
Sorption Mechanisms of Chemicals in Soils

Abstract
1. Historical Advancements in Adsorption Phenomenon in Soils
“…prove of great importance in modifying the theory and in confirming or improving the practice of many agricultural operations.”
- Chemical availability from soil solution for uptake by an organism or transport out of the soil is controlled by many distinct types of sorption processes that occur at the solid-solution interface, each with its own chemical energy that controls the distribution of the chemical between the solid and solution.
- Sorption amount can be evaluated indirectly by changes in the chemical composition of the solution, but accurate measurement of sorption mechanisms requires a multitude of investigation methods and is best supported using molecular-level tools that can directly measure sorbed chemical speciation [11,12].
2. Modern Concepts of Sorption
3. Factors Controlling Sorption Mechanisms in Soils
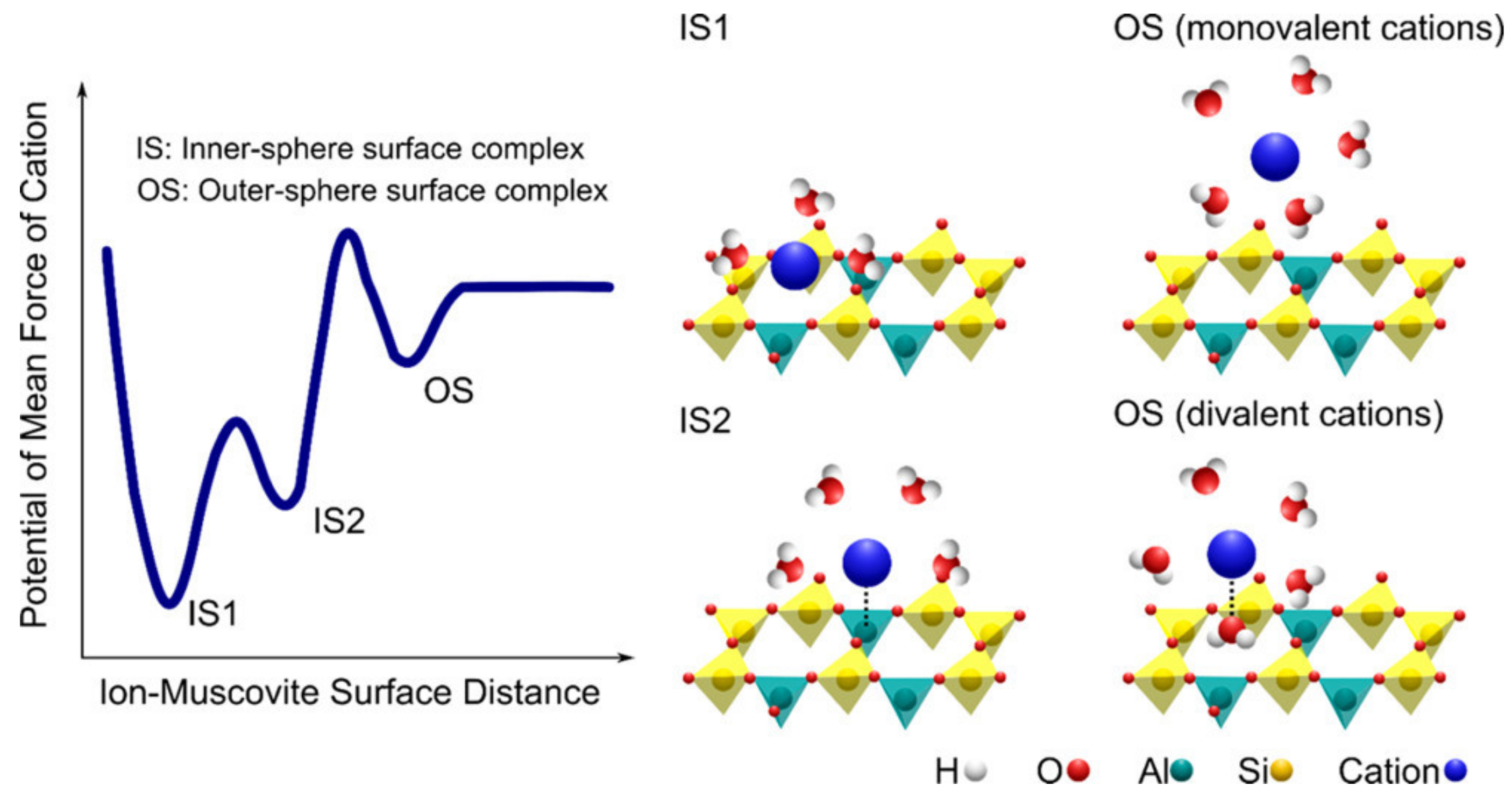
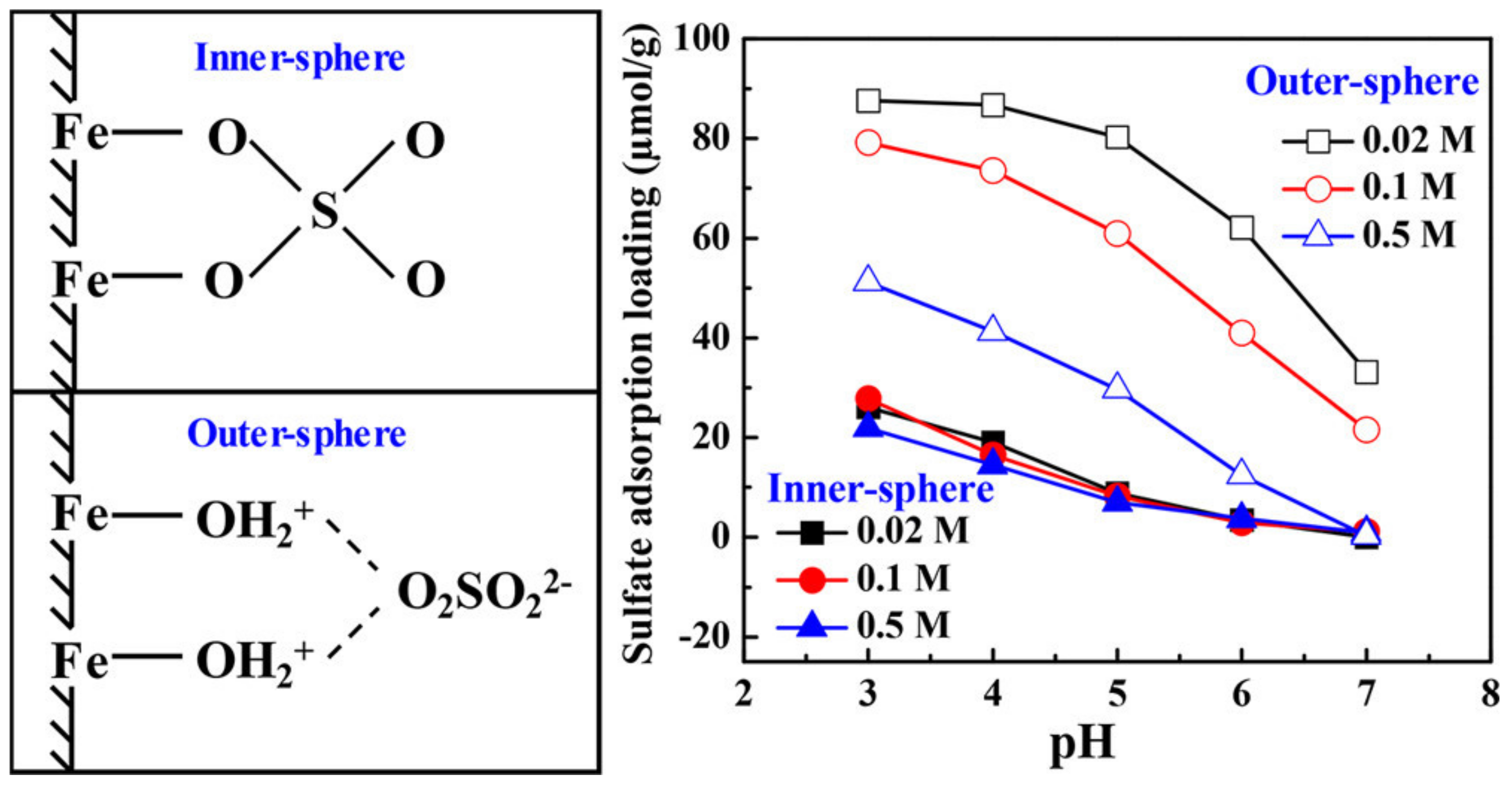
4. Outer-Sphere Adsorption
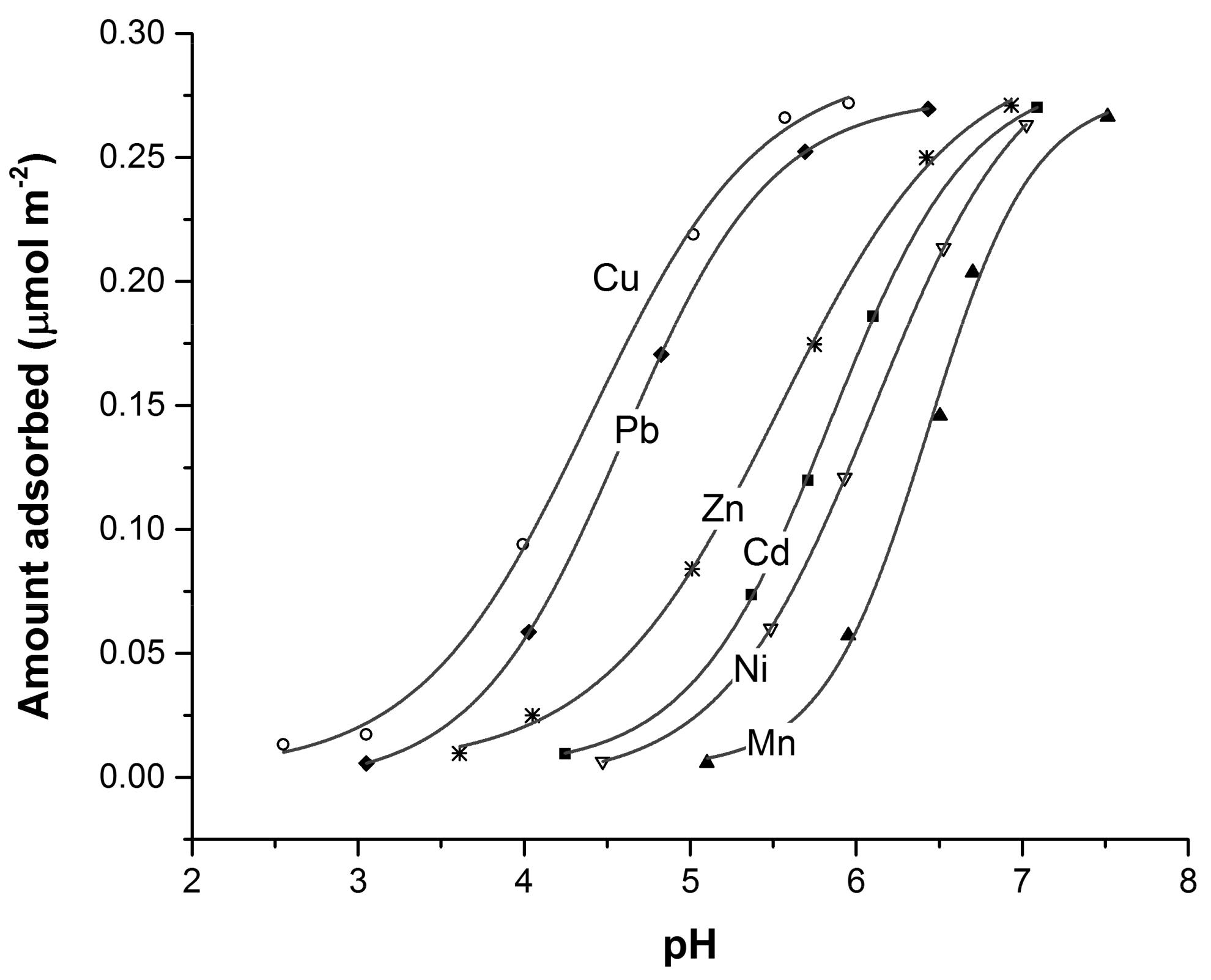
5. Inner-Sphere Adsorption
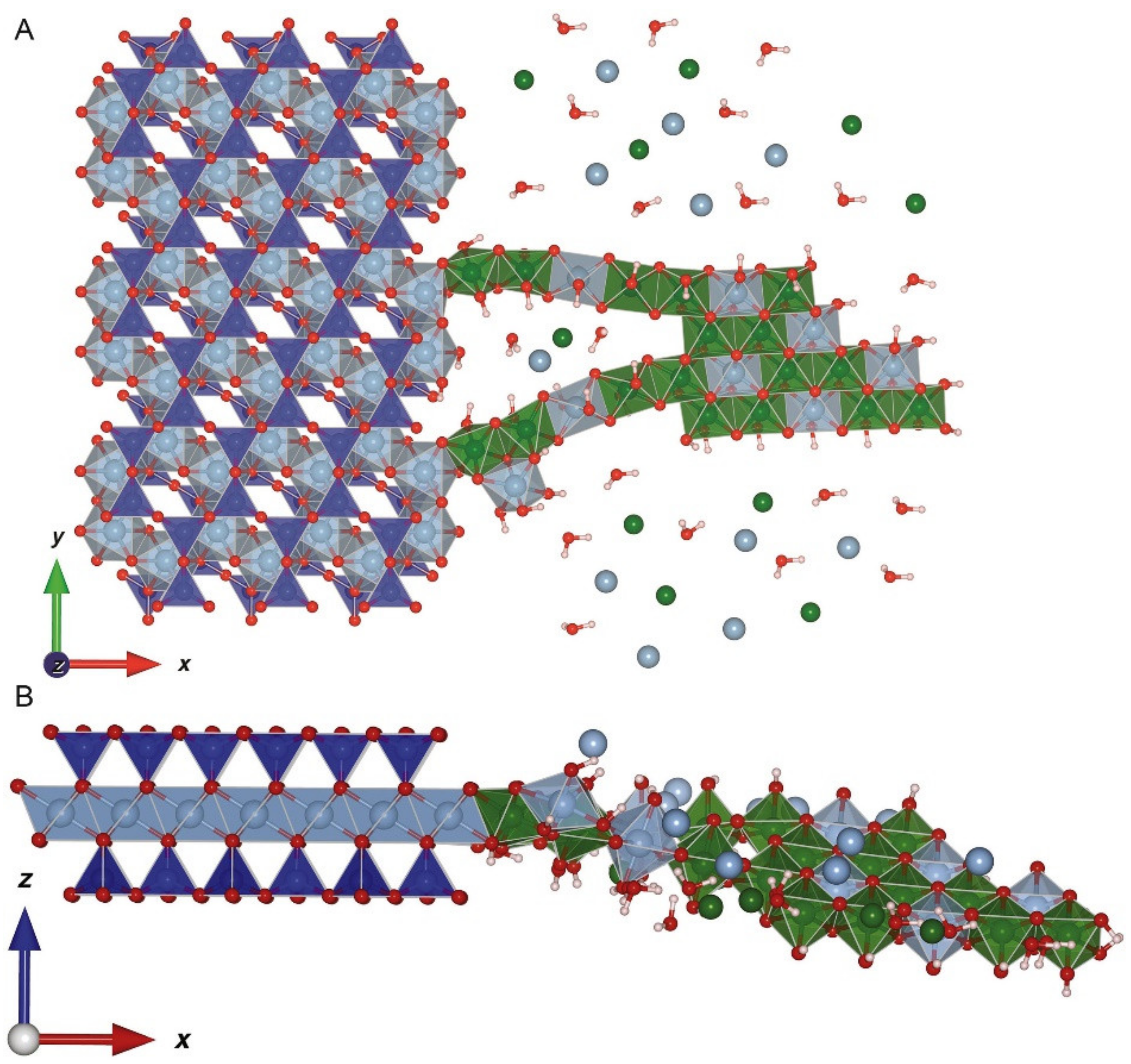
6. Multi-Nuclear Precipitation on Mineral Surfaces
“Molecular concepts can be studied only by molecular methods.”
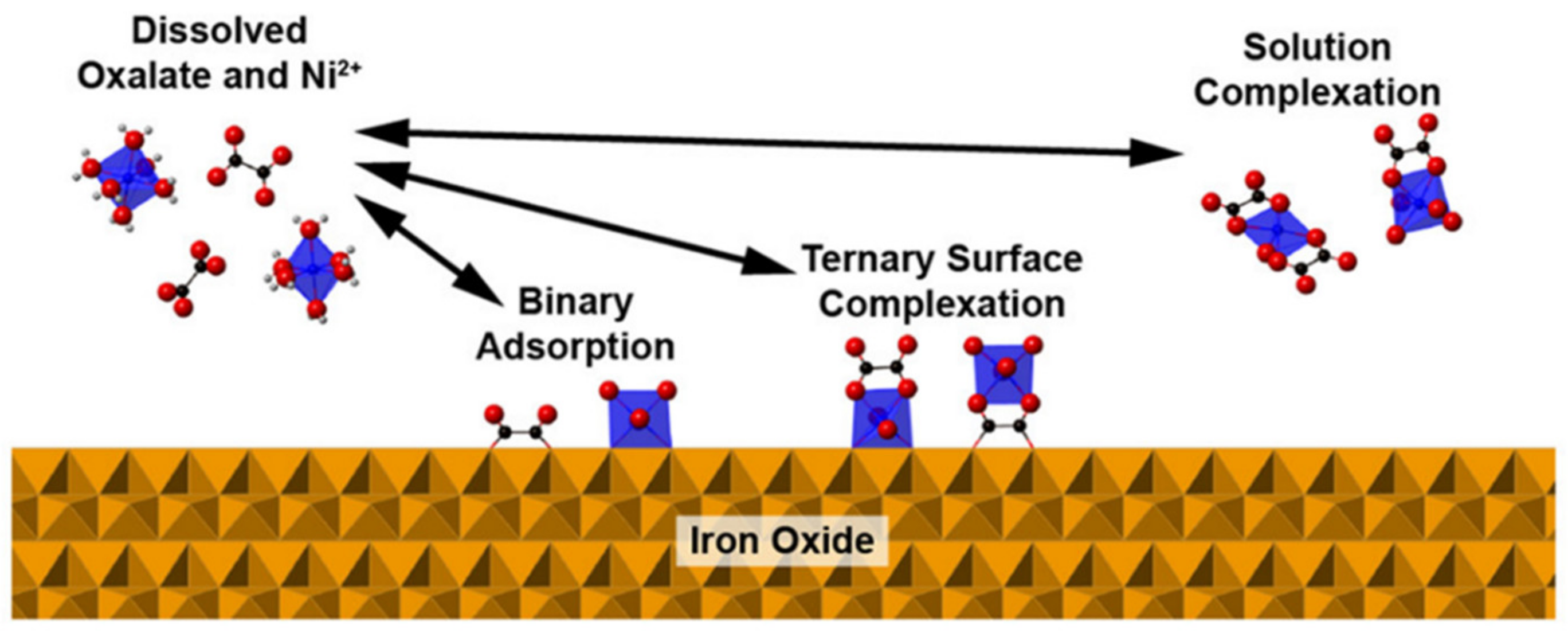
7. Ternary Surface Complexes
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lavoisier, A. Elements of Chemistry in New Systematic Order, Containing All Modern Discoveries, Illustrated with 13 Copperplates, Translated from the French by Robert Kerr, 1st ed.; William Creech: Edinburgh, UK, 1790; Volume 1, p. 511. [Google Scholar]
- Sparks, D.L. The origins of agricultural chemistry: The forerunner of soil chemistry. In Footprints in Soil; Warkentin, B., Ed.; Elsevier Science: Amsterdam, The Netherlands, 2006; p. 572. [Google Scholar]
- Van der Ploeg, R.R.; Böhm, W.; Kirkham, M.B. On the origin of the theory of mineral nutrition of plants and the law of the minimum. Soil Sci. Soc. Am. J. 1999, 63, 1055–1062. [Google Scholar] [CrossRef]
- Everett, D.H. Manual of symbols and terminology for physicochemical quantities and units, appendix II: Definitions, terminology and symbols in colloid and surface chemistry. Pure Appl. Chem. 1972, 31, 577–638. [Google Scholar] [CrossRef]
- Way, J.T. On the power of soils to absorb manure. J. R. Agric. Soc. Engl. 1850, 11, 313. [Google Scholar]
- Schuffelen, A.C. A few aspects of 50 years of soil chemistry. Geoderma 1974, 12, 281–297. [Google Scholar] [CrossRef]
- Thomas, G.W. Historical developments in soil chemistry—Ion-exchange. Soil Sci. Soc. Am. J. 1977, 41, 230–238. [Google Scholar] [CrossRef]
- Sposito, G. Cation exchange in soils: A historical and theoretical perspective. In Chemistry in the Soil Environment; American Society of Agronomy: Madison, WI, USA, 1981; pp. 13–30. [Google Scholar]
- Hendricks, S.B.; Fry, W.H. The results of X-ray and microscopical examinations of soil colloids. Soil Sci. 1930, 29, 457–479. [Google Scholar] [CrossRef]
- Kelley, W.P.; Dore, W.H.; Brown, S.M. The nature of the base-exchange material of bentonite, soils, and zeolites, as revealed by chemical investigation and X-ray analysis. Soil Sci. 1931, 31, 25–55. [Google Scholar] [CrossRef]
- Sposito, G. Distinguishing adsorption from surface precipitation. In Geochemical Processes at Mineral Surfaces; American Chemical Society: Washington, DC, USA, 1987; Volume 323, pp. 217–228. [Google Scholar]
- Veith, J.A.; Sposito, G. On the use of the Langmuir equation in the interpretation of “adsorption” phenomena. Soil Sci. Soc. Am. J. 1977, 41, 697–702. [Google Scholar] [CrossRef]
- Gebhardt, H.; Coleman, N.T. Anion adsorption by allophanic tropical soils: I. chloride adsorption. Soil Sci. Soc. Am. J. 1974, 38, 255–259. [Google Scholar] [CrossRef]
- Wang, P.G.; Ji, G.L.; Yu, T.R. Adsorption of chloride and nitrate by variable charge soils in relation to the electric charge of the soil. Z. Pflanz. Bodenkd. 1987, 150, 17–23. [Google Scholar] [CrossRef]
- Sokolova, T.A.; Alekseeva, S.A. Adsorption of sulfate ions by soils (A review). Eurasian Soil Sci. 2008, 41, 140–148. [Google Scholar] [CrossRef]
- Goldberg, S. Modeling selenite adsorption envelopes on oxides, clay minerals, and soils using the triple layer model. Soil Sci. Soc. Am. J. 2013, 77, 64–71. [Google Scholar] [CrossRef]
- Goldberg, S. Competitive adsorption of arsenate and arsenite on oxides and clay minerals. Soil Sci. Soc. Am. J. 2002, 66, 413–421. [Google Scholar] [CrossRef]
- Bradl, H.B. Adsorption of heavy metal ions on soils and soils constituents. J. Colloid Interface Sci. 2004, 277. [Google Scholar] [CrossRef]
- Peng, L.; Liu, P.; Feng, X.; Wang, Z.; Cheng, T.; Liang, Y.; Lin, Z.; Shi, Z. Kinetics of heavy metal adsorption and desorption in soil: Developing a unified model based on chemical speciation. Geochim. Cosmochim. Acta 2018, 224, 282–300. [Google Scholar] [CrossRef]
- Arai, Y.; Sparks, D.L. Phosphate reaction dynamics in soils and soil components: A multiscale approach. In Advances in Agronomy; Sparks, D.L., Ed.; Academic Press: Cambridge, MA, USA, 2007; Volume 94, pp. 135–179. [Google Scholar]
- Arai, Y.; Sparks, D.L.; Davis, J.A. Arsenate adsorption mechanisms at the allophane-water interface. Environ. Sci. Technol. 2005, 39, 2537–2544. [Google Scholar] [CrossRef]
- Manceau, A. The mechanism of anion adsorption on iron oxides: Evidence for the bonding of arsenate tetrahedra on free Fe(O, OH)6 edges. Geochim. Cosmochim. Acta 1995, 59, 3647–3653. [Google Scholar] [CrossRef]
- Sawhney, B.L. Selective sorption and fixation of cations by clay minerals: A review. Clays Clay Miner. 1972, 20, 93–100. [Google Scholar] [CrossRef]
- Siebecker, M.G.; Li, W.; Sparks, D.L. The important role of layered double hydroxides in soil chemical processes and remediation: What we have learned over the past 20 years. In Advances in Agronomy; Elsevier: Amsterdam, The Netherlands; Academic Press Inc: San Diego, CA, USA, 2018; Volume 147, pp. 1–59. [Google Scholar]
- Waychunas, G.A.; Rea, B.A.; Fuller, C.C.; Davis, J.A. Surface-chemistry of ferrihydrite.1. EXAFS studies of the geometry of coprecipitated and adsorbed arsenate. Geochim. Cosmochim. Acta 1993, 57, 2251–2269. [Google Scholar] [CrossRef]
- Voice, T.C.; Weber, W.J. Sorption of hydrophobic compounds by sediments, soils and suspended solids—I. Theory and background. Water Res. 1983, 17, 1433–1441. [Google Scholar] [CrossRef]
- Kravchenko, A.N.; Guber, A.K. Soil pores and their contributions to soil carbon processes. Geoderma 2017, 287, 31–39. [Google Scholar] [CrossRef]
- Strawn, D.; Bohn, H.L.; O’Connor, G.A. Soil Chemistry, 5th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2020. [Google Scholar]
- Allison, L.E.; Al, E. Diagnosis and Improvement of Saline and Alkali Soils; USDA, United States Salinity Laboratory: Riverside, CA, USA, 1954; Volume 60.
- Parfitt, R.L. Phosphate adsorption on an oxisol. Soil Sci. Soc. Am. J. 1977, 41, 1064–1067. [Google Scholar] [CrossRef]
- Tournassat, C.; Davis, J.A.; Chiaberge, C.; Grangeon, S.; Bourg, I.C. Modeling the acid-base properties of montmorillonite edge surfaces. Environ. Sci. Technol. 2016, 50, 13436–13445. [Google Scholar] [CrossRef] [PubMed]
- Bickmore, B.R.; Tadanier, C.J.; Rosso, K.M.; Monn, W.D.; Eggett, D.L. Bond-valence methods for pKa prediction: Critical reanalysis and a new approach. Geochim. Cosmochim. Acta 2004, 68, 2025–2042. [Google Scholar] [CrossRef]
- Hiemstra, T.; van Riemsdijk, W.H.; Bolt, G.H. Multisite proton adsorption modeling at the solid/solution interface of (hydr)oxides: A new approach: I. model description and evaluation of intrinsic reaction constants. J. Colloid Interface Sci. 1989, 133, 91–104. [Google Scholar] [CrossRef]
- Zachara, J.; Brantley, S.; Chorover, J.; Ewing, R.; Kerisit, S.; Liu, C.; Perfect, E.; Rother, G.; Stack, A.G. Internal domains of natural porous media revealed: Critical locations for transport, storage, and chemical reaction. Environ. Sci. Technol. 2016, 50, 2811–2829. [Google Scholar] [CrossRef] [PubMed]
- Posner, A.M. Humic acids extracted by various reagents from a soil yield inorganic components and titration curves. J. Soil Sci. 1966, 17, 65–78. [Google Scholar] [CrossRef]
- Lees, H. A note on the copper-retaining power of a humic acid from peat soil. Biochem. J. 1950, 46, 450–451. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, F.J. Humus Chemistry: Genesis, Composition, Reactions; Wiley: New York, NY, USA, 1982; p. 443. [Google Scholar]
- Xia, K.; Skyllberg, U.L.; Bleam, W.F.; Bloom, P.R.; Nater, E.A.; Helmke, P.A. X-ray absorption spectroscopic evidence for the complexation of Hg (II) by reduced sulfur in soil humic substances. Environ. Sci. Technol. 1999, 33, 257–261. [Google Scholar] [CrossRef]
- Sposito, G. Characterization of particle surface charge. In Environmental Particles; Buffle, J., van Leeuwen, H.P., Eds.; Lewis Publishers: Boca Raton, FL, USA, 1992; Volume 1, p. 291. [Google Scholar]
- Kleber, M.; Lehmann, J. Humic substances extracted by alkali are invalid proxies for the dynamics and functions of organic matter in terrestrial and aquatic ecosystems. J. Environ. Qual. 2019, 48, 207–216. [Google Scholar] [CrossRef]
- Helling, C.S.; Chesters, G.; Corey, R.B. Contribution of organic matter and clay to soil cation-exchange capacity as affected by the pH of the saturating solution. Soil Sci. Soc. Am. J. 1964, 28, 517–520. [Google Scholar] [CrossRef]
- Strawn, D.G.; Baker, L.L. Speciation of Cu in a contaminated agricultural soil measured by XAFS, μ-XAFS, and μ-XRF. Environ. Sci. Technol. 2008, 42, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Strawn, D.G.; Sparks, D.L. The use of XAFS to distinguish between inner- and outer-sphere lead adsorption complexes on montmorillonite. J. Colloid Interface Sci. 1999, 216, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Manceau, A.; Boisset, M.-C.; Sarret, G.; Hazemann, J.-L.; Mench, M.; Cambier, P.; Prost, R. Direct determination of lead speciation in contaminated soils by EXAFS spectroscopy. Environ. Sci. Technol. 1996, 30, 1540–1552. [Google Scholar] [CrossRef]
- Shi, Z.; Peltier, E.; Sparks, D.L. Kinetics of Ni sorption in soils: Roles of soil organic matter and Ni precipitation. Environ. Sci. Technol. 2012, 46, 2212–2219. [Google Scholar] [CrossRef]
- Anderson, S.J.; Sposito, G. Proton surface-charge density in soils with structural and pH-dependent charge. Soil Sci. Soc. Am. J. 1992, 56, 1437–1443. [Google Scholar] [CrossRef]
- Sparks, D.L. Environmental Soil Chemistry, 2nd ed.; Academic Press: Amsterdam, The Netherlands; Boston, MA, USA, 2003; p. 352. [Google Scholar]
- Zelazny, L.W.; He, L.; Vanwormhoudt, A. Charge analysis of soils and anion exchange. In Methods of Soil Analysis; Soil Science Society of America: Madison, WI, USA, 1996; pp. 1231–1253. [Google Scholar]
- Eick, M.J.; Brady, W.D.; Lynch, C.K. Charge properties and nitrate adsorption of some acid southeastern soils. J. Environ. Qual. 1999, 28, 138–144. [Google Scholar] [CrossRef]
- Singh, U.; Uehara, G. Electrochemistry of the double layer: Principles and applications to soils. In Soil Physical Chemistry, 2nd ed.; Sparks, D.L., Ed.; CRC Press: New York, NY, USA, 1988. [Google Scholar]
- Quirk, J.P. Interparticle forces: A basis for the interpretation of soil physical behavior. In Advances in Agronomy; Sparks, D.L., Ed.; Academic Press: Cambridge, MA, USA, 1994; Volume 53, pp. 121–183. [Google Scholar]
- Bolt, G.H. Chapter 1: The ionic distribution in the diffuse double layer. In Developments in Soil Science; Bolt, G.H., Ed.; Elsevier: Amsterdam, The Netherlands, 1979; Volume 5, pp. 1–26. [Google Scholar]
- Bolt, G.H.; van Riemsdijk, W.H. Chapter 13: Ion adsorption on inorganic variable charge constituents. In Developments in Soil Science; Bolt, G.H., Ed.; Elsevier: Amsterdam, The Netherlands, 1979; Volume 5, pp. 459–504. [Google Scholar]
- Barak, P. Double layer theory prediction of Al-Ca exchange on clay and soil. J. Colloid Interface Sci. 1989, 133, 479–490. [Google Scholar] [CrossRef]
- Quirk, J.P. Particle interaction and soil swelling. Isr. J. Chem. 1968, 6, 213–234. [Google Scholar] [CrossRef]
- Westall, J. Chemical equilibrium including adsorption on charged surfaces. In Particulates in Water; American Chemical Society: Washington, DC, USA, 1980; Volume 189, pp. 33–44. [Google Scholar]
- Goldberg, S. Constant Capacitance Model. In Emerging Technologies in Hazardous Waste Management III; American Chemical Society: Washington, DC, USA, 1993; Volume 518, pp. 278–307. [Google Scholar]
- Goldberg, S. Use of surface complexation models in soil chemical systems. In Advances in Agronomy; Sparks, D.L., Ed.; Soil Science Society of America: Madison, WI, USA, 1992; Volume 47, pp. 233–330. [Google Scholar]
- Goldberg, S.; Criscenti, L. Modeling adsorption of metals and metalloids by soil components. In Biophysico-Chemical Processes of Heavy Metals and Metalloids in Soil Environments; John Wiley and Sons Inc.: Hoboken, NJ, USA, 2007; pp. 215–264. [Google Scholar]
- Hiemstra, T.; van Riemsdijk, W.H. A surface structural approach to ion adsorption: The charge distribution (CD) model. J. Colloid Interface Sci. 1996, 179, 488–508. [Google Scholar] [CrossRef]
- Gustafsson, J.P. Modelling competitive anion adsorption on oxide minerals and an allophane-containing soil. Eur. J. Soil Sci. 2001, 52, 639–653. [Google Scholar] [CrossRef]
- Goldberg, S.; Corwin, D.L.; Shouse, P.J.; Suarez, D.L. Prediction of boron adsorption by field samples of diverse textures. Soil Sci. Soc. Am. J. 2005, 69, 1379–1388. [Google Scholar] [CrossRef]
- Goldberg, S.; Lesch, S.M.; Suarez, D.L. Predicting boron adsorption by soils using soil chemical parameters in the constant capacitance model. Soil Sci. Soc. Am. J. 2000, 64, 1356–1363. [Google Scholar] [CrossRef]
- Kelley, W.P. Cation Exchange in Soils; Reinhold Publishing Corporation: New York, NY, USA; Chapman & Hall: London, UK, 1948; p. 144. [Google Scholar]
- Kelley, W.P. Review of investigations on cation exchange and semiarid soils. Soil Sci. 1964, 97, 80–88. [Google Scholar] [CrossRef]
- Richards, L.A. Diagnosis and Improvement of Saline and Alkali Soils; US Department of Agriculture: Washington, DC, USA, 1954; p. 166.
- Norrish, K. The swelling of montmorillonite. Discuss. Faraday Soc. 1954, 18, 120–134. [Google Scholar] [CrossRef]
- Norrish, K.; Quirk, J.P. Crystalline swelling of montmorillonite—Use of electrolytes to control swelling. Nature 1954, 173, 255–256. [Google Scholar] [CrossRef]
- Moore, D.M.; Reynolds, R.C. X-ray Diffraction and the Identification and Analysis of Clay Minerals; Oxford University Press: Oxford, UK; New York, NY, USA, 1989; pp. 227–260. [Google Scholar]
- Harris, W.; Norman White, G. X-ray diffraction techniques for soil mineral identification. In Methods of Soil Analysis Part 5—Mineralogical Methods; Soil Science Society of America: Madison, WI, USA, 2008; pp. 81–115. [Google Scholar]
- Hensen, E.J.M.; Smit, B. Why clays swell. J. Phys. Chem. B 2002, 106, 12664–12667. [Google Scholar] [CrossRef]
- Whittaker, M.L.; Lammers, L.N.; Carrero, S.; Gilbert, B.; Banfield, J.F. Ion exchange selectivity in clay is controlled by nanoscale chemical-mechanical coupling. Proc. Natl. Acad. Sci. USA 2019, 116, 22052–22057. [Google Scholar] [CrossRef]
- Tester, C.C.; Aloni, S.; Gilbert, B.; Banfield, J.F. Short- and long-range attractive forces that influence the structure of montmorillonite osmotic hydrates. Langmuir 2016, 32, 12039–12046. [Google Scholar] [CrossRef]
- Fukuma, T. Water distribution at solid/liquid interfaces visualized by frequency modulation atomic force microscopy. Sci. Technol. Adv. Mater. 2010, 11, 033003. [Google Scholar] [CrossRef] [PubMed]
- Siretanu, I.; Ebeling, D.; Andersson, M.P.; Stipp, S.L.S.; Philipse, A.; Stuart, M.C.; van den Ende, D.; Mugele, F. Direct observation of ionic structure at solid-liquid interfaces: A deep look into the Stern Layer. Sci. Rep. 2014, 4, 4956. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Andersson, M.P.; van den Ende, D.; Mugele, F.; Siretanu, I. Probing the surface charge on the basal planes of kaolinite particles with high-resolution atomic force microscopy. Langmuir 2017, 33, 14226–14237. [Google Scholar] [CrossRef]
- Zhai, H.; Wang, L.; Putnis, C.V. Molecular-scale investigations reveal noncovalent bonding underlying the adsorption of environmental DNA on mica. Environ. Sci. Technol. 2019, 53, 11251–11259. [Google Scholar] [CrossRef] [PubMed]
- Martin-Jimenez, D.; Garcia, R. Identification of single adsorbed cations on mica-liquid interfaces by 3D force microscopy. J. Phys. Chem. Lett. 2017, 8, 5707–5711. [Google Scholar] [CrossRef]
- Araki, Y.; Satoh, H.; Okumura, M.; Onishi, H. Localization of cesium on montmorillonite surface investigated by frequency modulation atomic force microscopy. Surf. Sci. 2017, 665, 32–36. [Google Scholar] [CrossRef][Green Version]
- Xian, Z.; Hao, Y.; Zhao, Y.; Song, S. Quantitative determination of isomorphous substitutions on clay mineral surfaces through AFM imaging: A case of mica. Colloids Surf. A Physicochem. Eng. Asp. 2017, 533, 55–60. [Google Scholar] [CrossRef]
- Rotenberg, B.; Marry, V.; Malikova, N.; Turq, P. Molecular simulation of aqueous solutions at clay surfaces. J. Phys. Condens. Matter 2010, 22, 284114. [Google Scholar] [CrossRef] [PubMed]
- Le Crom, S.; Tournassat, C.; Robinet, J.-C.; Marry, V. Influence of polarizability on the prediction of the electrical double layer structure in a clay mesopore: A molecular dynamics study. J. Phys. Chem. C 2020, 124, 6221–6232. [Google Scholar] [CrossRef]
- Greathouse, J.A.; Refson, K.; Sposito, G. Molecular dynamics simulation of water mobility in magnesium-smectite hydrates. J. Am. Chem. Soc. 2000, 122, 11459–11464. [Google Scholar] [CrossRef]
- Teppen, B.J.; Rasmussen, K.; Bertsch, P.M.; Miller, D.M.; Schäfer, L. Molecular dynamics modeling of clay minerals. 1. Gibbsite, kaolinite, pyrophyllite, and beidellite. J. Phys. Chem. B 1997, 101, 1579–1587. [Google Scholar] [CrossRef]
- Chatterjee, A.; Iwasaki, T.; Ebina, T.; Miyamoto, A. A DFT study on clay-cation-water interaction in montmorillonite and beidellite. Comput. Mater. Sci. 1999, 14, 119–124. [Google Scholar] [CrossRef]
- Sposito, G.; Skipper, N.T.; Sutton, R.; Park, S.-h.; Soper, A.K.; Greathouse, J.A. Surface geochemistry of the clay minerals. Proc. Natl. Acad. Sci. USA 1999, 96, 3358–3364. [Google Scholar] [CrossRef]
- Kobayashi, K.; Oyabu, N.; Kimura, K.; Ido, S.; Suzuki, K.; Imai, T.; Tagami, K.; Tsukada, M.; Yamada, H. Visualization of hydration layers on muscovite mica in aqueous solution by frequency-modulation atomic force microscopy. J. Chem. Phys. 2013, 138, 184704. [Google Scholar] [CrossRef]
- Kubicki, J.D.; Ohno, T. Integrating density functional theory modeling with experimental data to understand and predict sorption reactions: Exchange of salicylate for phosphate on goethite. Soil Syst. 2020, 4, 27. [Google Scholar] [CrossRef]
- Ho, T.A.; Greathouse, J.A.; Lee, A.S.; Criscenti, L.J. Enhanced ion adsorption on mineral nanoparticles. Langmuir 2018, 34, 5926–5934. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, J.A.R.; Myneni, S.C.B.; Bourg, I.C. Molecular dynamics simulations of the adsorption of phthalate esters on smectite clay surfaces. J. Phys. Chem. C 2019, 123, 13624–13636. [Google Scholar] [CrossRef]
- Kobayashi, K.; Liang, Y.; Murata, S.; Matsuoka, T.; Takahashi, S.; Nishi, N.; Sakka, T. Ion distribution and hydration structure in the stern layer on muscovite surface. Langmuir 2017, 33, 3892–3899. [Google Scholar] [CrossRef]
- Rahromostaqim, M.; Sahimi, M. Molecular dynamics study of the effect of layer charge and interlayer cations on swelling of mixed-layer chlorite-montmorillonite clays. J. Phys. Chem. C 2020, 124, 2553–2561. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, X.; Zhang, C.; Lu, X. A combined first principles and classical molecular dynamics study of clay-soil organic matters (SOMs) interactions. Geochim. Cosmochim. Acta 2020, 291, 110–125. [Google Scholar] [CrossRef]
- Sowers, T.D.; Wani, R.P.; Coward, E.K.; Fischel, M.H.H.; Betts, A.R.; Douglas, T.A.; Duckworth, O.W.; Sparks, D.L. Spatially resolved organomineral interactions across a permafrost chronosequence. Environ. Sci. Technol. 2020, 54, 2951–2960. [Google Scholar] [CrossRef] [PubMed]
- Papelis, C.; Hayes, K.F. Distinguishing between interlayer and external sorption sites of clay minerals using X-ray absorption spectroscopy. Colloids Surf. A Physicochem. Eng. Asp. 1996, 107, 89–96. [Google Scholar] [CrossRef]
- Vasconcelos, I.F.; Haack, E.A.; Maurice, P.A.; Bunker, B.A. EXAFS analysis of cadmium (II) adsorption to kaolinite. Chem. Geol. 2008, 249, 237–249. [Google Scholar] [CrossRef]
- Vasconcelos, I.F.; Bunker, B.A.; Cygan, R.T. Molecular dynamics modeling of ion adsorption to the basal surfaces of kaolinite. J. Phys. Chem. C 2007, 111, 6753–6762. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Z.; Peak, D.; Tang, Y.; Feng, X.; Zhu, M. Quantification of coexisting inner- and outer-sphere complexation of sulfate on hematite surfaces. ACS Earth Space Chem. 2018, 2, 387–398. [Google Scholar] [CrossRef]
- Xu, T.; Stubbs, J.E.; Eng, P.J.; Catalano, J.G. Response of interfacial water to arsenate adsorption on corundum (0 0 1) surfaces: Effects of pH and adsorbate surface coverage. Geochim. Cosmochim. Acta 2018, 239, 198–212. [Google Scholar] [CrossRef]
- Roszmann, C.A. Retention of phosphorus by soil colloids. Soil Sci. 1927, 24, 465–474. [Google Scholar] [CrossRef]
- Ravikovitch, S. Anion exchange: I. adsorption of the phosphoric acid ions by soils. Soil Sci. 1934, 38, 219–240. [Google Scholar] [CrossRef]
- Sumner, M.E. Soil chemistry: Past, present and future. In Future Prospects for Soil Chemistry: Proceedings of a Symposium Sponsored by Division S-2, Soil Chemistry of the Soil Science Society of America in St. Louis, MO, USA, 30–31 October 1995; Huang, P.M., Sparks, D.L., Boyd, S.A., Eds.; Soil Science Society of America: Madison, WI, USA, 1998; p. 233. [Google Scholar]
- Leeper, G.W. Factors affecting availability of inorganic nutrients in soils with special reference to micronutrient metals. Annu. Rev. Plant Phys. 1952, 3. [Google Scholar] [CrossRef]
- Sumner, M.E. Effect of iron oxides on positive and negative charges in clays and soils. Clay Miner. 1963, 5, 218–226. [Google Scholar] [CrossRef]
- Hingston, F.J.; Atkinson, R.J.; Posner, A.M.; Quirk, J.P. Specific adsorption of anions. Nature 1967, 215, 1459–1461. [Google Scholar] [CrossRef]
- Stumm, W.; Huang, C.; Jenkins, S. Specific chemical interaction affecting the stability of dispersed systems. Croat. Chem. Acta 1970, 42, 223–245. [Google Scholar]
- Huang, C.-P.; Stumm, W. Specific adsorption of cations on hydrous γ-Al2O3. J. Colloid Interface Sci. 1973, 43, 409–420. [Google Scholar] [CrossRef]
- Schindler, P.W.; Fürst, B.; Dick, R.; Wolf, P.U. Ligand properties of surface silanol groups. I. Surface complex formation with Fe3+, Cu2+, Cd2+, and Pb2+. J. Colloid Interface Sci. 1976, 55, 469–475. [Google Scholar] [CrossRef]
- Forbes, E.A.; Posner, A.M.; Quirk, J.P. The specific adsorption of divalent Cd, Co, Cu, Pb, and Zn on goethite. J. Soil Sci. 1976, 27, 154–166. [Google Scholar] [CrossRef]
- Sauve, S.; Hendershot, W.; Allen, H.E. Solid-solution partitioning of metals in contaminated soils: Dependence on pH, total metal burden, and organic matter. Environ. Sci. Technol. 2000, 34, 1125–1131. [Google Scholar] [CrossRef]
- Manning, B.A.; Goldberg, S. Modeling competitive adsorption of arsenate with phosphate and molybdate on oxide minerals. Soil Sci. Soc. Am. J. 1996, 60, 121–131. [Google Scholar] [CrossRef]
- Cabrera, F.; Madrid, L.; de Arambarri, P. Adsorption of phosphate by various oxides: Theoretical treatment of the adsorption envelope. J. Soil Sci. 1977, 28, 306–313. [Google Scholar] [CrossRef]
- Hingston, F.J.; Posner, A.M.; Quirk, J.P. Competitive adsorption of negatively charged ligands on oxide surfaces. Discuss. Faraday Soc. 1971, 334–342. [Google Scholar] [CrossRef]
- Parfitt, R.L.; Russell, J.D. Adsorption on hydrous oxides. IV. Mechanisms of adsorption of various ions on goethite. J. Soil Sci. 1977, 28, 297–305. [Google Scholar] [CrossRef]
- Hiemstra, T.; van Riemsdijk, W.H. Surface structural ion adsorption modeling of competitive binding of oxyanions by metal (hydr)oxides. J. Colloid Interface Sci. 1999, 210, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Fukushi, K.; Sverjensky, D.A. A surface complexation model for sulfate and selenate on iron oxides consistent with spectroscopic and theoretical molecular evidence. Geochim. Cosmochim. Acta 2007, 71. [Google Scholar] [CrossRef]
- McKenzie, R. The adsorption of lead and other heavy metals on oxides of manganese and iron. Soil Res. 1980, 18, 61–73. [Google Scholar] [CrossRef]
- Hayes, K.F.; Roe, A.L.; Brown, G.E., Jr.; Hodgson, K.O.; Leckie, J.O.; Parks, G.A. In situ X-ray absorption study of surface complexes: Selenium oxyanions on α-FeOOH. Science 1987, 238, 783–786. [Google Scholar] [CrossRef] [PubMed]
- Manceau, A.; Charlet, L. The mechanism of selenate adsorption on goethite and hydrous ferric oxide. J. Colloid Interface Sci. 1994, 168, 87–93. [Google Scholar] [CrossRef]
- Peak, D.; Sparks, D.L. Mechanisms of selenate adsorption on iron oxdes and hyroxides. Environ. Sci. Technol. 2002, 36, 1460–1466. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, M.L.; Manceau, A.; Chateigner, D.; Charlet, L. Sorption of metal Ions on clay minerals: I. Polarized EXAFS evidence for the adsorption of Co on the edges of hectorite particles. J. Colloid Interface Sci. 1999, 215, 140–158. [Google Scholar] [CrossRef]
- Furnare, L.J.; Vailionis, A.; Strawn, D.G. Molecular-level investigation into copper complexes on vermiculite: Effect of reduction of structural iron on copper complexation. J. Colloid Interface Sci. 2005, 289. [Google Scholar] [CrossRef] [PubMed]
- Peacock, C.L.; Sherman, D.M. Copper (II) sorption onto goethite, hematite and lepidocrocite: A surface complexation model based on ab initio molecular geometries and EXAFS spectroscopy. Geochim. Cosmochim. Acta 2004, 68, 2623–2637. [Google Scholar] [CrossRef]
- Tiberg, C.; Sjöstedt, C.; Eriksson, A.K.; Klysubun, W.; Gustafsson, J.P. Phosphate competition with arsenate on poorly crystalline iron and aluminum (hydr)oxide mixtures. Chemosphere 2020, 255, 126937. [Google Scholar] [CrossRef] [PubMed]
- Barrow, N. The description of desorption of phosphate from soil. J. Soil Sci. 2006, 30, 259–270. [Google Scholar] [CrossRef]
- Corey, R.B. Sorption vs precipitaiton. In Adsorption of Inorgnaics at Solid-Liquid Interfaces; Anderson, M.A., Rubin, A.J., Eds.; Ann Arbor Science: Ann Arbor, MI, USA, 1980; pp. 161–182. [Google Scholar]
- Farley, K.J.; Dzombak, D.A.; Morel, F.M.M. A surface precipitation model for the sorption of cations on metal oxides. J. Colloid Interface Sci. 1985, 106, 226–242. [Google Scholar] [CrossRef]
- Comans, R.N.J.; Middelburg, J.J. Sorption of trace metals on calcite: Applicability of the surface precipitation model. Geochim. Cosmochim. Acta 1987, 51, 2587–2591. [Google Scholar] [CrossRef]
- Bleam, W.F.; McBride, M.B. Cluster formation versus isolated-site adsorption—A study of Mn (II) and Mg (II) adsorption on boehmite and goethite. J. Colloid Interface Sci. 1985, 103, 124–132. [Google Scholar] [CrossRef]
- Chisholmbrause, C.J.; Oday, P.A.; Brown, G.E.; Parks, G.A. Evidence for multinuclear metal-ion complexes at solid water interfaces from X-ray absorption-spectroscopy. Nature 1990, 348, 528–531. [Google Scholar] [CrossRef]
- Charlet, L.; Manceau, A. X-ray absorption spectroscopic study of the sorption of Cr (III) at the oxide water interface. 2. adsorption, coprecipitation, and surface precipitation on hydrous ferric-oxide. J. Colloid Interface Sci. 1992, 148, 443–458. [Google Scholar] [CrossRef]
- Charlet, L.; Manceau, A. Evidence for the neoformation of clays upon sorption of Co (II) and Ni (II) on silicates. Geochim. Cosmochim. Acta 1994, 58, 2577–2582. [Google Scholar] [CrossRef]
- Scheidegger, A.M.; Sparks, D.L. Kinetics of the formation and the dissolution of nickel surface precipitates on pyrophyllite. Chem. Geol. 1996, 132, 157–164. [Google Scholar] [CrossRef]
- Ford, R. Potential formation of secondary hydrotalcite-like precipitates during Zn and Cu sorption to pyrophyllite. Mineral. Mag. 1998, 462–463. [Google Scholar] [CrossRef]
- Zhu, C. Estimation of surface precipitation constants for sorption of divalent metals onto hydrous ferric oxide and calcite. Chem. Geol. 2002, 188, 23–32. [Google Scholar] [CrossRef]
- Peltier, E.; Allada, R.; Navrotsky, A.; Sparks, D.L. Nickel solubility and precipitation in soils: A thermodynamic study. Clays Clay Miner. 2006, 54, 153–164. [Google Scholar] [CrossRef]
- Zhu, J.; Fu, Q.; Qiu, G.; Liu, Y.; Hu, H.; Huang, Q.; Violante, A. Influence of low molecular weight anionic ligands on the sorption of heavy metals by soil constituents: A review. Environ. Chem. Lett. 2019, 17, 1271–1280. [Google Scholar] [CrossRef]
- Flynn, E.D.; Catalano, J.G. Competitive and cooperative effects during nickel adsorption to iron oxides in the presence of oxalate. Environ. Sci. Technol. 2017, 51, 9792–9799. [Google Scholar] [CrossRef] [PubMed]
- Mendez, J.C.; Hiemstra, T. Ternary complex formation of phosphate with Ca and Mg Ions binding to ferrihydrite: Experiments and mechanisms. ACS Earth Space Chem. 2020, 4, 545–557. [Google Scholar] [CrossRef]
- Tiberg, C.; Gustafsson, J.P. Phosphate effects on cadmium (II) sorption to ferrihydrite. J. Colloid Interface Sci. 2016, 471, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Strathmann, T.J.; Myneni, S.C.B. Effect of soil fulvic acid on nickel (II) sorption and bonding at the aqueous-boehmite (γ-AlOOH) interface. Environ. Sci. Technol. 2005, 39, 4027–4034. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sorption Mode | Molecular Mechanism | Soil Particles Involved | Examples 1 |
|---|---|---|---|
| Outer-sphere adsorption | Electrostatic attraction of opposite charges At least one water molecule exists between the surface and the adsorbed chemical | Clay minerals with permanent charge, edges of all soil minerals (esp. oxides) that become charged, charged functional groups on SOM | Cations in clay mineral interlayers, alkali and alkaline earth metals on mineral edges and SOM functional groups, anions Cl− [13], NO3− [14], SO42− [15], SeO42− [16], and AsO33− [17] |
| Inner-sphere adsorption—edges and SOM | Sharing of electrons in covalent-type bonds | Soil mineral edges with reactive O ligands and carboxylic acid and phenolic acid functional groups sites on SOM | Metals capable of forming covalent-type bonds [18,19], e.g., Pb2+, Cu2+, Ni2+, and Zn2+, and anions PO43− [20] and AsO43− [21,22] |
| Inner-sphere adsorption—basal planes | Ionic-type bond between dehydrated cations and permanently charged oxygen atoms on basal planes of clay minerals | Clay minerals with tetrahedral isomorphic substitution: vermiculite and illite | Alkali metals with weak hydration spheres [23] (K+, Cs+) and ammonium (NH4+) |
| Precipitation on mineral surfaces | Small multi-nuclear complexes formed on the surfaces of minerals | All soil minerals | Metals, especially transition metals [24], oxyanions AsO43− [25] |
| Hydrophobic partitioning | van der Waal forces between hydrophobic chemicals reduces interactions to polar water molecules reducing entropy | Uncharged regions of SOM and some uncharged mineral surfaces, e.g., basal plane of kaolinite | Organic compounds, e.g., pesticides, industrial compounds (DNAPL), hydrophobic molecules released from biota [26] |
| Absorption | Uptake of solutions into pore space, mainly hydrogen bonding and capillary forces, some Weak van der Waal forces | Porous solids (aggregates, crystallites, or SOM) in which solutes become entrapped | Aqueous and non-aqueous liquids that become physically isolated from the bulk solution [27] |
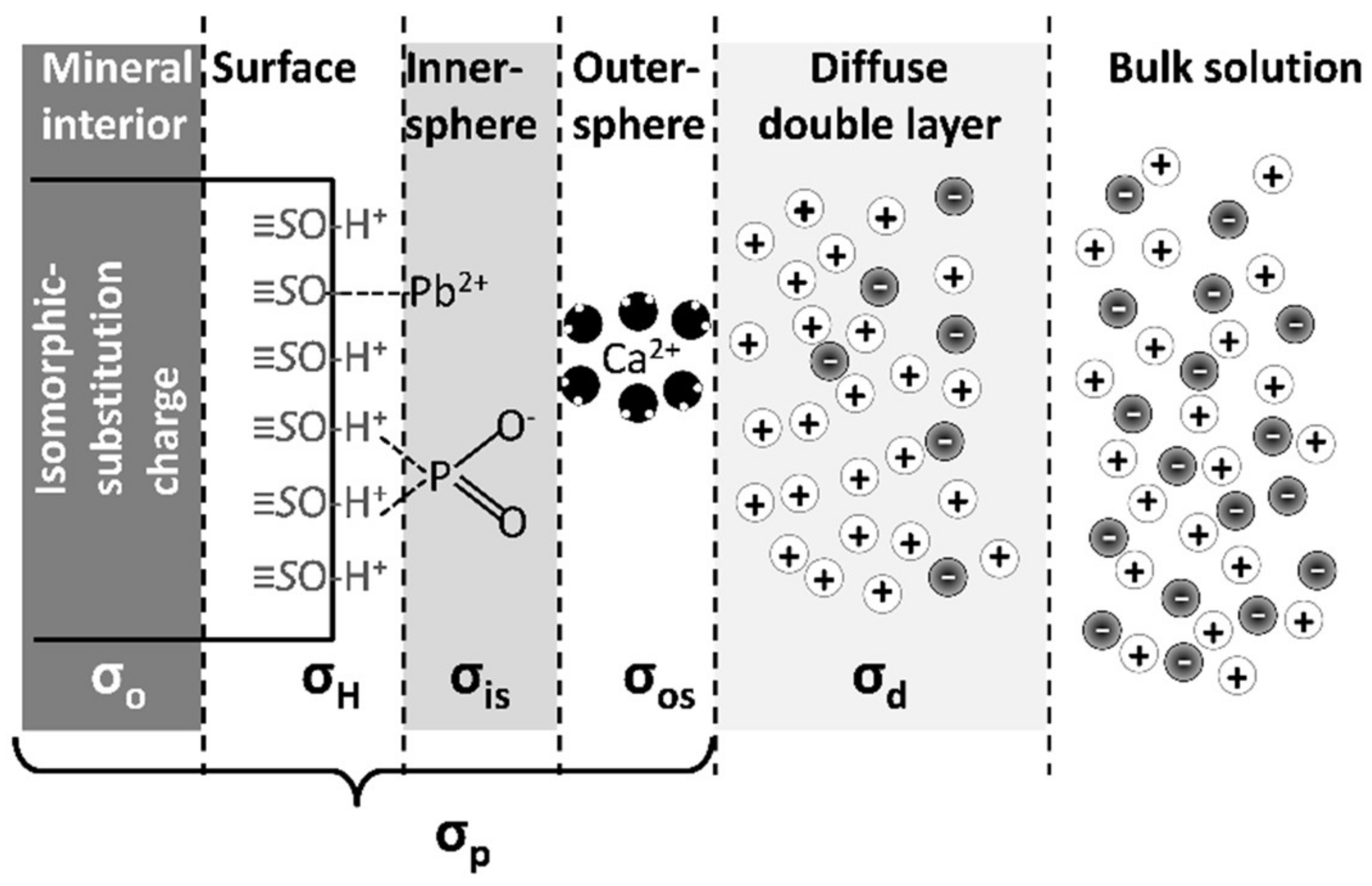
| Adsorption Surface Types | Details | Surface Charge Components 1 | Examples |
|---|---|---|---|
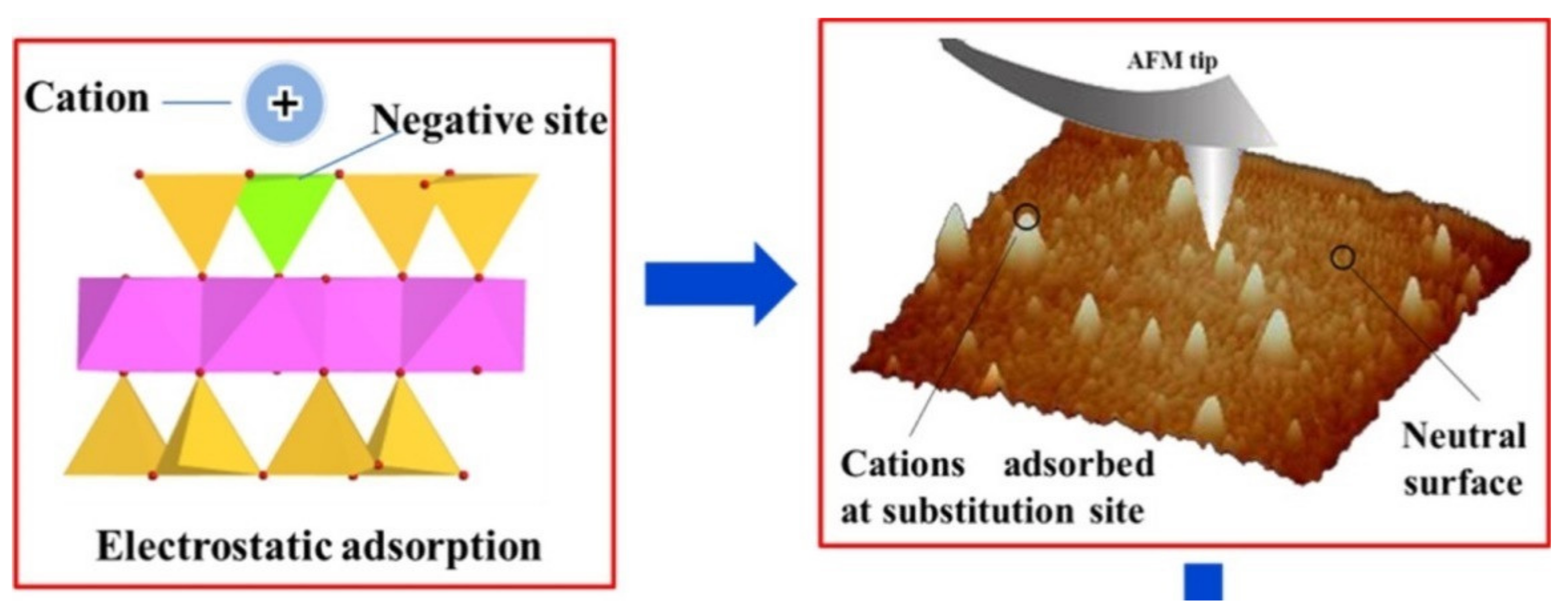
| Permanent negative charge in the interlayer of clay minerals created from isomorphic substitution | Delocalized electrostatic charge that attracts cations. In some cases, cations with weak hydration spheres dehydrate | σo + σOS | Common soil clay minerals, e.g., montmorillonitevermiculite, illite (frayed edges) |
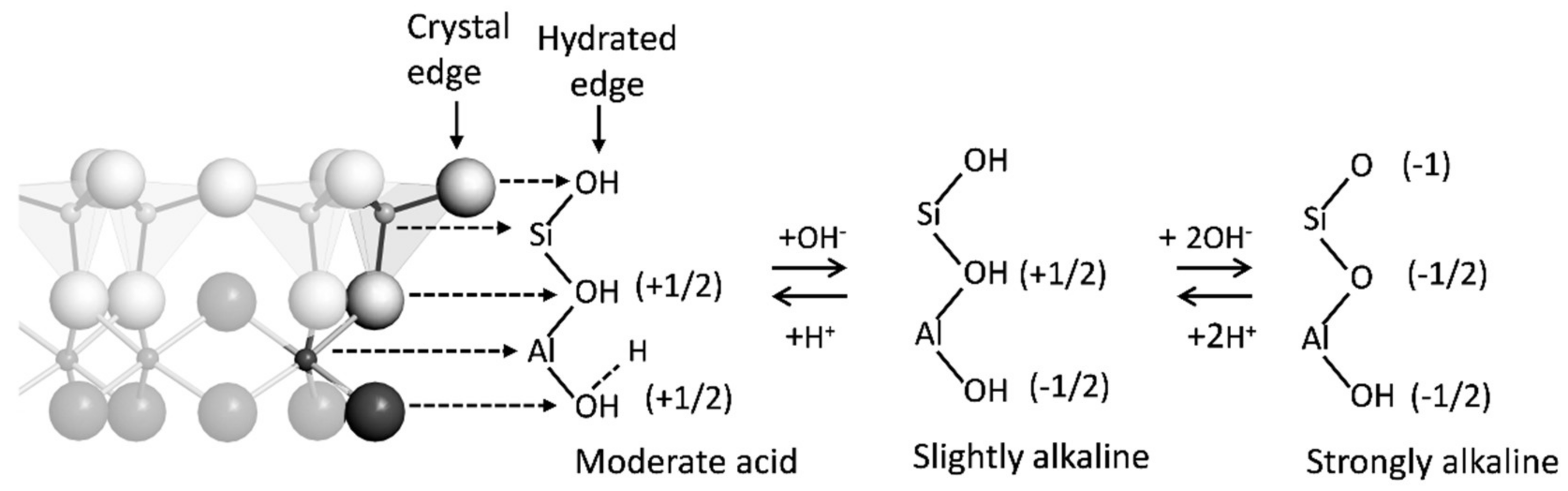
| Hydroxyl ligands on edges of minerals | Unsatisfied bonds on mineral edges are Bronsted acid and bases that gain or lose protons causing pH-dependent charge and adsorption | σH + σIS + σOS |
|
| Weak acid and base functional groups on SOM | Carboxyl, phenol, amine, and thiol functional groups | σH + σIS + σOS | SOM functional groups developed during biomolecule degradation |
| Hydrophobic regions on SOM | Uncharged regions of SOM such as alkanes and aromatic rings. | none |
|
| Surface Charge Component | Description |
|---|---|
| σp | Total charge on the surface of a particle, not including the ions in the diffuse double layer |
| σo | Permanent surface charge of the particle due to isomorphic substitution |
| σH | Charge on the surface of a particle due to protonation and deprotonation of oxygen functional groups on the edge of the mineral |
| σIS | Charge on the surface caused by adsorption of ions via inner-sphere adsorption |
| σOS | Charge on the surface caused by adsorption of ions via outer-sphere adsorption; not including diffuse layer ions |
| σd | Total charge of ions in the diffuse double layer |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strawn, D.G. Sorption Mechanisms of Chemicals in Soils. Soil Syst. 2021, 5, 13. https://doi.org/10.3390/soilsystems5010013
Strawn DG. Sorption Mechanisms of Chemicals in Soils. Soil Systems. 2021; 5(1):13. https://doi.org/10.3390/soilsystems5010013
Chicago/Turabian StyleStrawn, Daniel G. 2021. "Sorption Mechanisms of Chemicals in Soils" Soil Systems 5, no. 1: 13. https://doi.org/10.3390/soilsystems5010013
APA StyleStrawn, D. G. (2021). Sorption Mechanisms of Chemicals in Soils. Soil Systems, 5(1), 13. https://doi.org/10.3390/soilsystems5010013