C.; Brennan, B.; McCoy, A.P.; Bogan, J.; Brady, A.; Hughes, G. In Situ XPS Chemical Analysis of MnSiO3 Copper Diffusion Barrier Layer Formation and Simultaneous Fabrication of Metal Oxide Semiconductor Electrical Test MOS Structures. ACS Appl. Mater. Interfaces 2016, 8, 2470–2477. [Google Scholar] [CrossRef]
- Smith, G.C. Evaluation of a Simple Correction for the Hydrocarbon Contamination Layer in Quantitative Surface Analysis by XPS. J. Electron Spectrosc. Relat. Phenom. 2005, 148, 21–28. [Google Scholar] [CrossRef]
- Fairley, N. CasaXPS Manual 2.3.15 Introduction to XPS and AES; Casa Software: Valencia, Spain, 2009. [Google Scholar]
- Gupta, V.K.; Balzaretti, F.; Guo, P.; Köppen, S.; Frauenheim, T.; Dominguez, A. NO Degradation on the Anatase TiO2 (001) Surface in the Presence of Water. J. Phys. Chem. C 2022, 126, 17544–17553. [Google Scholar] [CrossRef]
- Pang, C.L.; Lindsay, R.; Thornton, G. Chemical Reactions on Rutile TiO2(110). Chem. Soc. Rev. 2008, 37, 2328–2353. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Payne, B.P.; Grosvenor, A.P.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. Resolving Surface Chemical States in XPS Analysis of First Row Transition Metals, Oxides and Hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 2011, 257, 2717–2730. [Google Scholar] [CrossRef]
- Gupta, R.P.; Sen, S.K. Calculation of Multiplet Structure of Core p-Vacancy Levels. Phys. Rev. B 1974, 10, 71–77. [Google Scholar] [CrossRef]
- Gupta, R.P.; Sen, S.K. Calculation of Multiplet Structure of Core p-Vacancy Levels. II. Phys. Rev. B 1975, 12, 15–19. [Google Scholar] [CrossRef]
- Chen, D.A.; Bartelt, M.C.; Seutter, S.M.; McCarty, K.F. Small, Uniform, and Thermally Stable Silver Particles on TiO2(110)-(1×1). Surf. Sci. 2000, 464, L708–L714. [Google Scholar] [CrossRef]
- Jiang, B.Q.; Wu, Z.B.; Liu, Y.; Lee, S.C.; Ho, W.K. DRIFT Study of the SO2 Effect on Low-Temperature SCR Reaction over Fe−Mn/TiO2. J. Phys. Chem. C 2010, 114, 4961–4965. [Google Scholar] [CrossRef]
- Matsumoto, S.; Ikeda, Y.; Suzuki, H.; Ogai, M.; Miyoshi, N. NOx Storage-Reduction Catalyst for Automotive Exhaust with Improved Tolerance against Sulfur Poisoning. Appl. Catal. B Environ. 2000, 25, 115–124. [Google Scholar] [CrossRef]
- Turner, N.H.; Murday, J.S.; Ramaker, D.E. Quantitative Determination of Surface Composition of Sulfur Bearing Anion Mixtures by Auger Electron Spectroscopy. Anal. Chem. 1980, 52, 84–92. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Liu, G.; Jirsak, T.; Hrbek, J.; Chang, Z.; Dvorak, J.; Maiti, A. Activation of Gold on Titania: Adsorption and Reaction of SO2 on Au/TiO2(110). J. Am. Chem. Soc. 2002, 124, 5242–5250. [Google Scholar] [CrossRef]
- Sayago, D.I.; Serrano, P.; Böhme, O.; Goldoni, A.; Paolucci, G.; Román, E.; Martín-Gago, J.A. Adsorption and Desorption SO2 on the TiO2(110) Surface: A Photoemission Study. Phys. Rev. B 2001, 64, 205402. [Google Scholar] [CrossRef]
- Sayago, D.I.; Serrano, P.; Böhme, O.; Goldoni, A.; Paolucci, G.; Román, E.; Martín-Gago, J.A. A Photoemission Study of the SO2 Adsorption on TiO2 (110) Surfaces. Surf. Sci. 2001, 482–485, 9–14. [Google Scholar] [CrossRef]
- Sun, K.; Zhang, Q.; Bock, D.C.; Tong, X.; Su, D.; Marschilok, A.C.; Takeuchi, K.J.; Takeuchi, E.S.; Gan, H. Interaction of TiS2 and Sulfur in Li-S Battery System. J. Electrochem. Soc. 2017, 164, A1291. [Google Scholar] [CrossRef]
- Franzen, H.F.; Umaña, M.X.; McCreary, J.R.; Thorn, R.J. XPS Spectra of Some Transition Metal and Alkaline Earth Monochalcogenides. J. Solid State Chem. 1976, 18, 363–368. [Google Scholar] [CrossRef]
- Sorescu, D.C.; Rusu, C.N.; Yates, J.T. Adsorption of NO on the TiO2(110) Surface: An Experimental and Theoretical Study. J. Phys. Chem. B 2000, 104, 4408–4417. [Google Scholar] [CrossRef]
- Briggs, D.; Seah, M.P. Practical Surface Analysis: Auger and Photoelectron Spectroscopy, 2nd ed.; John Wiley and Sons: Chichester, UK, 1990. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total Nads | NO3− | NO2− |
|---|---|---|---|
| s-TiO2(110) | 0.03 ± 0.01 | 0.03 ± 0.01 | N.D. * |
| r-TiO2(110) | 0.08 ± 0.02 | 0.08 ± 0.02 | N.D. |
| Mn-doped s-TiO2(110) | 0.06 ± 0.02 | 0.06 ± 0.01 | N.D. |
| Mn-doped r-TiO2(110) | 0.41 ± 0.05 | 0.16 ± 0.02 | 0.25 ± 0.05 |
| Sample | Total S | SO42− | SO32− | Sulphide |
|---|---|---|---|---|
| s-TiO2(110) | 0.08 ± 0.02 | 0.04 ± 0.01 | 0.04 ± 0.01 | 0.00 ± 0.01 |
| r-TiO2(110) | 0.08 ± 0.01 | 0.00 ± 0.01 | 0.08 ± 0.01 | 0.00 ± 0.01 |
| Mn-doped s-TiO2(110) | 0.09 ± 0.02 | 0.03 ± 0.01 | 0.06 ± 0.01 | 0.00 ± 0.01 |
| Mn-doped r-TiO2(110) | 0.40 ± 0.05 | 0.06 ± 0.01 | 0.25 ± 0.05 | 0.09 ± 0.01 |
| Sample | Total N | NO3− | NOads |
|---|---|---|---|
| Croda 1 | 0.35 ± 0.02 | 0.20 ± 0.01 | 0.15 ± 0.01 |
| Croda 1 + H2O | 0.99 ± 0.05 | 0.60 ± 0.05 | 0.39 ± 0.02 |
| Croda 2 | 0.33 ± 0.02 | 0.23 ± 0.01 | 0.10 ± 0.01 |
| Croda 2 + H2O | 0.35 ± 0.01 | 0.18 ± 0.01 | 0.17 ± 0.01 |
| Sample | Total S | SO42− | SO2 ads |
|---|---|---|---|
| Croda 1 | 0.49 ± 0.01 | 0.36 ± 0.01 | 0.13 ± 0.01 |
| Croda 2 | 0.28 ± 0.01 | 0.23 ± 0.01 | 0.05 ± 0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ke, J.C.-R.; Thomas, A.G.; Peake, J.; Sayer, R. The Effect of Mn Doping and Ti3+ Defects at TiO2 Surfaces in NO and SO2 Gas Capture Investigated Using Near-Ambient Pressure X-ray Photoelectron Spectroscopy. Surfaces 2024, 7, 26-43. https://doi.org/10.3390/surfaces7010003
Ke JC-R, Thomas AG, Peake J, Sayer R. The Effect of Mn Doping and Ti3+ Defects at TiO2 Surfaces in NO and SO2 Gas Capture Investigated Using Near-Ambient Pressure X-ray Photoelectron Spectroscopy. Surfaces. 2024; 7(1):26-43. https://doi.org/10.3390/surfaces7010003
Chicago/Turabian StyleKe, Jack Chun-Ren, Andrew Guy Thomas, Joseph Peake, and Robert Sayer. 2024. "The Effect of Mn Doping and Ti3+ Defects at TiO2 Surfaces in NO and SO2 Gas Capture Investigated Using Near-Ambient Pressure X-ray Photoelectron Spectroscopy" Surfaces 7, no. 1: 26-43. https://doi.org/10.3390/surfaces7010003
APA StyleKe, J. C.-R., Thomas, A. G., Peake, J., & Sayer, R. (2024). The Effect of Mn Doping and Ti3+ Defects at TiO2 Surfaces in NO and SO2 Gas Capture Investigated Using Near-Ambient Pressure X-ray Photoelectron Spectroscopy. Surfaces, 7(1), 26-43. https://doi.org/10.3390/surfaces7010003