Subsoil Microbial Diversity and Stability in Rotational Cotton Systems


Abstract
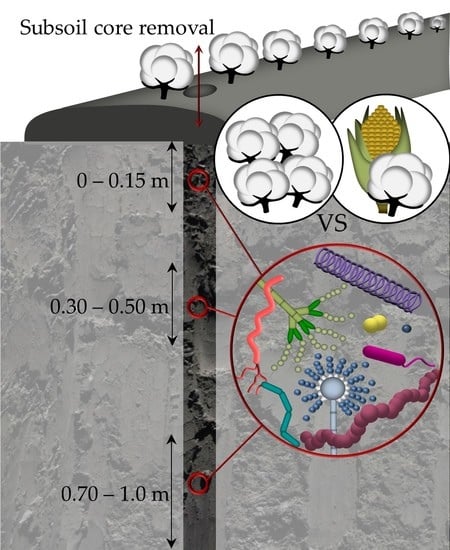
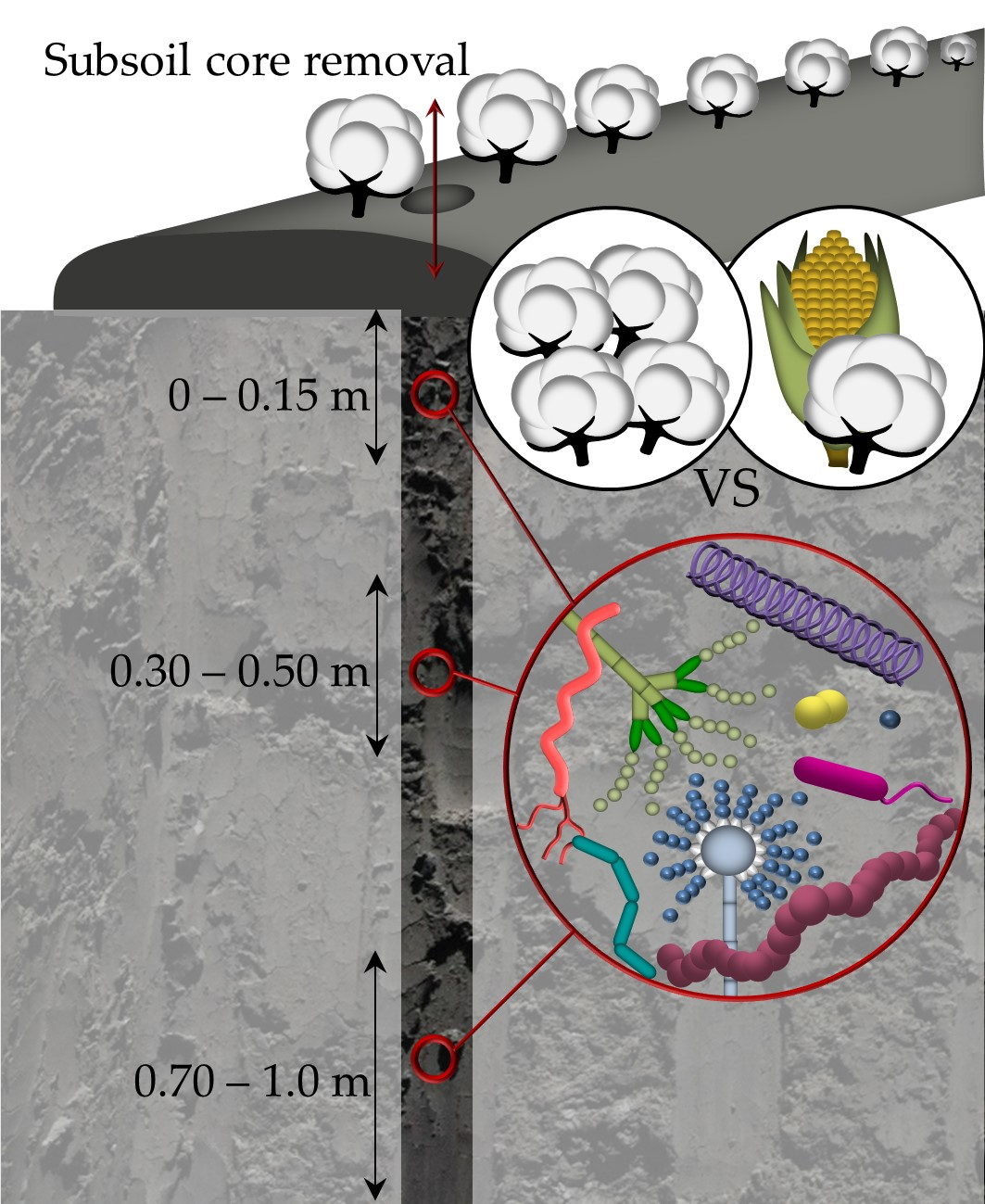
1. Introduction
2. Materials and Methods
3. Results
3.1. Quality Control and Microbial OTUs
3.2. Alpha-Diversity
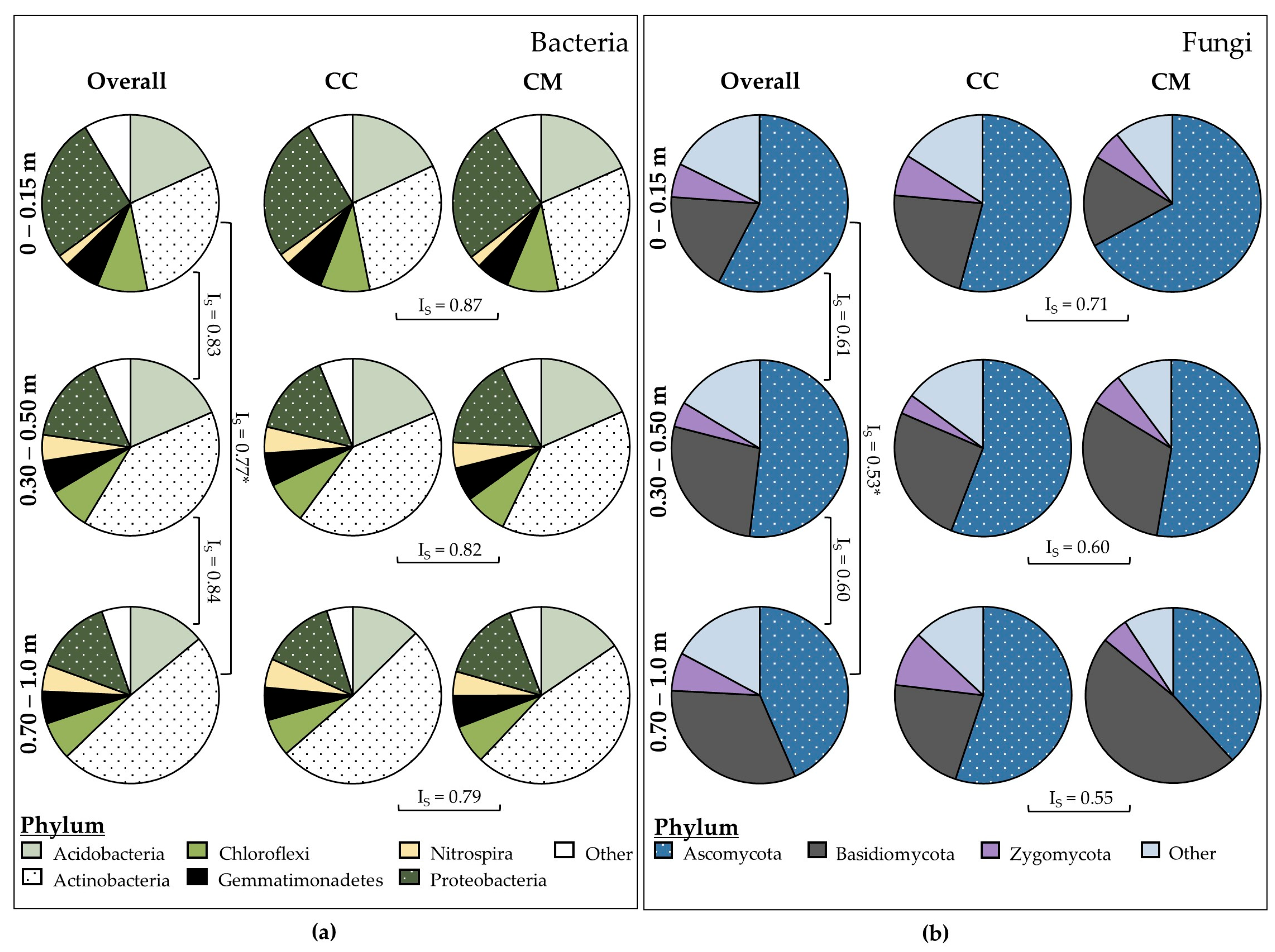
3.2.1. Bacteria (16S)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factors | Chao1 (SChao1) | Gini–Simpson Diversity Index (DGS) | Gini–Simpson Evenness Index (EGS) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 0–0.15 m | 0.30–0.50 m | 0.0–1.0 m | 0–0.15 m | 0.30–0.50 m | 0.0–1.0 m | 0–0.15 m | 0.30–0.50 m | 0.0–1.0 m | ||
| Depth | 2888 a | 4104 b | 3380 c | 0.996 a | 0.993 b | 0.989 c | 4 × 10−4 | 4 × 10−4 | 5 × 10−4 | |
| (p < 0.001) | (L.S.D = 421) | (p < 0.001) | (L.S.D = 0.001) | (p = 0.391) | (L.S.D = 2 × 10−4) | |||||
| System | ||||||||||
| CC | 3014 | 4083 | 3388 | 0.996 a | 0.993 ab | 0.987 c | 3 × 10−4 | 4 × 10−4 | 5 × 10−4 | |
| CM | 2761 | 4125 | 3371 | 0.996 a | 0.994 ab | 0.991 b | 5 × 10−4 | 4 × 10−4 | 5 × 10−4 | |
| (p = 0.727) | (L.S.D = 595) | (p = 0.01) | (L.S.D = 0.002) | (p = 0.244) | (L.S.D = 3 × 10−4) | |||||
| Time | ||||||||||
| S1 Pre | 3630 ab | 3058 ab | 3028 ab | 0.996 | 0.993 | 0.986 | 8 × 10−4 | 4 × 10−4 | 6 × 10−4 | |
| S1 In | 2367 b | 4292 ab | 3168 ab | 0.995 | 0.992 | 0.988 | 3 × 10−4 | 4 × 10−4 | 6 × 10−4 | |
| S2 Pre | 2459 b | 4434 a | 3478 ab | 0.996 | 0.994 | 0.989 | 3 × 10−4 | 4 × 10−4 | 5 × 10−4 | |
| S2 In | 3094 ab | 4633 a | 3845 ab | 0.997 | 0.994 | 0.992 | 3 × 10−4 | 3 × 10−4 | 4 × 10−4 | |
| (p = 0.01) | (L.S.D = 903) | (p = 0.277) | (L.S.D = 0.003) | (p = 0.430) | (L.S.D = 5 × 10−4) | |||||
| Time x System | ||||||||||
| S1 Pre | CC | 4556 | 3108 | 2662 | 0.996 | 0.993 | 0.981 | 3 × 10−4 | 4 × 10−4 | 6 × 10−4 |
| CM | 2704 | 3009 | 3394 | 0.996 | 0.994 | 0.991 | 1 × 10−3 | 4 × 10−4 | 5 × 10−4 | |
| S1 In | CC | 2371 | 4367 | 3211 | 0.995 | 0.992 | 0.987 | 3 × 10−4 | 4 × 10−4 | 6 × 10−4 |
| CM | 2363 | 4217 | 3124 | 0.995 | 0.992 | 0.988 | 3 × 10−4 | 4 × 10−4 | 5 × 10−4 | |
| S2 Pre | CC | 2079 | 4151 | 3699 | 0.995 | 0.994 | 0.987 | 3 × 10−4 | 4 × 10−4 | 6 × 10−4 |
| CM | 2840 | 4718 | 3256 | 0.997 | 0.994 | 0.992 | 3 × 10−4 | 4 × 10−4 | 4 × 10−4 | |
| S2 In | CC | 3052 | 4709 | 3979 | 0.997 | 0.994 | 0.992 | 3 × 10−4 | 3 × 10−4 | 4 × 10−4 |
| CM | 3137 | 4558 | 3711 | 0.996 | 0.995 | 0.992 | 3 × 10−4 | 3 × 10−4 | 4 × 10−4 | |
| (p = 0.144) | (L.S.D = 1277) | (p = 0.247) | (L.S.D = 0.004) | (p =0.349) | (L.S.D = 7 × 10−4) | |||||
3.2.2. Fungi (ITS)
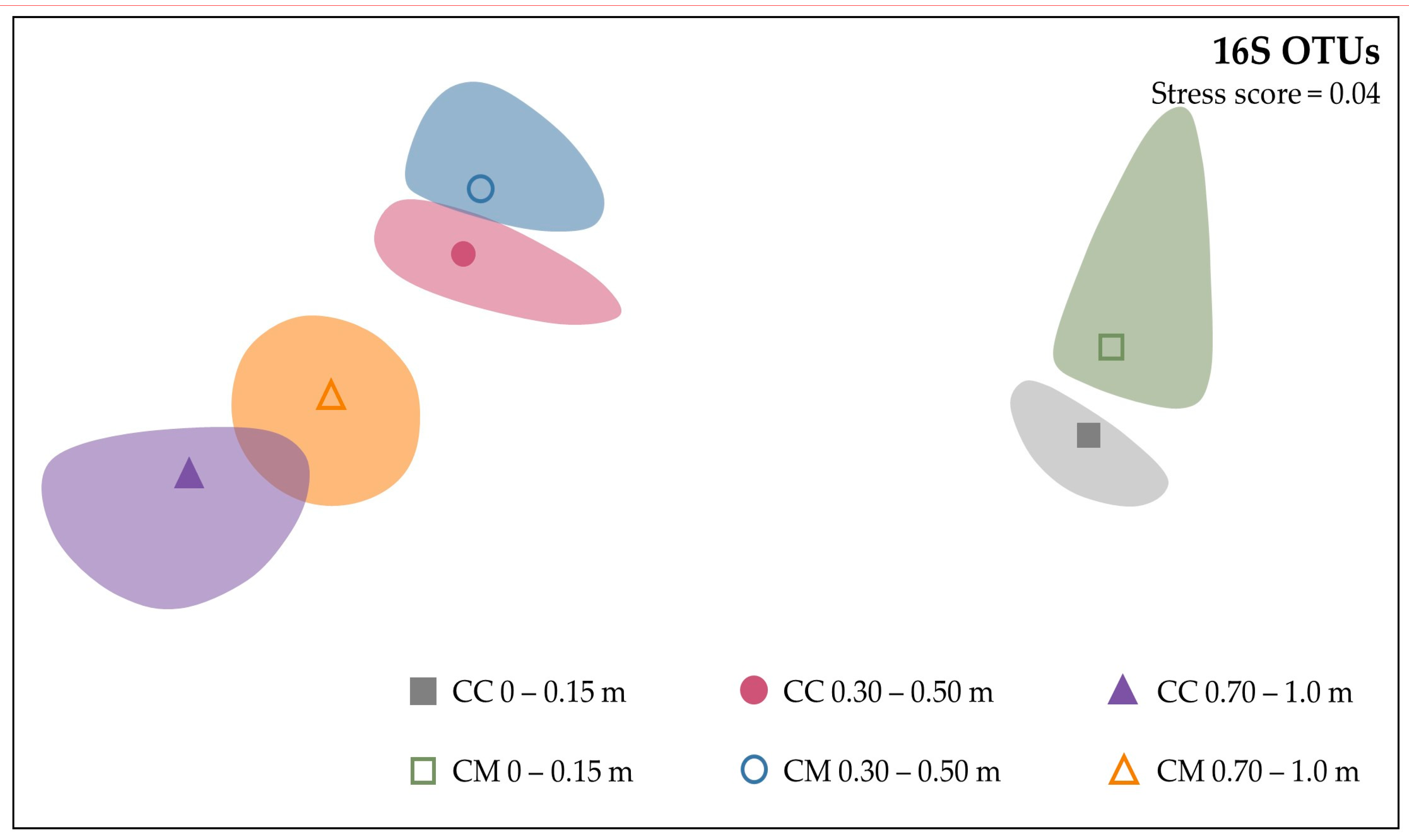
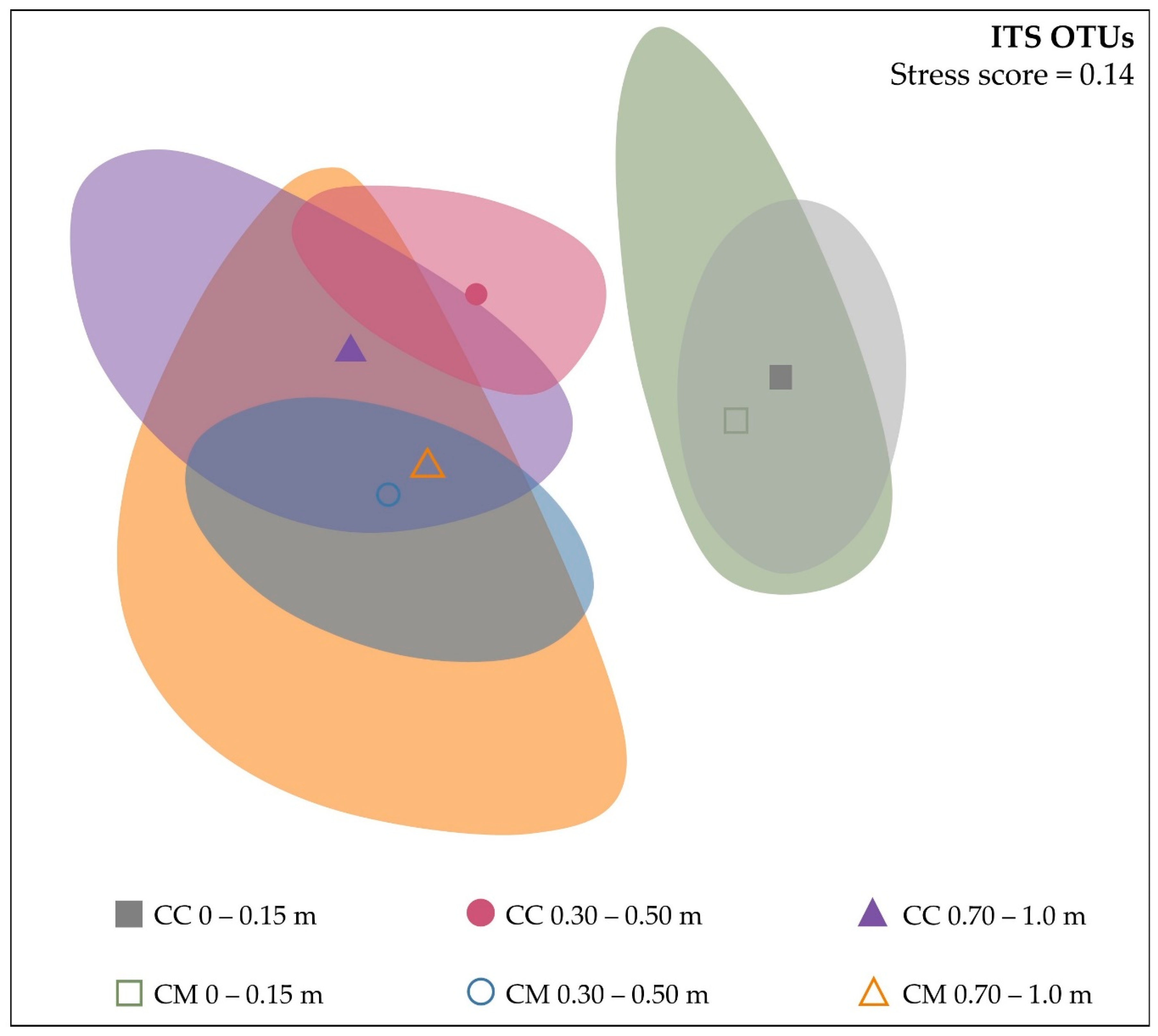
3.3. Beta-Diversity
3.3.1. Dissimilarities between Microbial Communities
3.3.2. Similarities between Microbial Communities
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gupta, V.V. Beneficial microorganisms for sustainable agriculture. Microbiol. Aust. 2012, 33, 113–115. [Google Scholar] [CrossRef]
- Gupta, V.; Neate, S.; Leonard, E. Life in the Soil-The Relationship between Agriculture and Soil Organisms; Cooperative Research Centre for Soil & Land Management: Glen Osmond, Adelaide, Australia, 2007. [Google Scholar]
- Bender, S.F.; Wagg, C.; van der Heijden, M.G. An underground revolution: Biodiversity and soil ecological engineering for agricultural sustainability. Trends Ecol. Evol. 2016, 31, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Lehman, R.M.; Acosta-Martinez, V.; Buyer, J.S.; Cambardella, C.A.; Collins, H.P.; Ducey, T.F.; Halvorson, J.J.; Jin, V.L.; Johnson, J.M.; Kremer, R.J. Soil biology for resilient, healthy soil. J. Soil Water Conserv. 2015, 70, 12A–18A. [Google Scholar] [CrossRef]
- Torsvik, V.; Øvreås, L. Microbial diversity and function in soil: From genes to ecosystems. Curr. Opin. Microbiol. 2002, 5, 240–245. [Google Scholar] [CrossRef]
- Tecon, R.; Or, D. Biophysical processes supporting the diversity of microbial life in soil. FEMS Microbiol. Rev. 2017, 41, 599–623. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.; McBratney, A.; Adams, M.; Field, D.; Hill, R.; Crawford, J.; Minasny, B.; Lal, R.; Abbott, L.; O’Donnell, A. Soil security: Solving the global soil crisis. Glob. Policy 2013, 4, 434–441. [Google Scholar] [CrossRef]
- Gupta, V. Intensive Cropping Starts with the Soil. Available online: https://grdc.com.au/resources-and-publications/groundcover/ground-cover-issue-48/intensive-cropping-starts-with-the-soil (accessed on 16 November 2016).
- Lal, R. Restoring soil quality to mitigate soil degradation. Sustainability 2015, 7, 5875–5895. [Google Scholar] [CrossRef]
- Shade, A.; Peter, H.; Allison, S.D.; Baho, D.; Berga, M.; Bürgmann, H.; Huber, D.H.; Langenheder, S.; Lennon, J.T.; Martiny, J.B. Fundamentals of microbial community resistance and resilience. Front. Microbiol. 2012, 3, 417. [Google Scholar] [CrossRef]
- Allison, S.D.; Martiny, J.B. Resistance, resilience, and redundancy in microbial communities. Proc. Natl. Acad. Sci. USA 2008, 105, 11512–11519. [Google Scholar] [CrossRef]
- Berg, G.; Smalla, K. Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbiol. Ecol. 2009, 68, 1–13. [Google Scholar] [CrossRef]
- Doornbos, R.F.; van Loon, L.C.; Bakker, P.A. Impact of root exudates and plant defense signaling on bacterial communities in the rhizosphere. A review. Agron. Sustain. Dev. 2012, 32, 227–243. [Google Scholar] [CrossRef]
- Chaparro, J.M.; Badri, D.V.; Vivanco, J.M. Rhizosphere microbiome assemblage is affected by plant development. ISME J. 2014, 8, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Hulugalle, N.R.; Broughton, K.J.; Tan, D.K.Y. Root growth of irrigated summer crops in cotton-based farming systems sown in Vertosols of northern New South Wales. Crop Pasture Sci. 2015, 66, 158–167. [Google Scholar] [CrossRef]
- Gunina, A.; Kuzyakov, Y. Sugars in soil and sweets for microorganisms: Review of origin, content, composition and fate. Soil Biol. Biochem. 2015, 90, 87–100. [Google Scholar] [CrossRef]
- Isbell, R. The Australian Soil Classification; CSIRO publishing: Collingwood, Australia; Clayton, Australia, 2016. [Google Scholar]
- IUSS Working Group WRB. World Reference Base for Soil Resources 2014, Update 2015: International Soil Classification System for Naming Soils and Creating Legends for Soil Maps, 106; FAO Rome: Roma, Italy, 2015; p. 192. [Google Scholar]
- Hullugalle, N.; Weaver, T.; Finlay, L.; Entwistle, P. Physical and chemical properties of soil near cracks in irrigated vertisols sown with cotton-wheat rotations. Arid Land Res. Manag. 2001, 15, 13–22. [Google Scholar] [CrossRef]
- Cattle, S.R.; Field, D.J. A review of the soil science research legacy of the triumvirate of cotton CRC. Crop Pasture Sci. 2014, 64, 1076–1094. [Google Scholar] [CrossRef]
- Dias, T.; Dukes, A.; Antunes, P.M. Accounting for soil biotic effects on soil health and crop productivity in the design of crop rotations. J. Sci. Food Agric. 2015, 95, 447–454. [Google Scholar] [CrossRef]
- Dalal, R.C.; Allen, D.E.; Chan, K.Y.; Singh, B.P. Soil organic matter, soil health and climate change. In Soil Health and Climate Change; Springer: Berlin, Germany, 2011; pp. 87–106. [Google Scholar]
- Lal, R. Soil health and carbon management. Food Energy Secur. 2016, 5, 212–222. [Google Scholar] [CrossRef]
- Koch, A.; Chappell, A.; Eyres, M.; Scott, E. Monitor soil degradation or triage for soil security? An Australian challenge. Sustainability 2015, 7, 4870–4892. [Google Scholar] [CrossRef]
- Osanai, Y.; Knox, O.; Nachimuthu, G.; Wilson, B. Increasing soil organic carbon with maize in cotton-based cropping systems: Mechanisms and potential. Agric. Ecosyst. Environ. 2020, 299, 106985. [Google Scholar] [CrossRef]
- Polain, K.; Guppy, C.; Knox, O.; Lisle, L.; Wilson, B.; Osanai, Y.; Siebers, N. Determination of Agricultural Impact on Soil Microbial Activity Using δ18OP HCl and Respiration Experiments. ACS Earth Space Chem. 2018, 2, 683–691. [Google Scholar] [CrossRef]
- Hulugalle, N.; Nachimuthu, G.; Kirkby, K.; Lonergan, P.; Heimoana, V.; Watkins, M.; Finlay, L. Sowing maize as a rotation crop in irrigated cotton cropping systems in a Vertosol: Effects on soil properties, greenhouse gas emissions, black root rot incidence, cotton lint yield and fibre quality. Soil Res. 2020, 58, 137–150. [Google Scholar] [CrossRef]
- Nachimuthu, G.; Hulugalle, N.R.; Watkins, M.D.; Finlay, L.A.; McCorkell, B. Irrigation induced surface carbon flow in a Vertisol under furrow irrigated cotton cropping systems. Soil Tillage Res. 2018, 183, 8–18. [Google Scholar] [CrossRef]
- Nachimuthu, G.; Watkins, M.D.; Hulugalle, N.R.; Weaver, T.B.; Finlay, L.A.; McCorkell, B.E. Leaching of dissolved organic carbon and nitrogen under cotton farming systems in a Vertisol. Soil Use Manag. 2019, 35, 443–452. [Google Scholar] [CrossRef]
- Eilers, K.G.; Debenport, S.; Anderson, S.; Fierer, N. Digging deeper to find unique microbial communities: The strong effect of depth on the structure of bacterial and archaeal communities in soil. Soil Biol. Biochem. 2012, 50, 58–65. [Google Scholar] [CrossRef]
- Hsiao, C.-J.; Sassenrath, G.F.; Zeglin, L.H.; Hettiarachchi, G.M.; Rice, C.W. Vertical changes of soil microbial properties in claypan soils. Soil Biol. Biochem. 2018, 121, 154–164. [Google Scholar] [CrossRef]
- Kramer, S.; Marhan, S.; Haslwimmer, H.; Ruess, L.; Kandeler, E. Temporal variation in surface and subsoil abundance and function of the soil microbial community in an arable soil. Soil Biol. Biochem. 2013, 61, 76–85. [Google Scholar] [CrossRef]
- Moll, J.; Hoppe, B.; König, S.; Wubet, T.; Buscot, F.; Krüger, D. Spatial distribution of fungal communities in an arable soil. PLoS ONE 2016, 11, e0148130. [Google Scholar] [CrossRef]
- Schlatter, D.C.; Kahl, K.; Carlson, B.; Huggins, D.R.; Paulitz, T. Fungal community composition and diversity vary with soil depth and landscape position in a no-till wheat-based cropping system. FEMS Microbiol. Ecol. 2018, 94, fiy098. [Google Scholar] [CrossRef]
- Ashworth, A.; DeBruyn, J.; Allen, F.; Radosevich, M.; Owens, P. Microbial community structure is affected by cropping sequences and poultry litter under long-term no-tillage. Soil Biol. Biochem. 2017, 114, 210–219. [Google Scholar] [CrossRef]
- Dorr de Quadros, P.; Zhalnina, K.; Davis-Richardson, A.; Fagen, J.R.; Drew, J.; Bayer, C.; Camargo, F.A.; Triplett, E.W. The effect of tillage system and crop rotation on soil microbial diversity and composition in a subtropical acrisol. Diversity 2012, 4, 375–395. [Google Scholar] [CrossRef]
- Mbuthia, L.W.; Acosta-Martinez, V.; DeBryun, J.; Schaeffer, S.; Tyler, D.; Odoi, E.; Mpheshea, M.; Walker, F.; Eash, N. Long term tillage, cover crop, and fertilization effects on microbial community structure, activity: Implications for soil quality. Soil Biol. Biochem. 2015, 89, 24–34. [Google Scholar] [CrossRef]
- Silvestro, L.B.; Biganzoli, F.; Stenglein, S.A.; Forjan, H.; Manso, L.; Moreno, M.V. Mixed cropping regimes promote the soil fungal community under zero tillage. Antonie van Leeuwenhoek 2018, 111, 1055–1064. [Google Scholar] [CrossRef]
- Tiemann, L.; Grandy, A.; Atkinson, E.; Marin-Spiotta, E.; McDaniel, M. Crop rotational diversity enhances belowground communities and functions in an agroecosystem. Ecol. Lett. 2015, 18, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Meterology, B.O. Climate Statistics for Australian Locations, 14 March 2019 ed.; Australian Government: Canberra, Australia, 2019.
- Rochester, I.J. Nutrient uptake and export from an Australian cotton field. Nutr. Cycl. Agroecosyst. 2007, 77, 213–223. [Google Scholar] [CrossRef]
- Staff, S.S. Keys to Soil Taxonomy; Department of Agriculture, Natural Resources Conservation Service: Washington, DC, USA, 2003.
- Hulugalle, N.; Weaver, T.; Finlay, L.; Luelf, N.; Tan, D. Potential contribution by cotton roots to soil carbon stocks in irrigated Vertosols. Soil Res. 2009, 47, 243–252. [Google Scholar] [CrossRef]
- Ceeney, S.; Williams, S.; Maas, S. Integrated Pest Management & Resistance Management; Cotton Research and Development Corporation: Queensland, Australia, 2016; pp. 54–62. [Google Scholar]
- Dorahy, C.G.; Rochester, I.J.; Blair, G.J. Response of field-grown cotton (Gossypium hirsutum L.) to phosphorus fertilisation on alkaline soils in eastern Australia. Soil Res. 2004, 42, 913–920. [Google Scholar] [CrossRef]
- CSIRO. CottASSIST. Available online: https://www.cottassist.com.au/ (accessed on 9 July 2019).
- Polain, K.; Joice, G.; Jones, D.; Pereg, L.; Nachimuthu, G.; Knox, O.G. Coring lubricants can increase soil microbial activity in Vertisols. J. Microbiol. Methods 2019. [Google Scholar] [CrossRef]
- AGRF. Australian Genomic Research Facility: Next Generation Sequencing Resources. Available online: http://www.agrf.org.au/resources/applications/-next-gen-sequencing#Diversity (accessed on 28 February 2017).
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef]
- Kõljalg, U.; Larsson, K.H.; Abarenkov, K.; Nilsson, R.H.; Alexander, I.J.; Eberhardt, U.; Erland, S.; Høiland, K.; Kjøller, R.; Larsson, E. UNITE: A database providing web-based methods for the molecular identification of ectomycorrhizal fungi. New Phytol. 2005, 166, 1063–1068. [Google Scholar] [CrossRef]
- Chao, A.; Chiu, C.H. Species richness: Estimation and comparison. Wiley StatsRef Stat. Ref. Online 2014. [Google Scholar] [CrossRef]
- Gotelli, N.J.; Chai, A. Measuring and Estimating Species Richness, Species Diversity, and Biotic Similarity from Sampling Data. In Encyclopedia of Biodiversity, 2nd ed.; Levin, S.A., Ed.; Academic Press: Waltham, MA, USA, 2013; Volume 5, pp. 195–211. [Google Scholar]
- Maurer, B.A.; McGill, B.J. Measurement of species diversity. In Biological Diversity: Frontiers in Measurement and Assessment; Maurer, B.A., Ed.; Oxfold University Press: Oxfold, UK, 2011; pp. 55–56. [Google Scholar]
- Kim, B.-R.; Shin, J.; Guevarra, R.B.; Lee, J.H.; Kim, D.W.; Seol, K.-H.; Lee, J.-H.; Kim, H.B.; Isaacson, R.E. Deciphering diversity indices for better understanding of the microbial communities. J. Microbiol. Biotechnol. 2017, 27, 2089–2093. [Google Scholar] [CrossRef]
- Tuomisto, H. An updated consumer’s guide to evenness and related indices. Oikos 2012, 121, 1203–1218. [Google Scholar] [CrossRef]
- Chao, A. Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Simpson, E.H. Measurement of diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Magurran, A.E. Biological diversity. Curr. Biol. 2005, 15, R116–R118. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.J.; Gorley, R.N.; Clarke, K.R. PERMANOVA+ for PRIMER: Guide to Software and Statistical Methods, 1st ed.; PRIMER-E: Plymouth, UK, 2008. [Google Scholar]
- Benizri, E.; Amiaud, B. Relationship between plants and soil microbial communities in fertilized grasslands. Soil Biol. Biochem. 2005, 37, 2055–2064. [Google Scholar] [CrossRef]
- Hugerth, L.W.; Andersson, A.F. Analysing microbial community composition through amplicon sequencing: From sampling to hypothesis testing. Front. Microbiol. 2017, 8, 1561. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, K.; Stevens, S.; Shrestha, P.; Adetutu, E.M.; Walsh, K.B.; Ball, A.S.; Midmore, D.J. Characterisation of the soil microbial community of cultivated and uncultivated vertisol in Australia under several management regimes. Agric. Ecosyst. Environ. 2015, 199, 418–427. [Google Scholar] [CrossRef]
- Song, X.; Tao, B.; Guo, J.; Li, J.; Chen, G. Changes in the microbial community structure and soil chemical properties of vertisols under different cropping systems in Northern China. Front. Environ. Sci. 2018, 6, 132. [Google Scholar] [CrossRef]
- Jiao, S.; Chen, W.; Wang, J.; Du, N.; Li, Q.; Wei, G. Soil microbiomes with distinct assemblies through vertical soil profiles drive the cycling of multiple nutrients in reforested ecosystems. Microbiome 2018, 6, 1–13. [Google Scholar] [CrossRef]
- Fierer, N.; Schimel, J.P.; Holden, P.A. Variations in microbial community composition through two soil depth profiles. Soil Biol. Biochem. 2003, 35, 167–176. [Google Scholar] [CrossRef]
- Hartmann, M.; Lee, S.; Hallam, S.J.; Mohn, W.W. Bacterial, archaeal and eukaryal community structures throughout soil horizons of harvested and naturally disturbed forest stands. Environ. Microbiol. 2009, 11, 3045–3062. [Google Scholar] [CrossRef]
- Gordon, H.; Haygarth, P.M.; Bardgett, R.D. Drying and rewetting effects on soil microbial community composition and nutrient leaching. Soil Biol. Biochem. 2008, 40, 302–311. [Google Scholar] [CrossRef]
- Goldfarb, K.C.; Karaoz, U.; Hanson, C.A.; Santee, C.A.; Bradford, M.A.; Treseder, K.K.; Wallenstein, M.D.; Brodie, E.L. Differential growth responses of soil bacterial taxa to carbon substrates of varying chemical recalcitrance. Front. Microbiol. 2011, 2, 94. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N.; Lauber, C.L.; Ramirez, K.S.; Zaneveld, J.; Bradford, M.A.; Knight, R. Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. ISME J. 2012, 6, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Finn, D.; Kopittke, P.M.; Dennis, P.G.; Dalal, R.C. Microbial energy and matter transformation in agricultural soils. Soil Biol. Biochem. 2017, 111, 176–192. [Google Scholar] [CrossRef]
- Nelson, P.; Dictor, M.; Soulas, G. Availability of organic carbon in soluble and particle-size fractions from a soil profile. Soil Biol. Biochem. 1994, 26, 1549–1555. [Google Scholar] [CrossRef]
- Lundquist, E.J.; Jackson, L.; Scow, K. Wet–dry cycles affect dissolved organic carbon in two California agricultural soils. Soil Biol. Biochem. 1999, 31, 1031–1038. [Google Scholar] [CrossRef]
- McCarthy, A.J.; Williams, S.T. Actinomycetes as agents of biodegradation in the environment—A review. Gene 1992, 115, 189–192. [Google Scholar] [CrossRef]
- Li, X.; Sun, J.; Wang, H.; Li, X.; Wang, J.; Zhang, H. Changes in the soil microbial phospholipid fatty acid profile with depth in three soil types of paddy fields in China. Geoderma 2017, 290, 69–74. [Google Scholar] [CrossRef]
- Michéli, E.; Schad, P.; Spaargaren, O. World Reference Base for Soil Resources 2006: A Framework for International Classification, Correlation and Communication; Food and agriculture organization of the United nations (FAO): Rome, Italy, 2006. [Google Scholar]
- Strickland, M.S.; Rousk, J. Considering fungal: Bacterial dominance in soils–methods, controls, and ecosystem implications. Soil Biol. Biochem. 2010, 42, 1385–1395. [Google Scholar] [CrossRef]
- Taylor, D.L.; Sinsabaugh, R.L. The soil Fungi: Occurrence, phylogeny, and ecology. Soil Microbiol. Ecol. Biochem. 2015, 4, 77–109. [Google Scholar]
- Rasse, D.P.; Smucker, A.J. Root recolonization of previous root channels in corn and alfalfa rotations. Plant Soil 1998, 204, 203–212. [Google Scholar] [CrossRef]
- Hulugalle, N.R.; Broughton, K.J.; Tan, D.K.Y. Fine root production and mortality in irrigated cotton, maize and sorghum sown in vertisols of northern New South Wales, Australia. Soil Tillage Res. 2015, 146 Pt B, 313–322. [Google Scholar] [CrossRef]
- Hulugalle, N.R.; Weaver, T.B.; Finlay, L.A. Carbon inputs by irrigated corn roots to a Vertisol. Plant Root 2010, 4, 18–21. [Google Scholar] [CrossRef]
- Boer, W.d.; Folman, L.B.; Summerbell, R.C.; Boddy, L. Living in a fungal world: Impact of fungi on soil bacterial niche development. FEMS Microbiol. Rev. 2005, 29, 795–811. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.A.E.; Coleman, W.; Dube, M.; Rodrigues, A.E.; Pinto, P.C.R. Assessment of key features of lignin from lignocellulosic crops: Stalks and roots of corn, cotton, sugarcane, and tobacco. Ind. Crops Prod. 2016, 92, 136–148. [Google Scholar] [CrossRef]
- Houlden, A.; Timms-Wilson, T.M.; Day, M.J.; Bailey, M.J. Influence of plant developmental stage on microbial community structure and activity in the rhizosphere of three field crops. FEMS Microbiol. Ecol. 2008, 65, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Q.; Wang, F.; Zhang, J.; Chen, Y.; Zhang, C.; Liu, G.; Zhang, H.; Ma, C.; Zhang, J. The variation in the rhizosphere microbiome of cotton with soil type, genotype and developmental stage. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Ai, C.; Zhang, S.; Zhang, X.; Guo, D.; Zhou, W.; Huang, S. Distinct responses of soil bacterial and fungal communities to changes in fertilization regime and crop rotation. Geoderma 2018, 319, 156–166. [Google Scholar] [CrossRef]
- Hilton, S.; Bennett, A.J.; Chandler, D.; Mills, P.; Bending, G.D. Preceding crop and seasonal effects influence fungal, bacterial and nematode diversity in wheat and oilseed rape rhizosphere and soil. Appl. Soil Ecol. 2018, 126, 34–46. [Google Scholar] [CrossRef]
- Kuzyakov, Y.; Blagodatskaya, E. Microbial hotspots and hot moments in soil: Concept & review. Soil Biol. Biochem. 2015, 83, 184–199. [Google Scholar]
- Sikorski, J. The prokaryotic biology of soil. Soil Org. 2015, 87, 1–28. [Google Scholar]
- Chen, X.; Henriksen, T.M.; Svensson, K.; Korsaeth, A. Long-term effects of agricultural production systems on structure and function of the soil microbial community. Appl. Soil Ecol. 2019. [Google Scholar] [CrossRef]
- Fierer, N. Embracing the unknown: Disentangling the complexities of the soil microbiome. Nat. Rev. Microbiol. 2017, 15, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Baquerizo, M.; Maestre, F.T.; Reich, P.B.; Jeffries, T.C.; Gaitan, J.J.; Encinar, D.; Berdugo, M.; Campbell, C.D.; Singh, B.K. Microbial diversity drives multifunctionality in terrestrial ecosystems. Nat. Commun. 2016, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Polain, K.; Knox, O.; Wilson, B.; Guppy, C.; Lisle, L.; Nachimuthu, G.; Osanai, Y.; Siebers, N. Distribution of subsoil microbial activity and biomass under Australian rotational cotton as influenced by system, crop status and season. Soil Res. 2020. accepted publication. [Google Scholar]



| Target | Primer | Sequence |
|---|---|---|
| 16S | 341F | CCTAYGGGRBGCASCAG |
| 806R | GGACTACNNGGGTATCTAAT | |
| ITS | 1F | CTTGGTCATTTAGAGGAAGTAA |
| 2R | GCTGCGTTCTTCATCGATGC |
| Factors | Chao1 (SChao1) | Gini–Simpson Diversity Index (DGS) | Gini–Simpson Evenness Index (EGS) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 0–0.15 m | 0.30–0.50 m | 0.0–1.0 m | 0–0.15 m | 0.30–0.50 m | 0.0–1.0 m | 0–0.15 m | 0.30–0.50 m | 0.0–1.0 m | ||
| Depth | 411 a | 238 b | 177 b | 0.895 | 0.925 | 0.889 | 5 × 10−3 | 5 × 10−3 | 6 × 10−3 | |
| (p < 0.001) | (L.S.D = 56) | (p = 0.637) | (L.S.D = 0.09) | (p = 0.715) | (L.S.D = 2 × 10−3) | |||||
| System | ||||||||||
| CC | 414 | 259 | 181 | 0.916 | 0.932 | 0.904 | 5 × 10−3 | 6 × 10−3 | 6 × 10−3 | |
| CM | 409 | 217 | 174 | 0.874 | 0.919 | 0.874 | 6 × 10−3 | 5 × 10−3 | 6 × 10−3 | |
| (p = 0.738) | (L.S.D = 80) | (p = 0.938) | (L.S.D = 0.13) | (p = 0.556) | (L.S.D = 3 × 10−3) | |||||
| Time | ||||||||||
| S1 Pre | 499 | 290 | 189 | 0.818 | 0.916 | 0.861 | 3 × 10−3 | 6 × 10−3 | 5 × 10−3 | |
| S1 In | 430 | 179 | 143 | 0.922 | 0.912 | 0.865 | 6 × 10−3 | 7 × 10−3 | 8 × 10−3 | |
| S2 Pre | 310 | 230 | 176 | 0.908 | 0.932 | 0.901 | 8 × 10−3 | 5 × 10−3 | 6 × 10−3 | |
| S2 In | 406 | 253 | 201 | 0.931 | 0.942 | 0.929 | 5 × 10−3 | 4 × 10−3 | 4 × 10−3 | |
| (p = 0.054) | (L.S.D = 92) | (p = 0.791) | (L.S.D = 0.16) | (p = 0.281) | (L.S.D = 4 × 10−3) | |||||
| Time x System | ||||||||||
| S1 Pre | CC | 584 | 331 | 166 | 0.970 | 0.934 | 0.937 | 3 × 10−3 | 8 × 10−3 | 5 × 10−3 |
| CM | 414 | 249 | 212 | 0.665 | 0.897 | 0.785 | 2 × 10−3 | 4 × 10−3 | 6 × 10−3 | |
| S1 In | CC | 395 | 177 | 154 | 0.896 | 0.920 | 0.869 | 6 × 10−3 | 7 × 10−3 | 7 × 10−3 |
| CM | 465 | 180 | 131 | 0.948 | 0.904 | 0.861 | 6 × 10−3 | 6 × 10−3 | 9 × 10−3 | |
| S2 Pre | CC | 333 | 246 | 167 | 0.890 | 0.934 | 0.866 | 7 × 10−3 | 5 × 10−3 | 7 × 10−3 |
| CM | 288 | 215 | 185 | 0.926 | 0.930 | 0.936 | 1 × 10−2 | 5 × 10−3 | 5 × 10−3 | |
| S2 In | CC | 343 | 281 | 236 | 0.906 | 0.939 | 0.944 | 4 × 10−3 | 4 × 10−3 | 4 × 10−3 |
| CM | 468 | 225 | 167 | 0.957 | 0.945 | 0.915 | 6 × 10−3 | 4 × 10−3 | 5 × 10−3 | |
| (p = 0.057) | (L.S.D = 130) | (p = 0.486) | (L.S.D =0.23) | (p = 0.744) | (L.S.D = 5 × 10−3) | |||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polain, K.; Knox, O.; Wilson, B.; Pereg, L. Subsoil Microbial Diversity and Stability in Rotational Cotton Systems. Soil Syst. 2020, 4, 44. https://doi.org/10.3390/soilsystems4030044
Polain K, Knox O, Wilson B, Pereg L. Subsoil Microbial Diversity and Stability in Rotational Cotton Systems. Soil Systems. 2020; 4(3):44. https://doi.org/10.3390/soilsystems4030044
Chicago/Turabian StylePolain, Katherine, Oliver Knox, Brian Wilson, and Lily Pereg. 2020. "Subsoil Microbial Diversity and Stability in Rotational Cotton Systems" Soil Systems 4, no. 3: 44. https://doi.org/10.3390/soilsystems4030044
APA StylePolain, K., Knox, O., Wilson, B., & Pereg, L. (2020). Subsoil Microbial Diversity and Stability in Rotational Cotton Systems. Soil Systems, 4(3), 44. https://doi.org/10.3390/soilsystems4030044