
A Bench-Scale Demonstration of Direct Air Capture Using an Enhanced Electrochemical System

 , and
, and
Abstract
1. Introduction
2. Experimental Section
2.1. Materials
2.2. Fabrication of Electrochemical Regenerator
2.3. Integration of Absorber and Electrochemical Regenerator
3. Results and Discussion
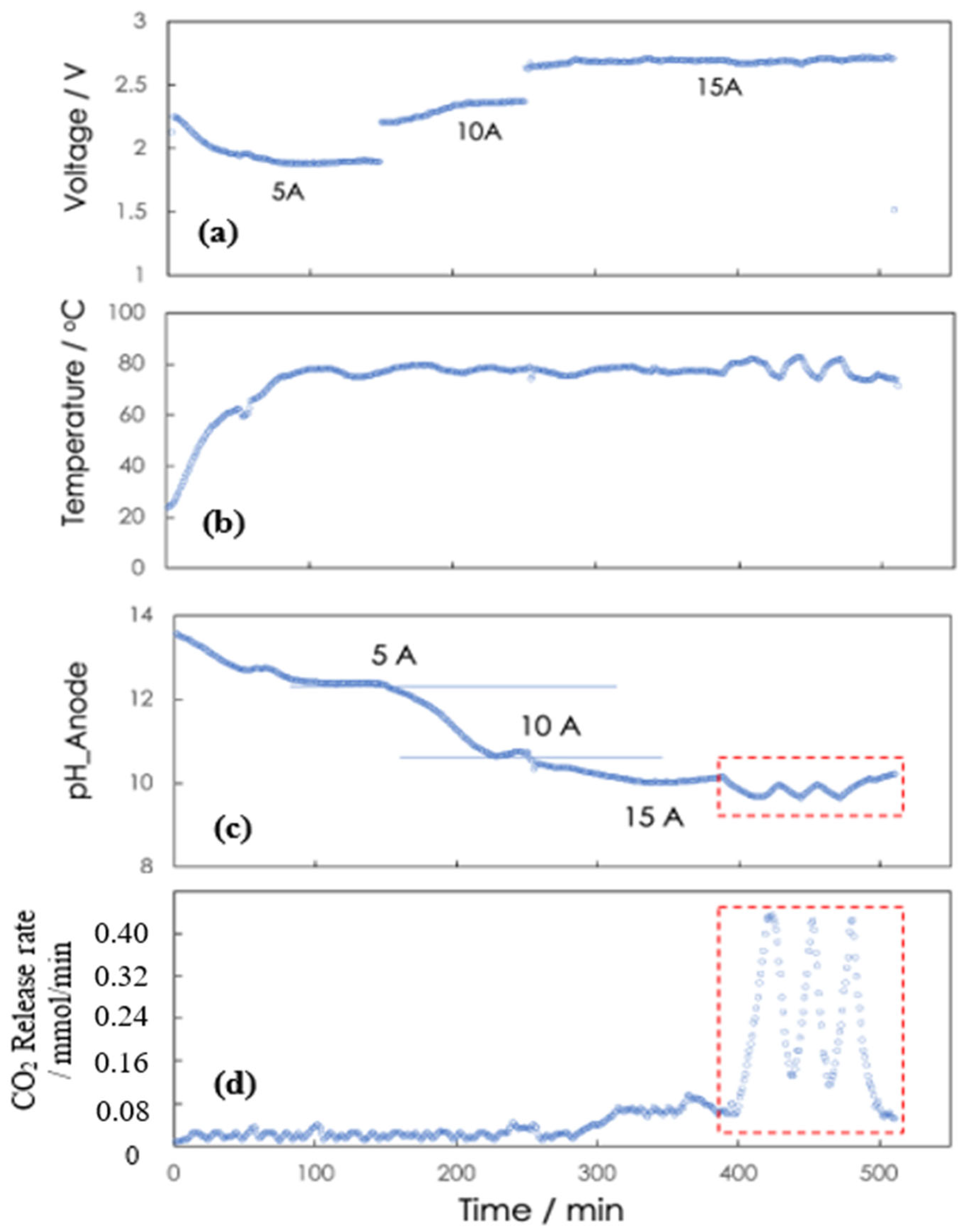
3.1. Operation of the Electrochemical Regenerator
3.2. Selection of Electrode Catalysts
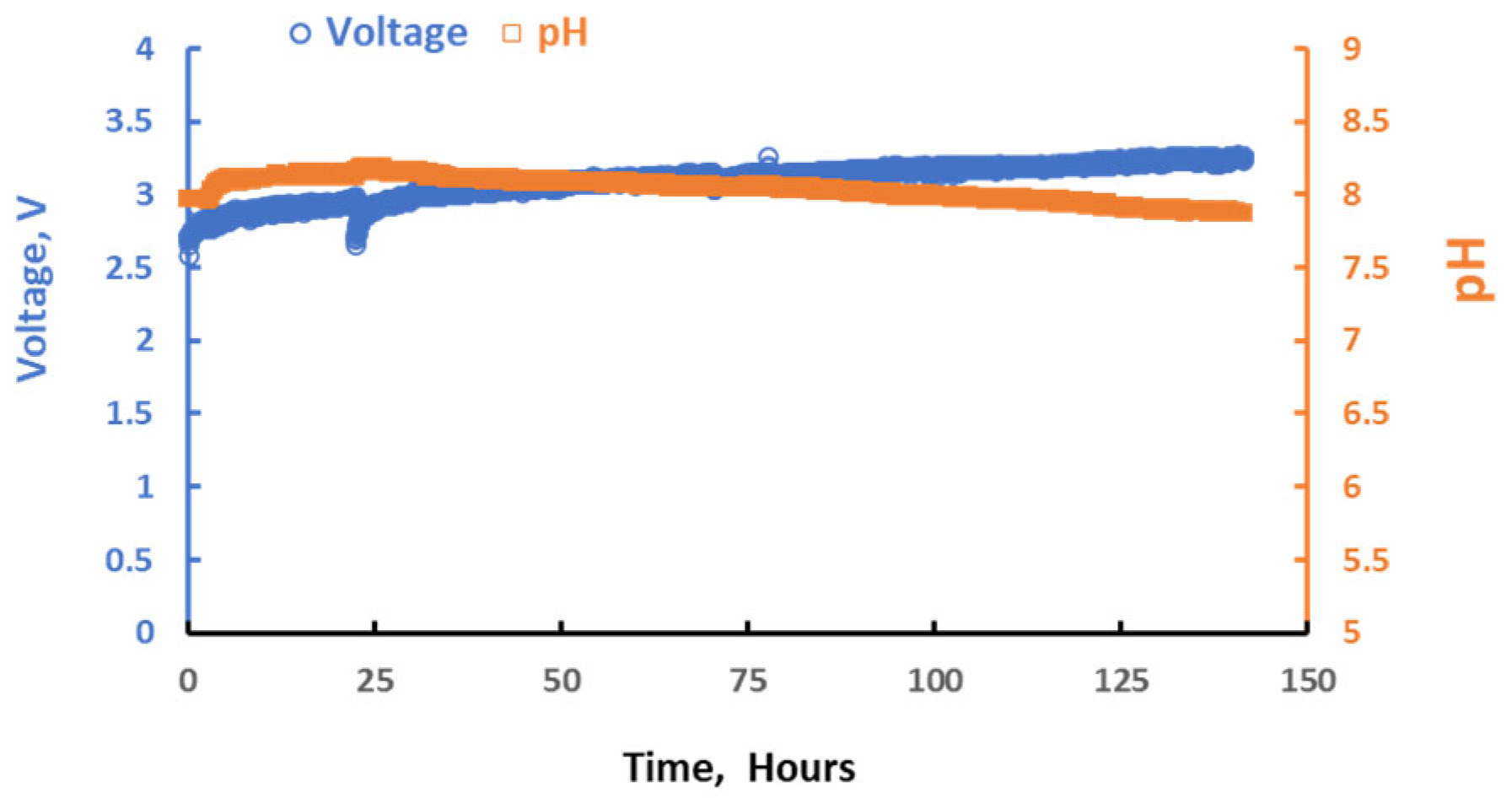
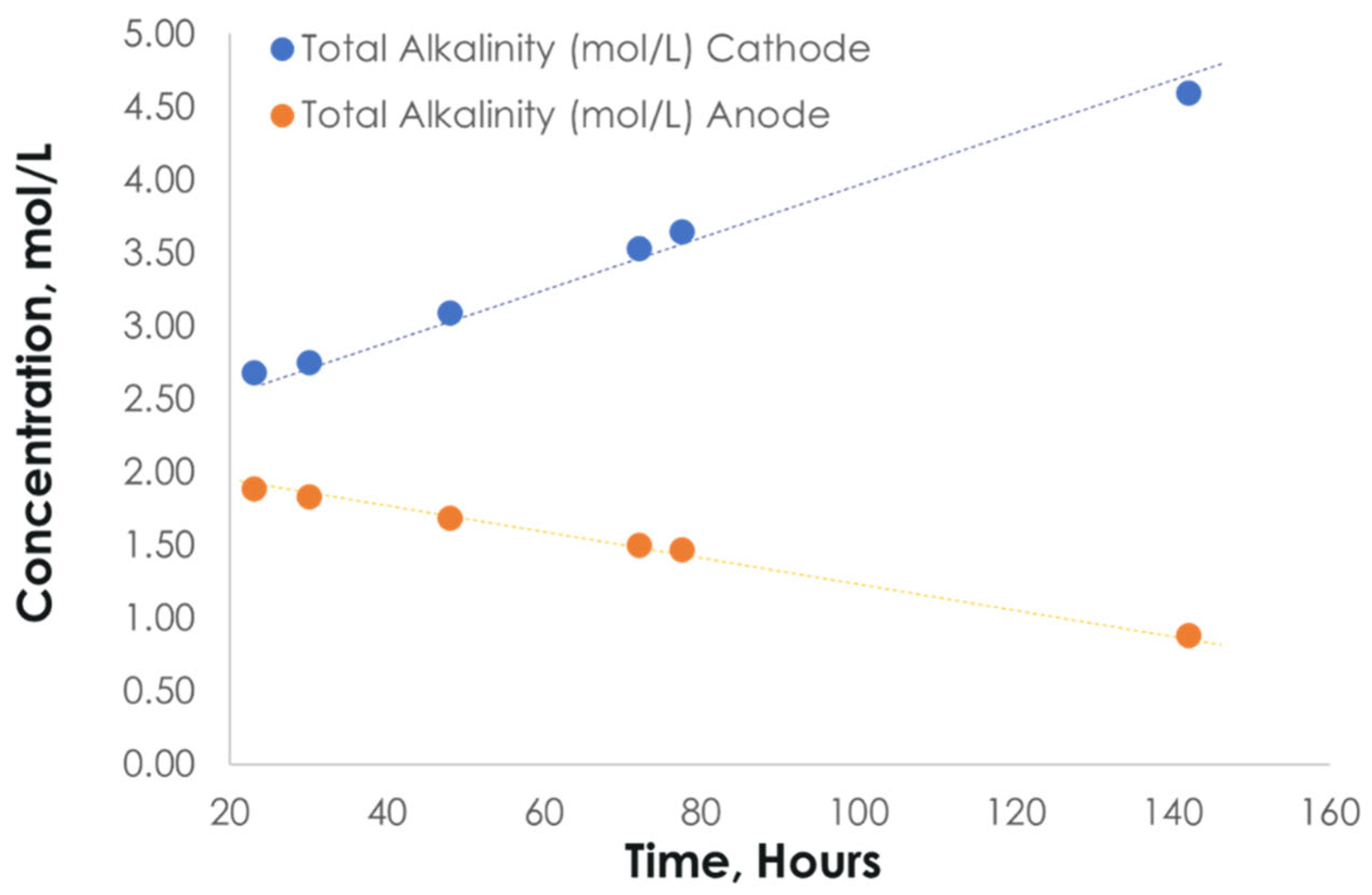
3.3. Integration of MA and ER
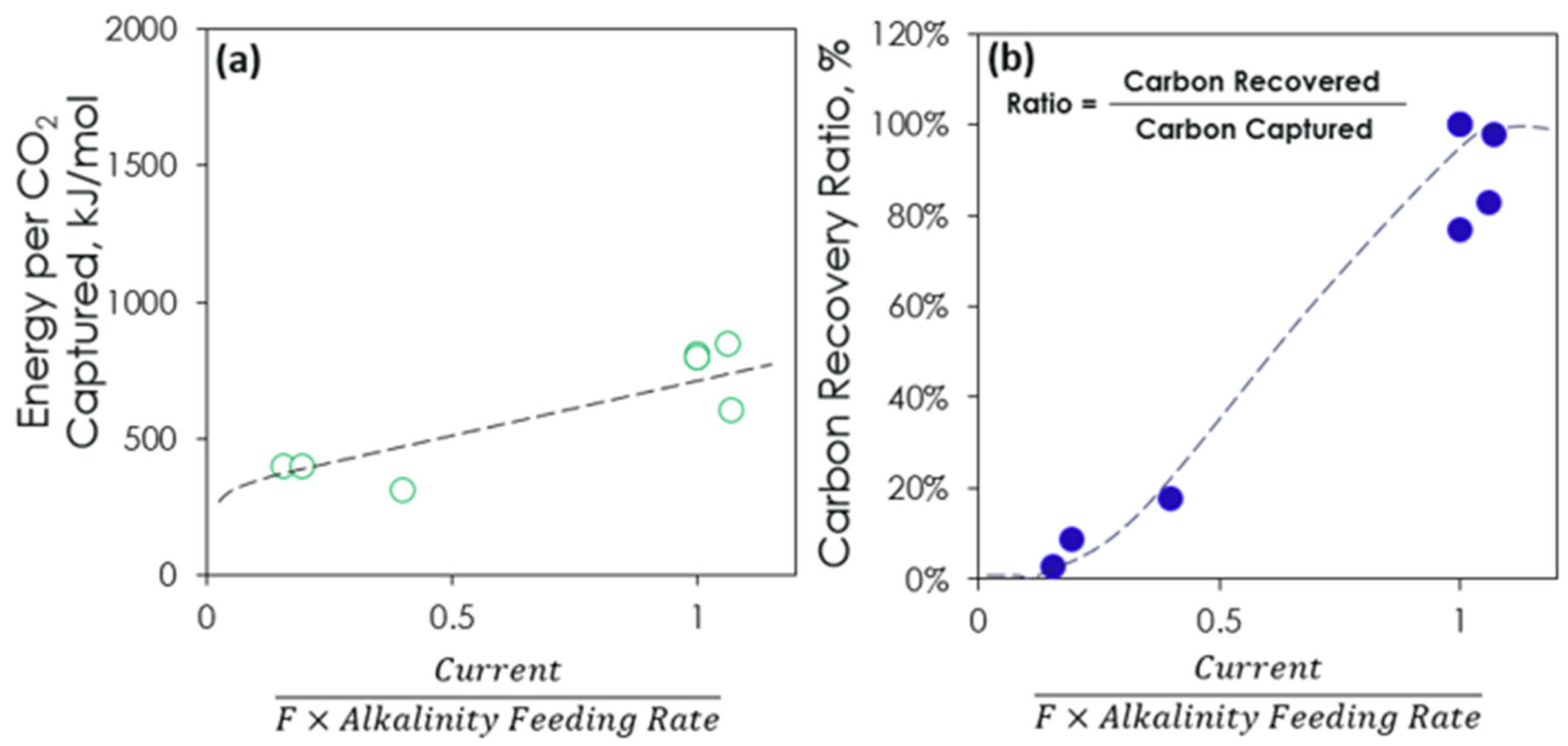
3.4. TEA and LCA of IDEA Process
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fasihi, M.; Efimova, O.; Breyer, C. Techno-economic assessment of CO2 direct air capture plants. J. Cleaner Prod. 2019, 224, 957. [Google Scholar] [CrossRef]
- Sholl, D.S.; Lively, R.P. Seven chemical separations to change the world. Nature 2016, 532, 435. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Pe, E.S.; Murdock, C.R.; Didas, S.A.; Jones, C.W. Direct capture of CO2 from ambient air. Chem. Rev. 2016, 116, 11840. [Google Scholar] [CrossRef] [PubMed]
- Holmes, G.; Nold, K.; Walsh, T.; Heidel, K.; Henderson, M.A.; Ritchie, J.; Klavins, P.; Singh, A.; Keith, D.W. Outdoor prototype results for direct atmospheric capture of carbon dioxide. Energy Procedia 2013, 37, 6079. [Google Scholar] [CrossRef]
- Kulkarni, A.R.; Sholl, D.S. Analysis of equilibrium-based TSA processes for direct capture of CO2 from air. Ind. Eng. Chem. Res. 2012, 51, 8631. [Google Scholar] [CrossRef]
- Socolow, R.; Desmond, M.; Aines, R.; Blackstock, J.; Bolland, O.; Kaarsberg, T.; Lewis, N.; Mazzotti, M.; Pfeffer, A.; Sawyer, K. Direct Air Capture of CO2 with Chemicals: A Technology Assessment for the APS Panel on Public Affairs; American Physical Society: College Park, MD, USA, 2011. [Google Scholar]
- Lackner, K.S.; Ziock, H.-J.; Grimes, P. Carbon Dioxide Extraction from Air: Is It an Option? Technical Report LA-UR-99–583; Los Alamos National Laboratory: Los Alamos, NM, USA, 1999. [Google Scholar]
- Sievert, K.; Schmidt, T.S.; Steffen, B. Considering technology characteristics to project future costs of direct air capture. Joule 2024, 8, 979. [Google Scholar] [CrossRef]
- Rosen, N.; Welter, A.; Schwankl, M.; Plumeré, N.; Staudt, J.; Burger, J. Assessment of the potential of electrochemical steps in direct air capture through techno-economic analysis. Energy Fuels 2024, 38, 15469. [Google Scholar] [CrossRef]
- Sodiq, A.; Abdullatif, Y.; Aissa, B.; Ostovar, A.; Nassar, N.; El-Naas, M.; Amhamed, A. A review on progress made in direct air capture of CO2. Environ. Technol. Innov. 2023, 29, 102991. [Google Scholar] [CrossRef]
- McQueen, N.; Gomes, K.V.; McCormick, C.; Blumanthal, K.; Pisciotta, M.; Wilcox, J. A review of direct air capture (DAC): Scaling up commercial technologies and innovating for the future. Prog. Energy 2021, 3, 032001. [Google Scholar] [CrossRef]
- Sadiq, M.M.; Batten, M.P.; Mulet, X.; Freeman, C.; Konstas, K.; Mardel, J.I.; Tanner, J.; Ng, D.; Wang, X.; Howard, S.; et al. A pilot-scale demonstration of mobile direct air capture using metal-organic frameworks. Adv. Sustain. Syst. 2020, 4, 2000101. [Google Scholar] [CrossRef]
- Keith, D.W.; Holmes, G.; Angelo, D.S.; Heidel, K. A process for capturing CO2 from the atmosphere. Joule 2018, 2, 1573. [Google Scholar] [CrossRef]
- Liu, P.; Liu, H.; Li, K.; Fan, Z.; Lu, Q.; Sun, B.; Hu, L.; Yin, L.; Wang, X.; Liu, L. Recent advances in integrating solvent-based CO2 capture with electrochemical regeneration process: A review. Fuel 2025, 385, 133943. [Google Scholar] [CrossRef]
- Sun, K.; Tebyetekerwa, M.; Zhang, H.; Zeng, X.; Wang, Z.; Ru, T.E.; Zhang, X. Electrode, electrolyte, and membrane materials for electrochemical CO2 capture. Adv. Energy Mater. 2024, 14, 2400625. [Google Scholar] [CrossRef]
- Pei, Y.; Zhang, B.; Lu, Y. Carbon capture and utilization via electrochemistry, what’s next? Next Nanotechnol. 2023, 3, 100020. [Google Scholar] [CrossRef]
- Voskian, S.; Hatton, T.A. Faradaic electro-swing reactive adsorption for CO2 capture. Energy Environ. Sci. 2019, 12, 3530. [Google Scholar] [CrossRef]
- Shu, Q.; Sin, C.S.; Tedesco, M.; Hamelers, H.V.M.; Kuntke, P. Optimization of an electrochemical direct air capture process with decreased CO2 desorption pressure and addition of background electrolyte. Chem. Eng. J. 2023, 470, 144251. [Google Scholar] [CrossRef]
- Amini, K.; Cochard, T.; Jing, Y.; Sosa, J.D.; Xi, D.; Alberts, M.; Emanuel, M.S.; Kerr, E.F.; Gordon, R.G.; Aziz, M.J. In situ techniques for aqueous quinone-mediated electrochemical carbon capture and release. Nat. Chem. Eng. 2024, 1, 774. [Google Scholar] [CrossRef]
- Jin, S.; Wu, M.; Jing, Y.; Gordon, R.G.; Aziz, M.J. Low energy carbon capture via electrochemically induced pH swing with electrochemical rebalancing. Nat. Commun. 2022, 13, 2140. [Google Scholar] [CrossRef]
- Seo, H.; Hatton, T.A. Electrochemical direct air capture of CO2 using neutral red as reversible redox-active material. Nat. Commun. 2023, 14, 313. [Google Scholar] [CrossRef]
- Khoiruddin, K.; Wenten, I.G.; Siagian, U.W.R. Advancements in bipolar membrane electrodialysis techniques for carbon capture. Langmuir 2024, 40, 9362. [Google Scholar] [CrossRef]
- An, K.; Li, K.; Yang, C.-M.; Brechtl, J.; Nawaz, K. A comprehensive review on regeneration strategies for direct air capture. J. CO2 Util. 2023, 76, 102587. [Google Scholar] [CrossRef]
- Rivero, J.R.; Panagakos, G.; Lieber, A.; Hornbostel, K. Hollow fiber membrane contactors for post-combustion carbon capture: A review of modeling approaches. Membranes 2020, 10, 382. [Google Scholar] [CrossRef] [PubMed]
- Bazhenov, S.D.; Bildyukevich, A.V.; Volkov, A.V. Gas-liquid hollow fiber membrane contactors for different applications. Fibers 2018, 6, 76. [Google Scholar] [CrossRef]
- Nogalska, A.; Trojanowska, A.; Garcia-Valls, R. Membrane contactors for CO2 capture processes—Critical review. Phys. Sci. Rev. 2017, 2, 20170059. [Google Scholar] [CrossRef]
- Dindore, V.Y.; Brilman, D.W.F.; Feron, P.H.M.; Versteeg, G.F. CO2 absorption at elevated pressures using a hollow fibre membrane contactor. J. Membr. Sci. 2004, 235, 99. [Google Scholar] [CrossRef]
- Gabelman, A.; Hwang, S.T. Hollow fiber membrane contactors. J. Membr. Sci. 1999, 159, 61. [Google Scholar] [CrossRef]
- Wang, J.; Gao, X.; Omosebi, A.; Liu, K. Electrochemically Regenerated Solvent for Direct Air Capture with Co-Generation of Hydrogen at Bench-Scale. DOE Report: DOE-UKIDEA-32125-1; 2024, the U.S. Department of Energy Office of Scientific and Technical Information. Available online: https://www.osti.gov/biblio/2349461 (accessed on 9 June 2025).
- Shu, Q.; Legrand, L.; Kuntke, P.; Tedesco, M.; Hamelers, H.V.M. Electrochemical regeneration of spent alkaline absorbent from direct air capture. Environ. Sci. Technol. 2020, 54, 8990. [Google Scholar] [CrossRef]
- Park, J.E.; Lee, H.; Oh, S.; Kang, S.Y.; Choi, I.; Cho, Y.; Sung, Y. Electrodeposited mesh-type dimensionally stable anode for oxygen evolution reaction in acidic and alkaline media. Chem. Eng. Sci. 2019, 206, 424. [Google Scholar] [CrossRef]
- Trasatti, S. Physical electrochemistry of ceramic oxides. Electrochim. Acta 1991, 36, 225. [Google Scholar] [CrossRef]
- Zeng, K.; Zhang, D. Recent progress in alkaline water electrolysis for hydrogen production and applications. Prog. Energy Combust. Sci. 2010, 36, 307. [Google Scholar] [CrossRef]
- Lin, M.; Ehret, C.; Hamelers, H.V.M.; Heijne, A.T.; Kuntke, P. Energy efficient carbon capture through electrochemical pH swing regeneration of amine solution. ACS Sustain. Chem. Eng. 2024, 12, 7309. [Google Scholar] [CrossRef] [PubMed]
- Bouaboula, H.; Belmabkhout, Y.; Zaabout, A. Integrated CO2 capture and green hydrogen production: A promising approach for energy and cost reductions. Chem. Eng. J. 2025, 509, 161361. [Google Scholar] [CrossRef]
- Deutz, S.; Bardow, A. Life-cycle assessment of an industrial direct air capture process based on temperature–vacuum swing adsorption. Nat. Energy 2021, 6, 203. [Google Scholar] [CrossRef]
- Realmonte, G.; Drouet, L.; Gambhir, A.; Tavoni, M.; Longden, T.; Smith, S.M.; Glynn, J.R. An inter-model assessment of the role of direct air capture in deep mitigation pathways. Nat. Commun. 2019, 10, 3277. [Google Scholar] [CrossRef]
- Shin, W.J.; Lee, Y.; Yu, Y.; Ko, M.; Lee, C.; Song, H.H. Comparative life cycle greenhouse gas analysis of clean hydrogen pathways: Assessing domestic production and overseas import in South Korea. J. Clean. Prod. 2023, 425, 138907. [Google Scholar] [CrossRef]
- Terlouw, T.; Treyer, K.; Bauer, C.; Mazzotti, M. Life cycle assessment of direct air carbon capture and storage with low-carbon energy sources. Environ. Sci. Technol. 2021, 55, 11397. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TEA Scale | Electricity/Hydrogen Credit Scenario | ||
|---|---|---|---|
| Current U.S. Grid Mix | |||
| No H2 Credit | H2 SMR Credit | H2 Electrolysis Credit | |
| 1X | N/A | N/A | 5032 |
| 14X | N/A | N/A | 3090 |
| 2050 U.S. Grid Mix | |||
| No H2 Credit | H2 SMR Credit | H2 Electrolysis Credit | |
| 1X | N/A | N/A | 3443 |
| 14X | N/A | N/A | 2115 |
| Fossil Power with CCS | |||
| No H2 Credit | H2 SMR Credit | H2 Electrolysis Credit | |
| 1X | 1366 | 1811 | 1600 |
| 14X | 839 | 1112 | 983 |
| Renewables | |||
| No H2 Credit | H2 SMR Credit | H2 Electrolysis Credit | |
| 1X | 1340 | 818 | 1188 |
| 14X | 823 | 502 | 730 |
| Electricity Scenario | Hydrogen Credit Scenario | ||
|---|---|---|---|
| DAC with No H2 | DAC with H2 | DAC with H2 | |
| Credit | SMR Credit | Electrolysis Credit | |
| Current U.S. Grid Mix | 1.394 | 0.862 | −0.220 |
| 2050 U.S. Grid Mix | 1.078 | 0.546 | −0.321 |
| Fossil Power with CCS | −0.081 | −0.614 | −0.695 |
| Renewables | −0.830 | −1.362 | −0.935 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Gao, X.; Berger, A.; Omosebi, A.; Chen, T.; Patrick, A.; Liu, K. A Bench-Scale Demonstration of Direct Air Capture Using an Enhanced Electrochemical System. Clean Technol. 2025, 7, 50. https://doi.org/10.3390/cleantechnol7020050
Wang J, Gao X, Berger A, Omosebi A, Chen T, Patrick A, Liu K. A Bench-Scale Demonstration of Direct Air Capture Using an Enhanced Electrochemical System. Clean Technologies. 2025; 7(2):50. https://doi.org/10.3390/cleantechnol7020050
Chicago/Turabian StyleWang, Jinwen, Xin Gao, Adam Berger, Ayokunle Omosebi, Tingfei Chen, Aron Patrick, and Kunlei Liu. 2025. "A Bench-Scale Demonstration of Direct Air Capture Using an Enhanced Electrochemical System" Clean Technologies 7, no. 2: 50. https://doi.org/10.3390/cleantechnol7020050
APA StyleWang, J., Gao, X., Berger, A., Omosebi, A., Chen, T., Patrick, A., & Liu, K. (2025). A Bench-Scale Demonstration of Direct Air Capture Using an Enhanced Electrochemical System. Clean Technologies, 7(2), 50. https://doi.org/10.3390/cleantechnol7020050