Formic Acid Oxidation on Pd Thin Film Coated Au Nanocrystals



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
2.1. Materials
2.2. Synthesis of Au Seeds
2.3. Synthesis of Au Nanocrystals
2.4. Electrochemical Measurement
2.5. Instrumentation
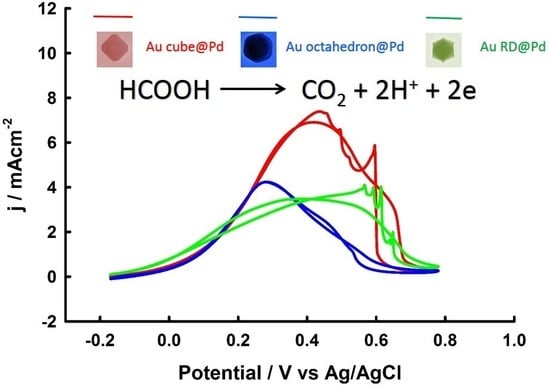
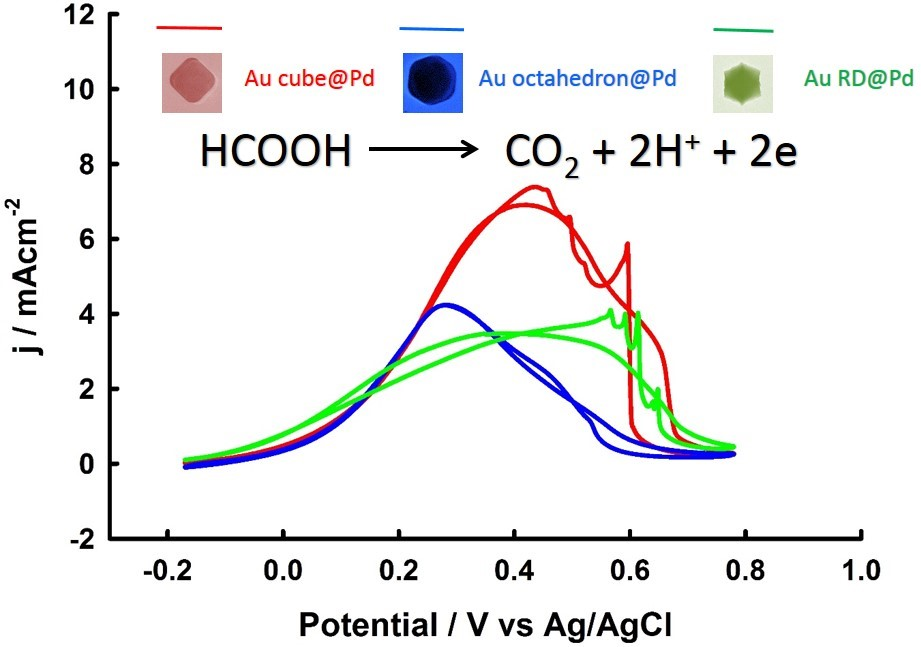
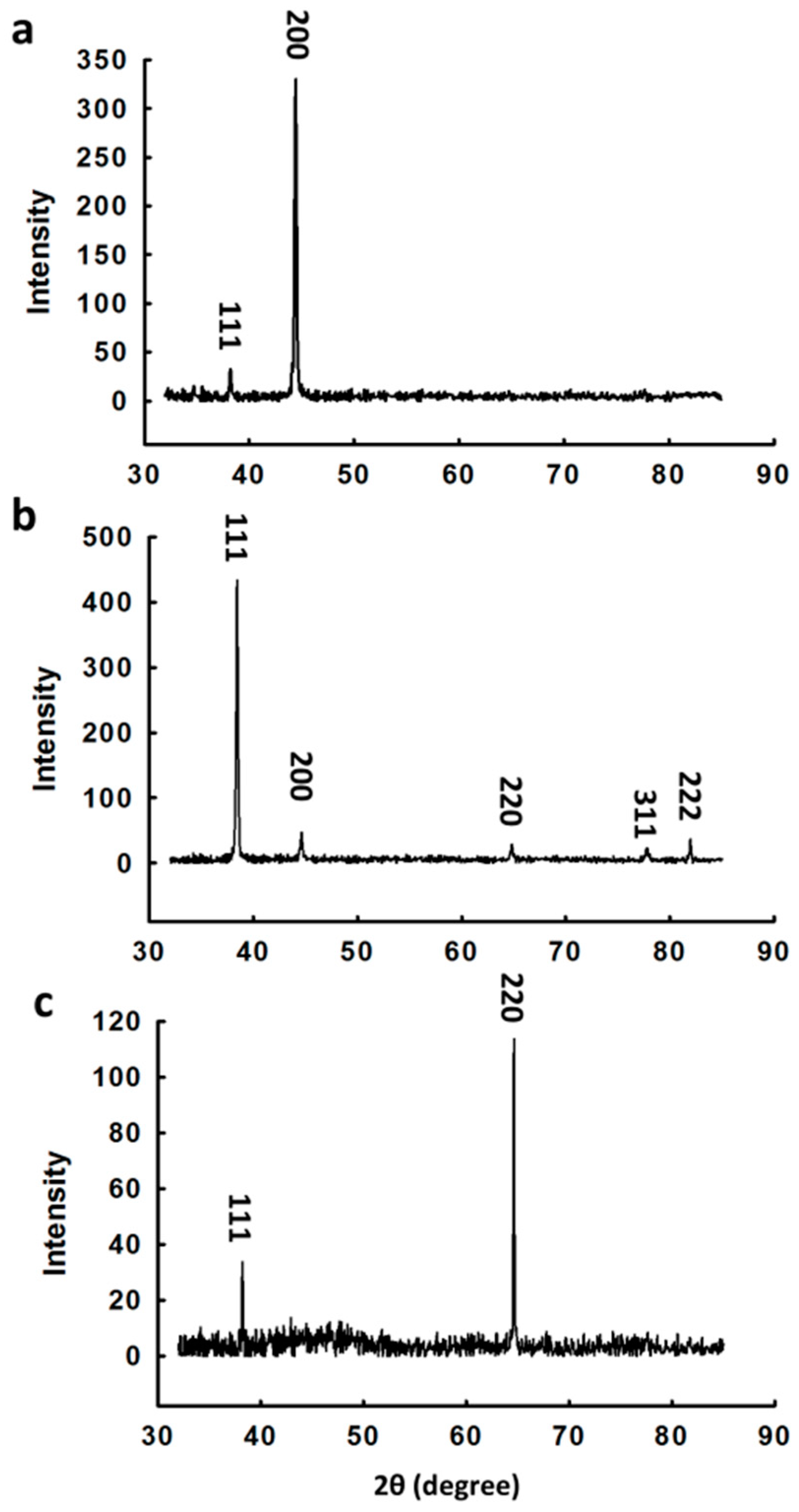
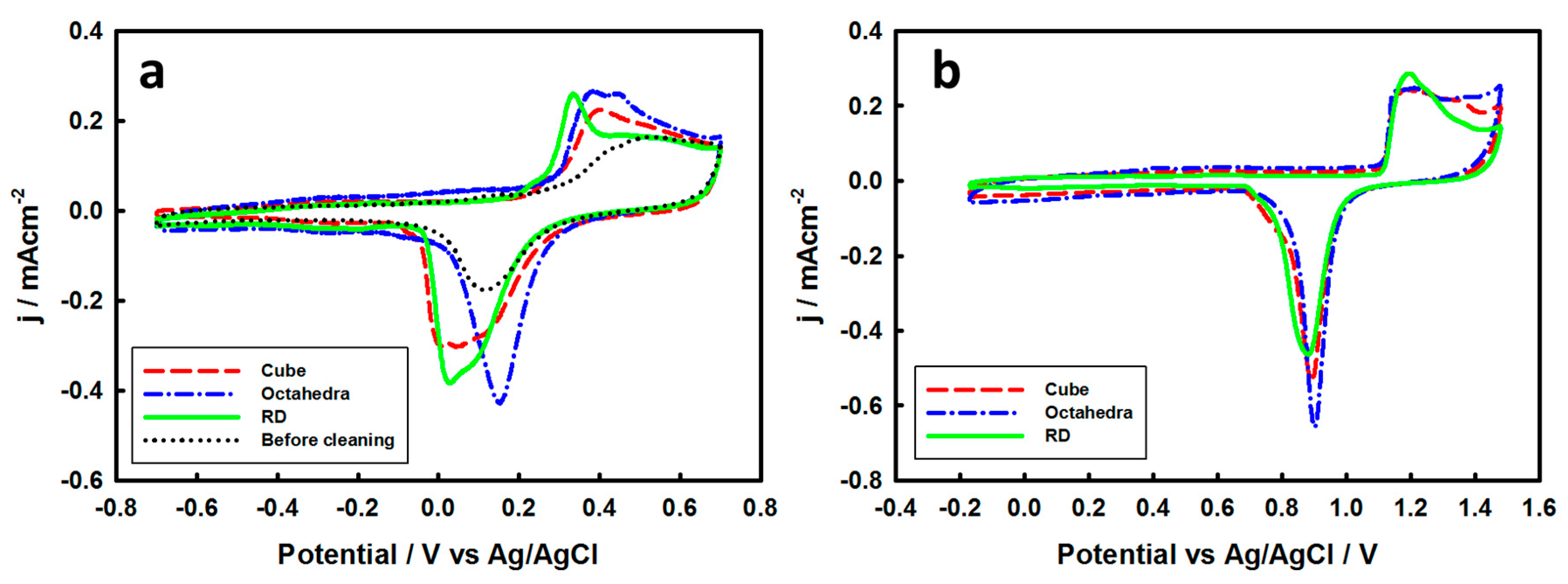
3. Results and Discussion
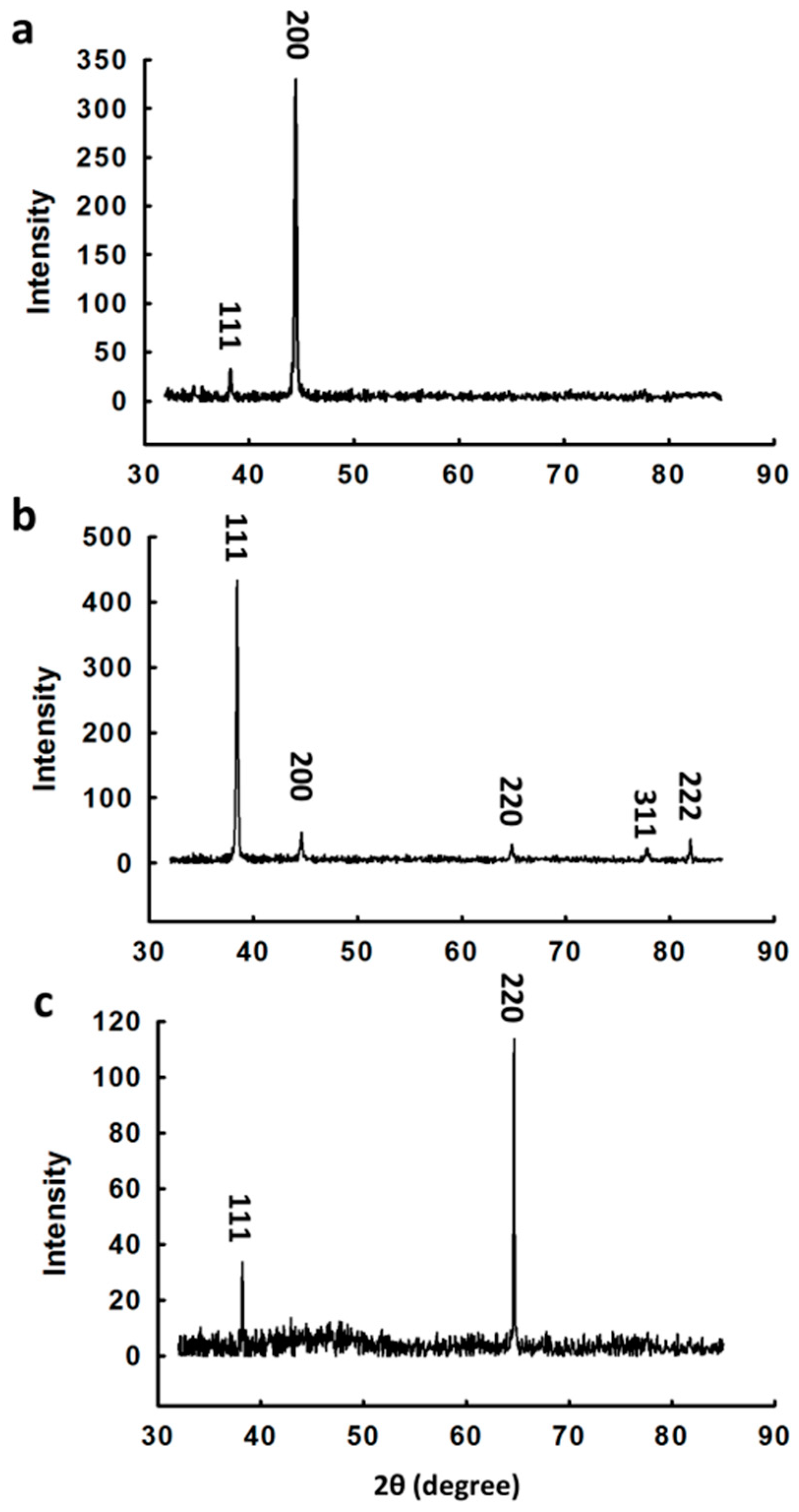
3.1. Synthesis and Characterization of Au Nanocrystals
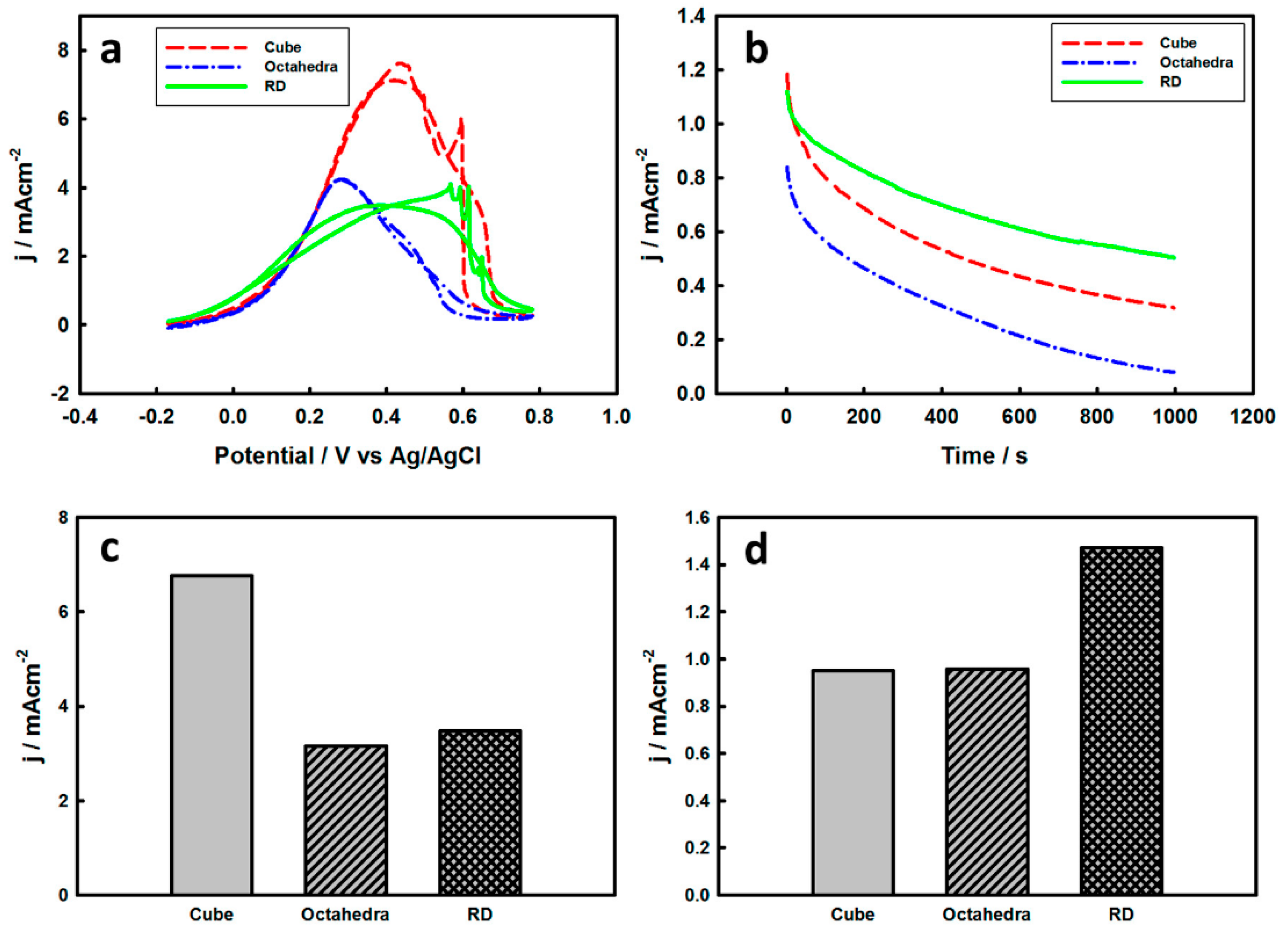
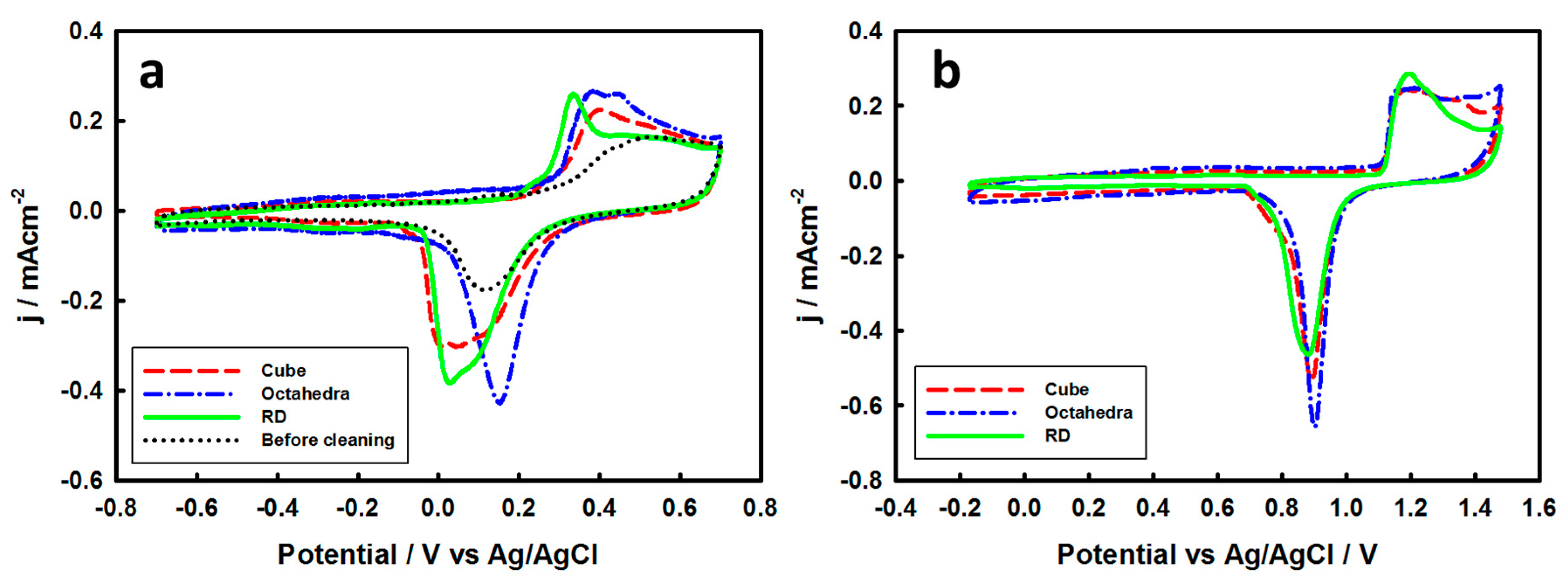
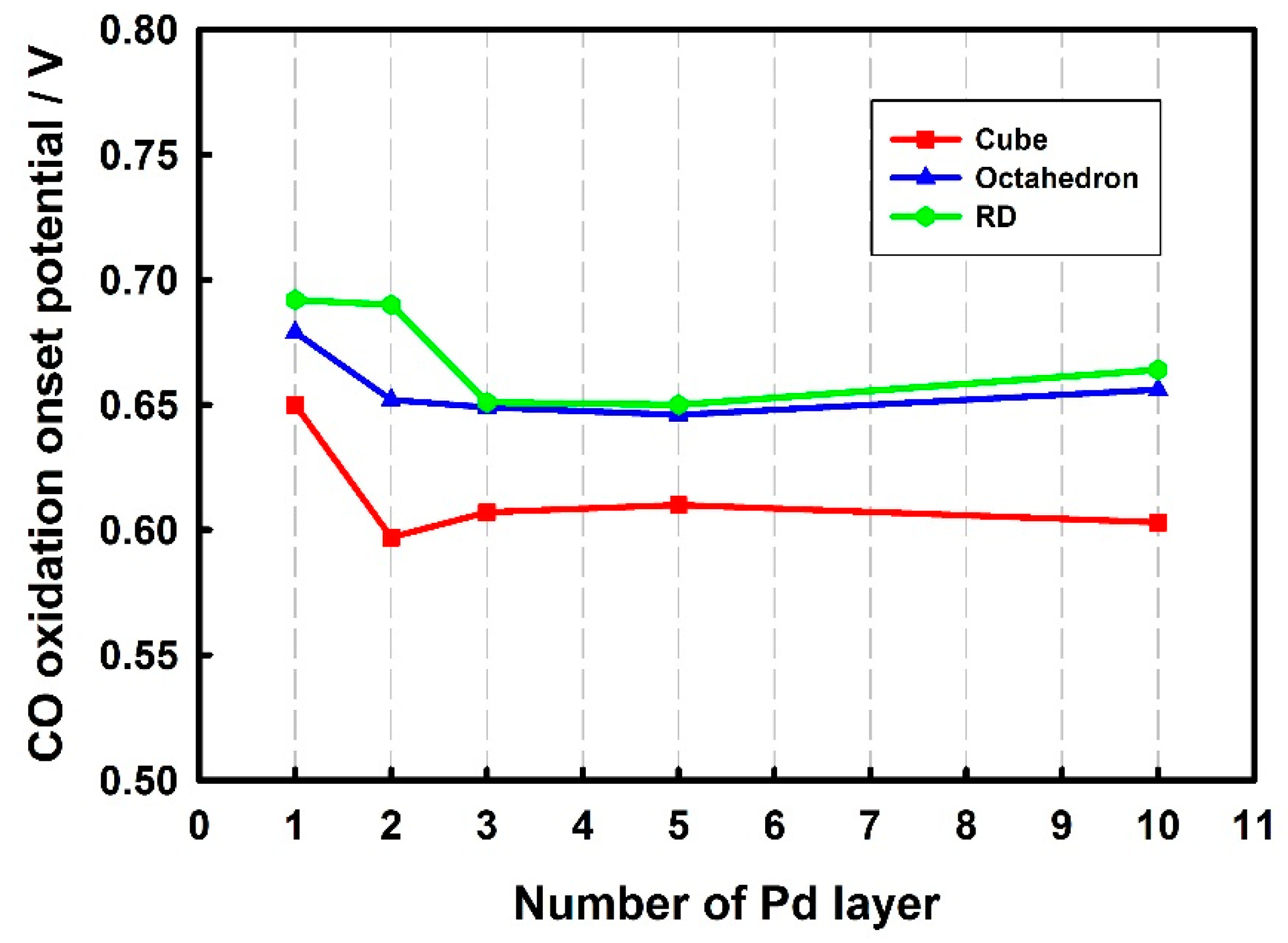
3.2. Electrochemical Properties of Pd-Monolayer-Coated Au Nanocrystals
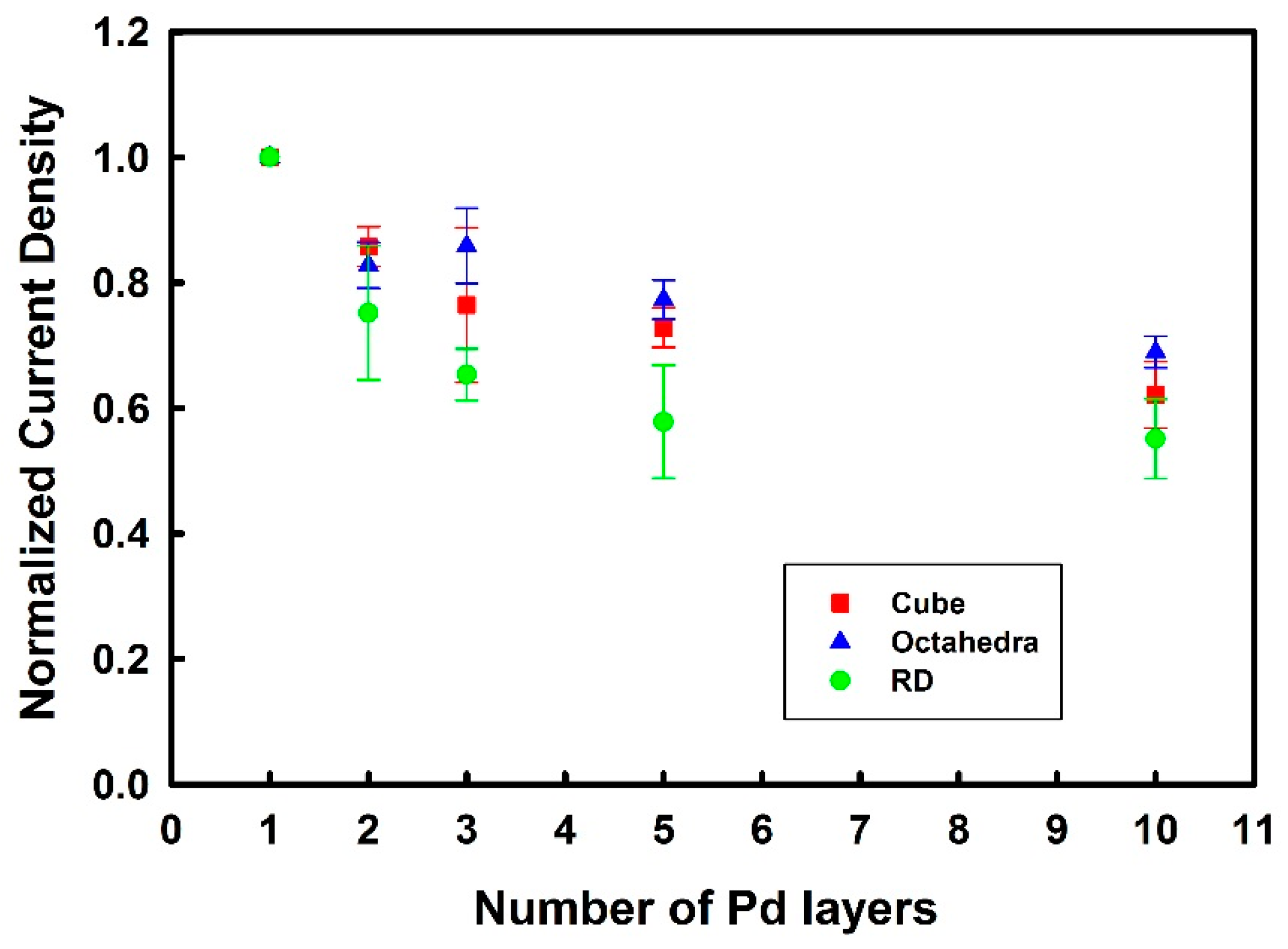
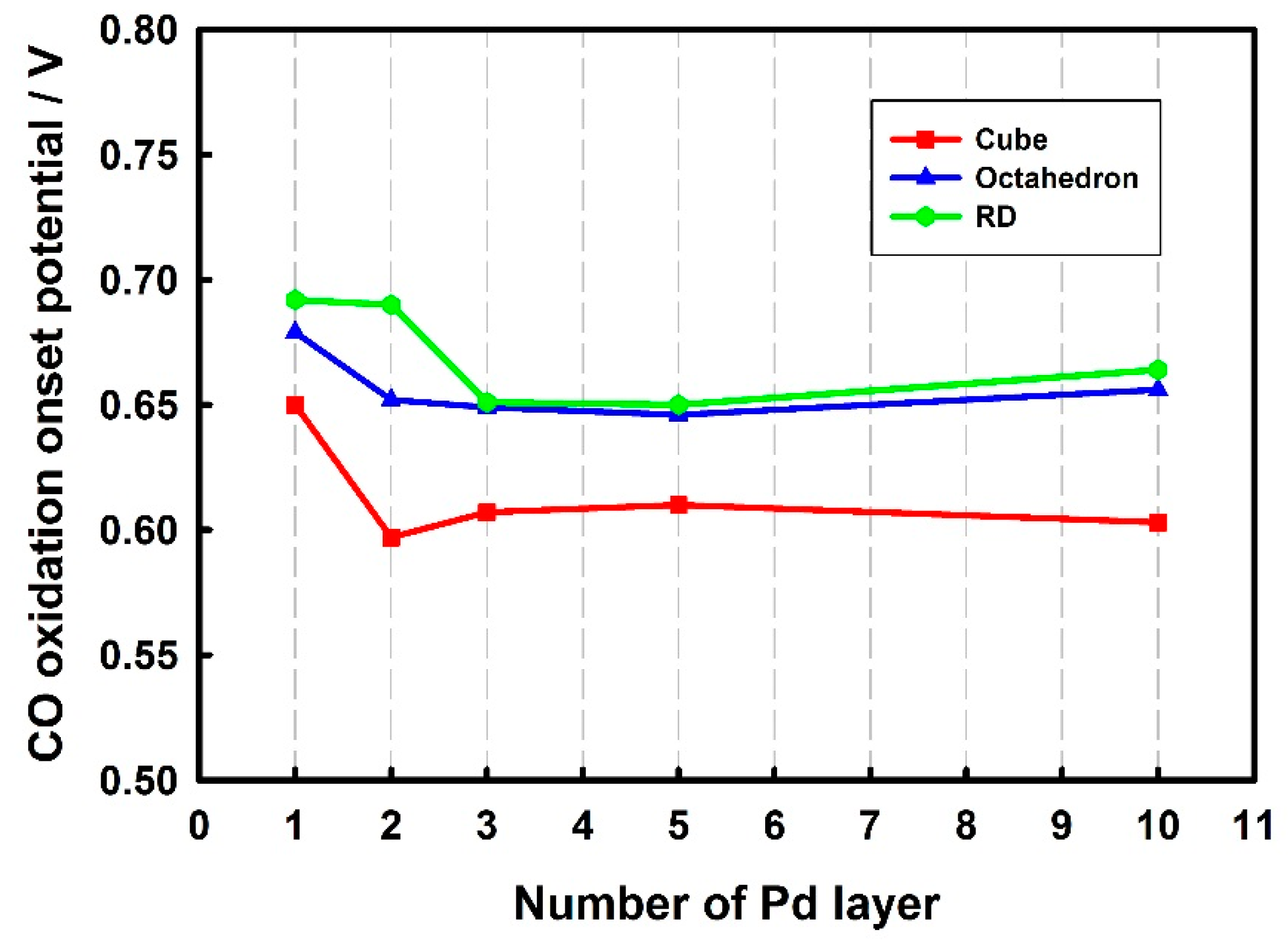
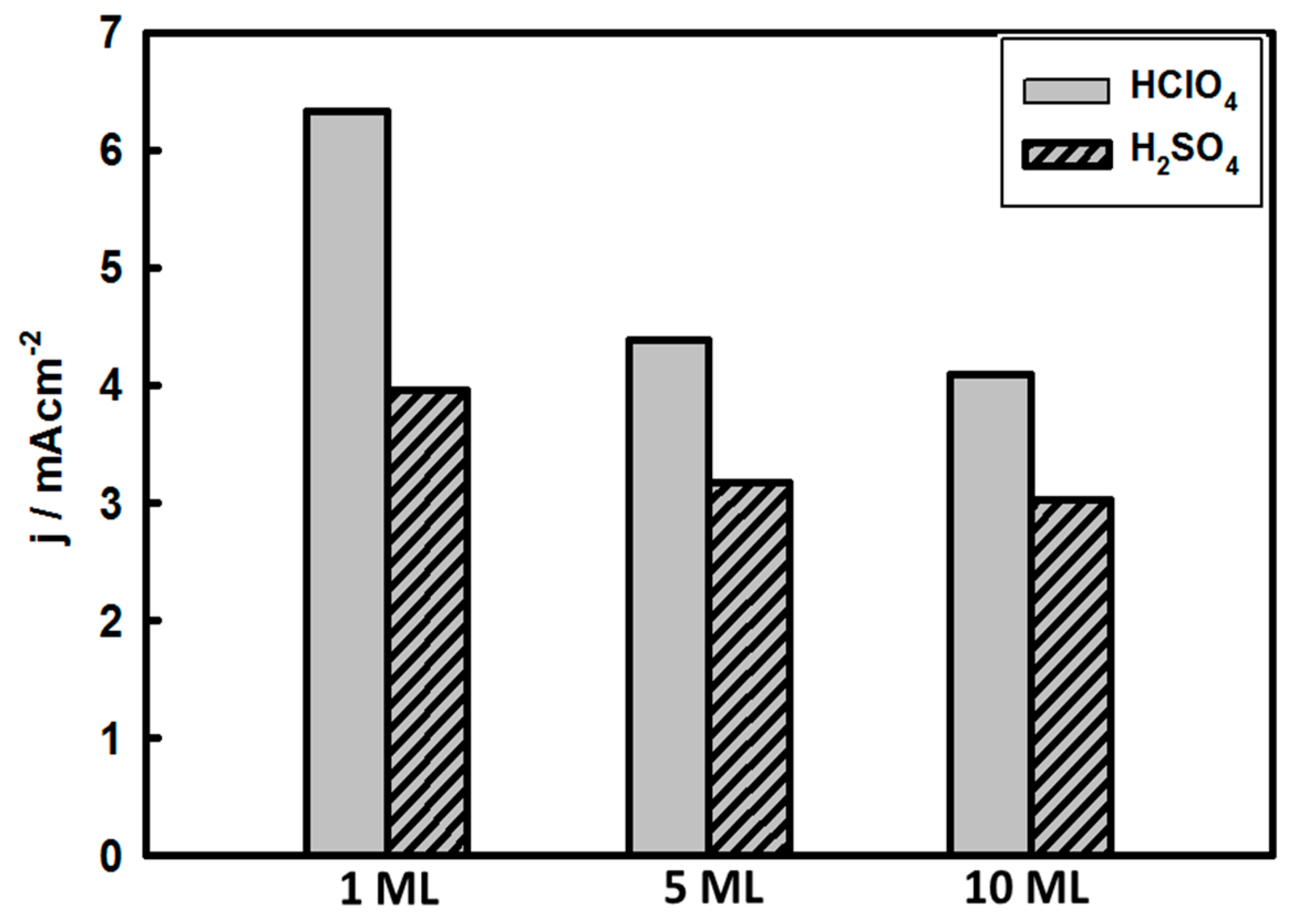
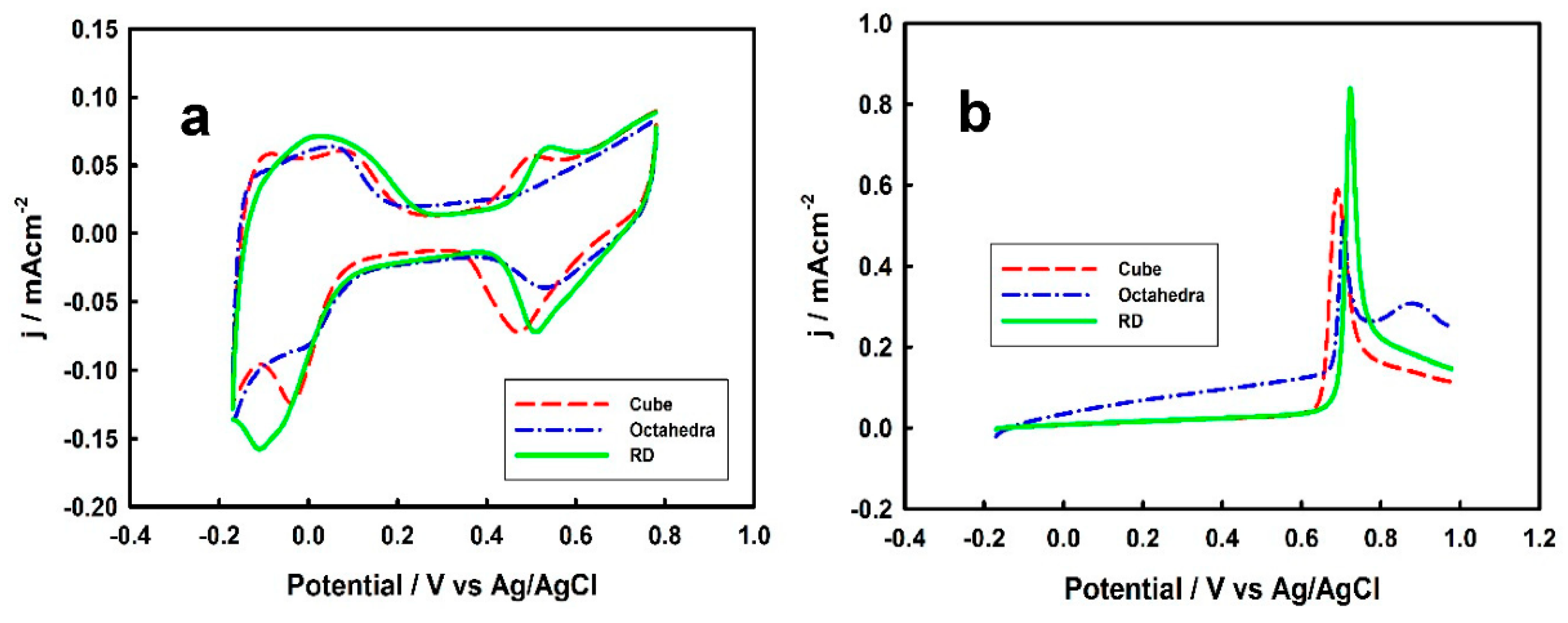
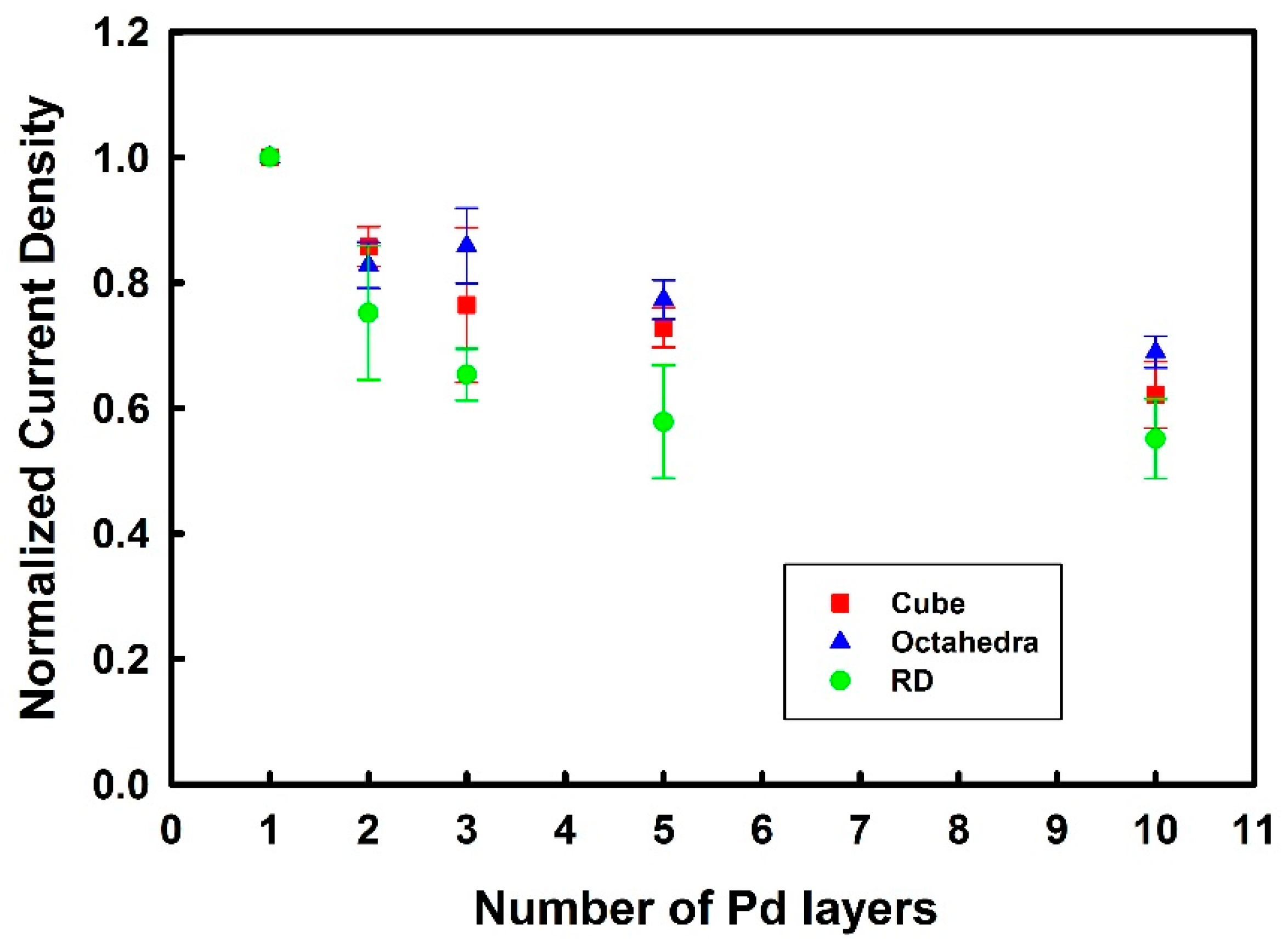
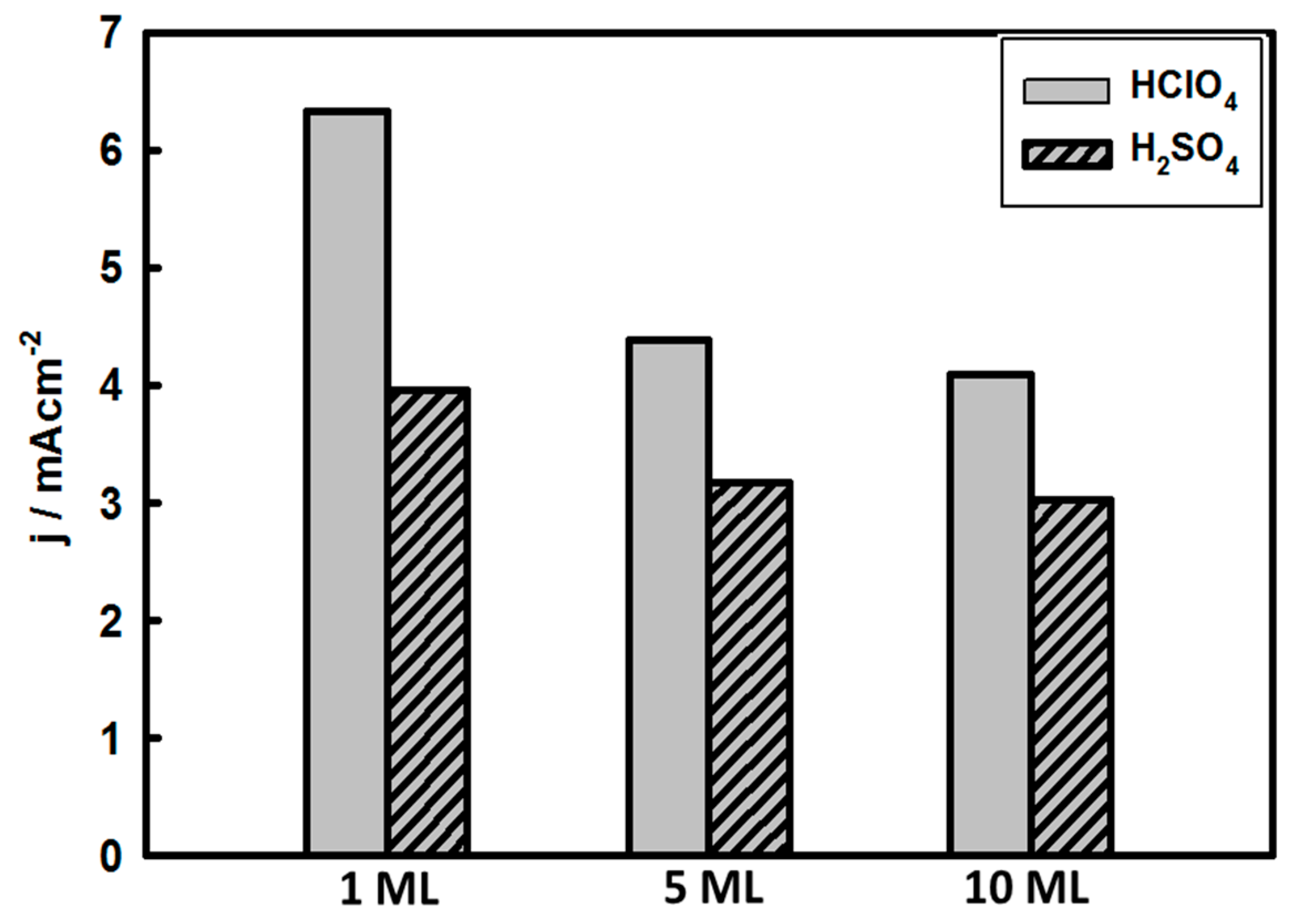
3.3. Thickness-Dependent Formic Acid Oxidation on Pd Thin Layer Coated Au Nanocrystals
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Jeong, K.-J.; Miesse, C.M.; Choi, J.-H.; Lee, J.; Han, J.; Yoon, S.P.; Nam, S.W.; Lim, T.-H.; Lee, T.G. Fuel Crossover in Direct Formic Acid Fuel Cells. J. Power Sources 2007, 168, 119–125. [Google Scholar] [CrossRef]
- Jiang, K.; Zhang, H.-X.; Zou, S.; Cai, W.-B. Electrocatalysis of Formic Acid on Palladium and Platinum Surfaces: From Fundamental Mechanisms to Fuel Cell Applications. Phys. Chem. Chem. Phys. 2014, 16, 20360–20376. [Google Scholar] [CrossRef] [PubMed]
- Uhm, S.; Lee, H.J.; Lee, J. Understanding Underlying Processes in Formic Acid Fuel Cells. Phys. Chem. Chem. Phys. 2009, 11, 9326–9336. [Google Scholar] [CrossRef]
- Wang, X.; Hu, J.-M.; Hsing, I.M. Electrochemical Investigation of Formic Acid Electro-Oxidation and Its Crossover through a Nafion® Membrane. J. Electroanal. Chem. 2004, 562, 73–80. [Google Scholar] [CrossRef]
- Yu, X.W.; Pickup, P.G. Recent Advances in Direct Formic Acid Fuel Cells (Dfafc). J. Power Sources 2008, 182, 124–132. [Google Scholar] [CrossRef]
- Capon, A.; Parsons, R. The Oxidation of Formic Acid at Noble Metal Electrodes Part Iii. Intermediates and Mechanism on Platinum Electrodes. J. Electroanal. Chem. Interfacial Electrochem. 1973, 45, 205–231. [Google Scholar] [CrossRef]
- Baldauf, M.; Kolb, D.M. Formic Acid Oxidation on Ultrathin Pd Films on Au(hkl) and Pt(hkl) Electrodes. J. Phys. Chem. 1996, 100, 11375–11381. [Google Scholar] [CrossRef]
- Ha, S.; Larsen, R.; Masel, R.I. Performance Characterization of Pd/C Nanocatalyst for Direct Formic Acid Fuel Cells. J. Power Sources 2005, 144, 28–34. [Google Scholar] [CrossRef]
- Zhu, Y.; Khan, Z.; Masel, R.I. The Behavior of Palladium Catalysts in Direct Formic Acid Fuel Cells. J. Power Sources 2005, 139, 15–20. [Google Scholar] [CrossRef]
- Solla-Gullon, J.; Montiel, V.; Aldaz, A.; Clavilier, J. Electrochemical and Electrocatalytic Behavior of Platinum–Palladium Nanoparticle Alloys. Electrochem. Commun. 2002, 4, 716–721. [Google Scholar] [CrossRef]
- Rand, D.A.J.; Woods, R. Determination of the Surface Composition of Smooth Noble Metal Alloys by Cyclic Voltammetry. J. Electroanal. Chem. Interfacial Electrochem. 1972, 36, 57–69. [Google Scholar] [CrossRef]
- Antolini, E. Palladium in Fuel Cell Catalysis. Energy Environ. Sci. 2009, 2, 915–931. [Google Scholar] [CrossRef]
- Zhou, W.; Lee, J.Y. Highly Active Core–Shell Au@Pd Catalyst for Formic Acid Electrooxidation. Electrochem. Commun. 2007, 9, 1725–1729. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Wang, G.; Deng, C.; Wu, B.; Gao, Y. High Active Carbon Supported PdAu Catalyst for Formic Acid Electrooxidation and Study of the Kinetics. J. Phys. Chem. C 2010, 114, 21417–21422. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, Y.; Wang, X.; Chen, Y.; Zhou, Y.; Tang, Y.; Lu, L.; Bao, J.; Lu, T. Preparation of Pd–Au/C Catalysts with Different Alloying Degree and Their Electrocatalytic Performance for Formic Acid Oxidation. Appl. Catal. B Environ. 2011, 102, 614–619. [Google Scholar] [CrossRef]
- Lee, S.Y.; Jung, N.; Cho, J.; Park, H.Y.; Ryu, J.; Jang, I.; Kim, H.J.; Cho, E.; Park, Y.H.; Ham, H.C.; et al. Surface-Rearranged Pd3Au/C Nanocatalysts by Using Co-Induced Segregation for Formic Acid Oxidation Reactions. ACS Catal. 2014, 4, 2402–2408. [Google Scholar] [CrossRef]
- Hong, J.W.; Kim, D.; Lee, Y.W.; Kim, M.; Kang, S.W.; Han, S.W. Atomic-Distribution-Dependent Electrocatalytic Activity of Au–Pd Bimetallic Nanocrystals. Angew. Chem. Int. Ed. 2011, 50, 8876–8880. [Google Scholar] [CrossRef] [PubMed]
- Montes de Oca, M.G.; Plana, D.; Celorrio, V.; Lazaro, M.J.; Fermín, D.J. Electrocatalytic Properties of Strained Pd Nanoshells at Au Nanostructures: CO and HCOOH Oxidation. J. Phys. Chem. C 2012, 116, 692–699. [Google Scholar] [CrossRef]
- Celorrio, V.N.; Quaino, P.M.; Santos, E.; Flórez-Montaño, J.; Humphrey, J.J.L.; Guillén-Villafuerte, O.; Plana, D.; Lázaro, M.J.; Pastor, E.; Fermín, D.J. Strain Effects on the Oxidation of CO and HCOOH on Au−Pd Core−Shell Nanoparticles. ACS Catal. 2017, 7, 1673–1680. [Google Scholar] [CrossRef]
- Hsu, C.; Huang, C.; Hao, Y.; Liu, F. Au/Pd Core–Shell Nanoparticles for Enhanced Electrocatalytic Activity and Durability. Electrochem. Commun. 2012, 23, 133–136. [Google Scholar] [CrossRef]
- Obradovic, M.D.; Gojkovic, S.L. HCOOH Oxidation on Thin Pd Layers on Au: Self-Poisoning by the Subsequent Reaction of the Reaction Product. Electrochim. Acta 2013, 88, 384–389. [Google Scholar] [CrossRef]
- Kibler, L.A.; El-Aziz, A.M.; Hoyer, R.; Kolb, D.M. Tuning Reaction Rates by Lateral Strain in a Palladium Monolayer. Angew. Chem. Int. Ed. 2005, 44, 2080–2084. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, N.; Kida, K.; Nakamura, M.; Nakada, M.; Osada, K. Structural Effects of Electrochemical Oxidation of Formic Acid on Single Crystal Electrodes of Palladium. J. Phys. Chem. B 2006, 110, 12480–12484. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.X.; Wang, H.; Re, Y.S.; Cai, W.B. Palladium Nanocrystals Bound by {110} or {100} Facets: From One Pot Synthesis to Electrochemistry. Chem. Commun. 2012, 48, 8362–8364. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.F.; Feng, C.; Deng, Y.D.; Liu, L.; Wu, Y.T.; Shen, B.; Zhong, C.; Hu, W.B. Shape-Controlled Synthesis of Palladium Single-Crystalline Nanoparticles: The Effect of HCl Oxidative Etching and Facet-Dependent Catalytic Properties. Chem. Mater. 2014, 26, 1213–1218. [Google Scholar] [CrossRef]
- Zhang, X.W.; Yin, H.J.; Wang, J.F.; Chang, L.; Gao, Y.; Liu, W.; Tang, Z.Y. Shape-Dependent Electrocatalytic Activity of Monodispersed Palladium Nanocrystals toward Formic Acid Oxidation. Nanoscale 2013, 5, 8392–8397. [Google Scholar] [CrossRef]
- Brankovic, S.R.; Wang, J.X.; Adzic, R.R. Metal Monolayer Deposition by Replacement of Metal Adlayers on Electrode Surfaces. Surf. Sci. 2001, 474, 173–179. [Google Scholar] [CrossRef]
- Sheridan, L.B.; Kim, Y.-G.; Perdue, B.R.; Jagannathan, K.; Stickney, J.L.; Robinson, D.B. Hydrogen Adsorption, Absorption, and Desorption at Palladium Nanofilms Formed on Au(111) by Electrochemical Atomic Layer Deposition (E-ALD): Studies Using Voltammetry and in Situ Scanning Tunneling Microscopy. J. Phys. Chem. C 2013, 117, 15728–15740. [Google Scholar] [CrossRef]
- Niu, W.; Zheng, S.; Wang, D.; Liu, X.; Li, H.; Han, S.; Chen, J.; Tang, Z.; Xu, G. Selective Synthesis of Single-Crystalline Rhombic Dodecahedral, Octahedral, and Cubic Gold Nanocrystals. J. Am. Chem. Soc. 2008, 131, 697–703. [Google Scholar] [CrossRef]
- Nikoobakht, B.; El-Sayed, M.A. Preparation and Growth Mechanism of Gold Nanorods (NRs) Using Seed-Mediated Growth Method. Chem. Mater. 2003, 15, 1957–1962. [Google Scholar] [CrossRef]
- Sau, T.K.; Murphy, C.J. Seeded High Yield Synthesis of Short Au Nanorods in Aqueous Solution. Langmuir 2004, 20, 6414–6420. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Fernandez, J.; Perez-Juste, J.; Mulvaney, P.; Liz-Marzan, L.M. Spatially-Directed Oxidation of Gold Nanoparticles by Au(III)-CTAB Complexes. J. Phys. Chem. B 2005, 109, 14257–14261. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.; Linke, U.; Wandlowski, T. Preparation and Electrochemical Characterization of Palladium Single Crystal Electrodes in 0.1 M H2SO4 and HClO4 Part I. Low-Index Phases. Electrochim. Acta 2007, 52, 5733–5748. [Google Scholar] [CrossRef]
- Kim, F.; Connor, S.; Song, H.; Kuykendall, T.; Yang, P.D. Platonic Gold Nanocrystals. Angew. Chem. Int. Ed. 2004, 43, 3673–3677. [Google Scholar] [CrossRef]
- Seo, D.; Park, J.C.; Song, H. Polyhedral Gold Nanocrystals with O-H Symmetry: From Octahedra to Cubes. J. Am. Chem. Soc. 2006, 128, 14863–14870. [Google Scholar] [CrossRef]
- Chen, Y.; Milenkovic, S.; Hassel, A.W. Reactivity of Gold Nanobelts with Unique {110} Facets. ChemPhysChem 2010, 11, 2838–2843. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Zhang, Y.; Zhang, L.; Zhang, Y.; Wang, J.X.; Adzic, R.R.; Stach, E.A.; Gang, O. Truncated Ditetragonal Gold Prisms as Nanofacet Activators of Catalytic Platinum. J. Am. Chem. Soc. 2011, 133, 18074–18077. [Google Scholar] [CrossRef] [PubMed]
- Tian, N.; Zhou, Z.Y.; Yu, N.F.; Wang, L.Y.; Sun, S.G. Direct Electrodeposition of Tetrahexahedral Pd Nanocrystals with High-Index Facets and High Catalytic Activity for Ethanol Electrooxidation. J. Am. Chem. Soc. 2010, 132, 7580–7581. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Iglesias, F.J.; Aran-Ais, R.M.; Solla-Gullon, J.; Herrero, E.; Feliu, J.M. Electrochemical Characterization of Shape-Controlled Pt Nanoparticles in Different Supporting Electrolytes. ACS Catal. 2012, 2, 901–910. [Google Scholar] [CrossRef]
- Yang, H.; Tang, Y.; Zou, S. Electrochemical Removal of Surfactants from Pt Nanocubes. Electrochem. Commun. 2014, 38, 134–137. [Google Scholar] [CrossRef]
- Vidal-Iglesias, F.J.; Solla-Gullon, J.; Herrero, E.; Montiel, V.; Aldaz, A.; Feliu, J.M. Evaluating the Ozone Cleaning Treatment in Shape-Controlled Pt Nanoparticles: Evidences of Atomic Surface Disordering. Electrochem. Commun. 2011, 13, 502–505. [Google Scholar] [CrossRef]
- Tang, Y.; Edelmann, R.E.; Zou, S. Length Tunable Penta-Twinned Palladium Nanorods: Seedless Synthesis and Electrooxidation of Formic Acid. Nanoscale 2014, 6, 5630–5633. [Google Scholar] [CrossRef]
- Chang, S.C.; Hamelin, A.; Weaver, M.J. Reactive and Inhibiting Adsorbates for the Catalytic Electrooxidation of Carbon-Monoxide on Gold (210) as Characterized by Surface Infrared-Spectroscopy. Surf. Sci. 1990, 239, L543–L547. [Google Scholar] [CrossRef]
- Chang, S.C.; Hamelin, A.; Weaver, M.J. Dependence of the Electrooxidation Rates of Carbon-Monoxide at Gold on the Surface Crystallographic Orientation—A Combined Kinetic-Surface Infrared Spectroscopic Study. J. Phys. Chem. 1991, 95, 5560–5567. [Google Scholar] [CrossRef]
- Lee, Y.; Loew, A.; Sun, S.H. Surface- and Structure-Dependent Catalytic Activity of Au Nanoparticles for Oxygen Reduction Reaction. Chem. Mater. 2010, 22, 755–761. [Google Scholar] [CrossRef]
- Hamelin, A.; Sottomayor, M.J.; Silva, F.; Chang, S.-C.; Weaver, M.J. Cyclic Voltammetric Characterization of Oriented Monocrystalline Gold Surfaces in Aqueous Alkaline Solution. J. Electroanal. Chem. Interfacial Electrochem. 1990, 295, 291–300. [Google Scholar] [CrossRef]
- Hamelin, A. Cyclic Voltammetry at Gold Single-Crystal Surfaces. Part 1. Behaviour at Low-Index Faces. J. Electroanal. Chem. 1996, 407, 1–11. [Google Scholar] [CrossRef]
- Schmidt, T.J.; Stamenkovic, V.; Arenz, M.; Markovic, N.M.; Ross, P.N., Jr. Oxygen Electrocatalysis in Alkaline Electrolyte: Pt(hkl), Au(hkl) and the Effect of Pd-Modification. Electrochim. Acta 2002, 47, 3765–3776. [Google Scholar] [CrossRef]
- Sashikata, K.; Matsui, Y.; Itaya, K.; Soriaga, M.P. Adsorbed-Iodine-Catalyzed Dissolution of Pd Single-Crystal Electrodes: Studies by Electrochemical Scanning Tunneling Microscopy. J. Phys. Chem. 1996, 100, 20027–20034. [Google Scholar] [CrossRef]
- Zou, S.Z.; Gomez, R.; Weaver, M.J. Infrared Spectroscopy of Carbon Monoxide at the Ordered Palladium (110)-Aqueous Interface: Evidence for Adsorbate-Induced Surface Reconstruction. Surf. Sci. 1998, 399, 270–283. [Google Scholar] [CrossRef]
- Zou, S.Z.; Gomez, R.; Weaver, M.J. Infrared Spectroscopy of Carbon Monoxide and Nitric Oxide on Palladium(111) in Aqueous Solution: Unexpected Adlayer Structural Differences between Electrochemical and Ultrahigh-Vacuum Interfaces. J. Electroanal. Chem. 1999, 474, 155–166. [Google Scholar] [CrossRef]
- Zou, S.Z.; Gomez, R.; Weaver, M.J. Coverage-Dependent Infrared Spectroscopy of Carbon Monoxide on Palladium(100) in Aqueous Solution: Adlayer Phase Transitions and Electrooxidation Pathways. Langmuir 1999, 15, 2931–2939. [Google Scholar] [CrossRef]
- Jin, M.S.; Zhang, H.; Xie, Z.X.; Xia, Y.N. Palladium Nanocrystals Enclosed by {100} and {111} Facets in Controlled Proportions and Their Catalytic Activities for Formic Acid Oxidation. Energy Environ. Sci. 2012, 5, 6352–6357. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Zhang, H.-X.; Jiang, K.; Cai, W.-B. From HCOOH to CO at Pd Electrodes: A Surface-Enhanced Infraredspectroscopy Study. J. Am. Chem. Soc. 2011, 133, 14876–14879. [Google Scholar] [CrossRef]
- Zhang, H.-X.; Wang, S.-H.; Jiang, K.; André, T.; Cai, W.-B. In Situ Spectroscopic Investigation of CO Accumulation and Poisoning on Pd Black Surfaces in Concentrated HCOOH. J. Power Sources 2012, 199, 165–169. [Google Scholar] [CrossRef]
- Ruban, A.; Hammer, B.; Stoltze, P.; Skriver, H.L.; Nørskov, J.K. Surface Electronic Structure and Reactivity of Transition and Noble Metals. J. Mol. Catal. A Chem. 1997, 115, 421–429. [Google Scholar] [CrossRef]
- Hammer, B.; Norskov, J.K. Electronic Factors Determining the Reactivity of Metal Surfaces. Surf. Sci. 1995, 343, 211–220. [Google Scholar] [CrossRef]
- Mavrikakis, M.; Hammer, B.; Norskov, J.K. Effect of Strain on the Reactivity of Metal Surfaces. Phys. Rev. Lett. 1998, 81, 2819–2822. [Google Scholar] [CrossRef]
- Roudgar, A.; Gross, A. Local Reactivity of Thin Pd Overlayers on Au Single Crystals. J. Electroanal. Chem. 2003, 548, 121–130. [Google Scholar] [CrossRef]
- Shao, M.H.; Huang, T.; Liu, P.; Zhang, J.; Sasaki, K.; Vukmirovic, M.B.; Adzic, R.R. Palladium Monolayer and Palladium Alloy Electrocatalysts for Oxygen Reduction†. Langmuir 2006, 22, 10409–10415. [Google Scholar] [CrossRef]
- Kibler, L.A.; Kleinert, M.; Kolb, D.M. Initial Stages of Pd Deposition on Au(hkl) Part II: Pd on Au(100). Surf. Sci. 2000, 461, 155–167. [Google Scholar] [CrossRef]
- Kibler, L.A.; Kleinert, M.; Lazarescu, V.; Kolb, D.M. Initial Stages of Palladium Deposition on Au(hkl) Part III: Pd on Au(110). Surf. Sci. 2002, 498, 175–185. [Google Scholar] [CrossRef]
- Kibler, L.A.; Kleinert, M.; Randler, R.; Kolb, D.M. Initial Stages of Pd Deposition on Au(hkl)—Part I: Pd on Au(111). Surf. Sci. 1999, 443, 19–30. [Google Scholar] [CrossRef]
- Takahasi, M.; Tamura, K.; Mizuki, J.; Kondo, T.; Uosaki, K. Orientation Dependence of Pd Growth on Au Electrode Surfaces. J. Phys. Condens. Matter. 2010, 22, 474002. [Google Scholar] [CrossRef]
- Vidal-Iglesias, F.J.; Arán-Ais, R.M.; Solla-Gullón, J.; Garnier, E.; Herrero, E.; Aldaz, A.; Feliu, J.M. Shape-Dependent Electrocatalysis: Formic Acid Electrooxidation on Cubic Pd Nanoparticles. Phys. Chem. Chem. Phys. 2012, 14, 10258–10265. [Google Scholar] [CrossRef]
- Hoshi, N.; Kuroda, M.; Koga, O.; Hori, Y. Infrared Reflection Absorption Spectroscopy of the Sulfuric Acid Anion on Low and High Index Planes of Palladium. J. Phys. Chem. B 2002, 102, 9107–9113. [Google Scholar] [CrossRef]
- Pronkin, S.; Wandlowski, T. ATR-SEIRAS—An Approach to Probe the Reactivity of Pd-Modified Quasi-Single Crystal Gold Film Electrodes. Surf. Sci. 2004, 573, 109–127. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, Y.; Zou, S. Formic Acid Oxidation on Pd Thin Film Coated Au Nanocrystals. Surfaces 2019, 2, 372-386. https://doi.org/10.3390/surfaces2020027
Tang Y, Zou S. Formic Acid Oxidation on Pd Thin Film Coated Au Nanocrystals. Surfaces. 2019; 2(2):372-386. https://doi.org/10.3390/surfaces2020027
Chicago/Turabian StyleTang, Yongan, and Shouzhong Zou. 2019. "Formic Acid Oxidation on Pd Thin Film Coated Au Nanocrystals" Surfaces 2, no. 2: 372-386. https://doi.org/10.3390/surfaces2020027
APA StyleTang, Y., & Zou, S. (2019). Formic Acid Oxidation on Pd Thin Film Coated Au Nanocrystals. Surfaces, 2(2), 372-386. https://doi.org/10.3390/surfaces2020027