A Novel Approach for High-Frequency in-situ Quantification of Methane Oxidation in Peatlands

 , , and
, , and 
Abstract
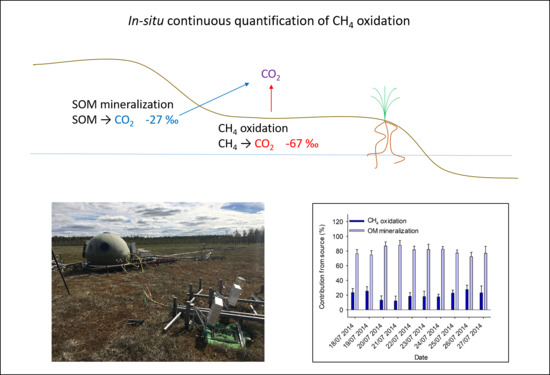
1. Introduction
2. Materials and Methods
2.1. Site Description
2.2. Experimental Setup
2.3. Measurements of Mass and Isotopes of CH4 and CO2
2.4. Isotopic Signature of Soil Organic Matter and Pore Water CH4
2.5. Incubation Experiment to Determine the Fractionation Factor for CH4 Oxidation
2.6. Flux Calculation and Estimation of Flux Isotopic Signature
2.7. Calculation of Fractionation Factor for CH4 Oxidation
2.8. Mixing Model
2.9. Data Presentation and Statistics
3. Results and Discussion
3.1. Isotopic Signatures of CO2 Fluxes
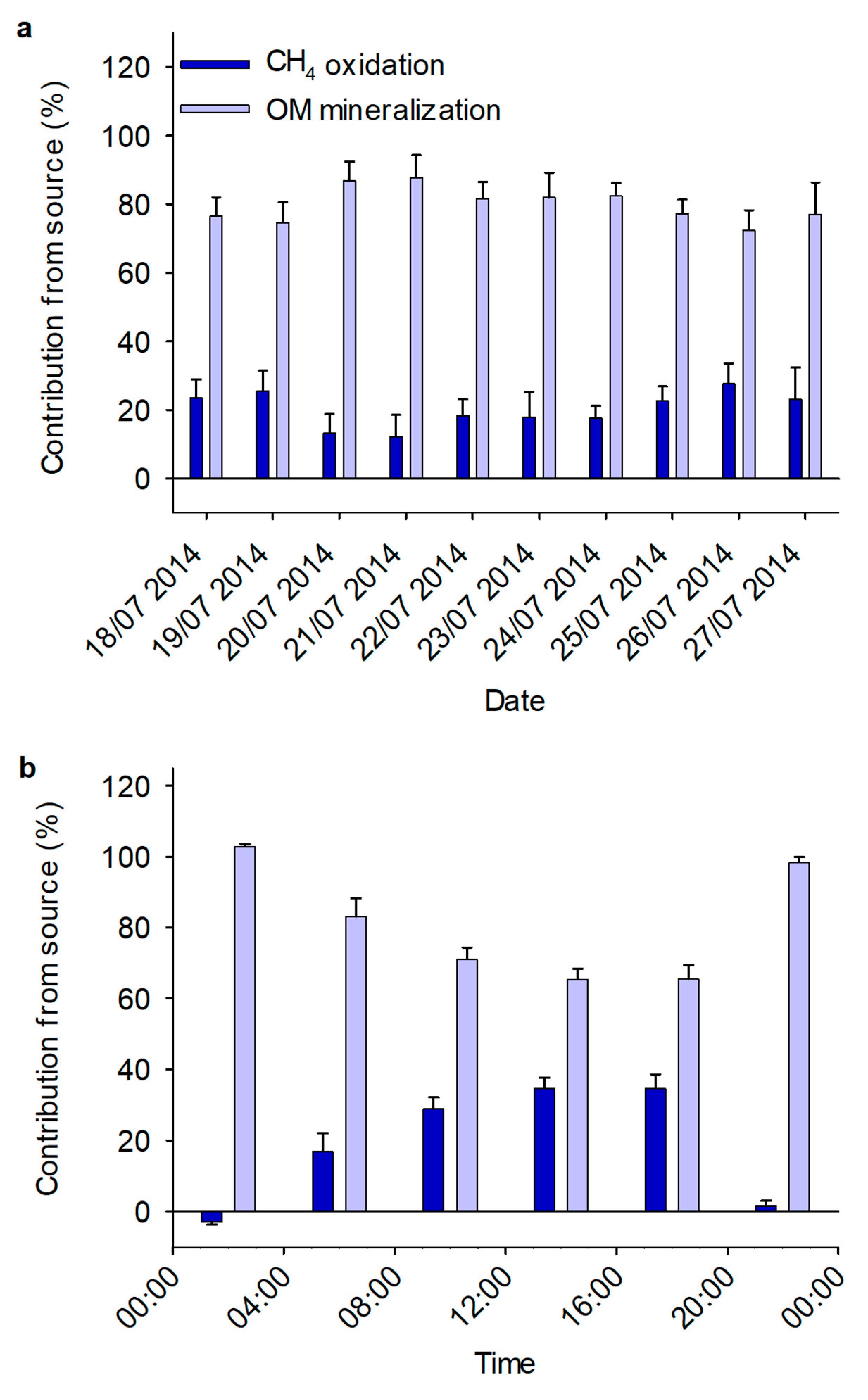
3.2. Mixing Model
3.3. Diurnal Variation
3.4. Methodological Limitations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Loisel, J.; Yu, Z.C.; Beilman, D.W.; Camill, P.; Alm, J.; Amesbury, M.J.; Anderson, D.; Andersson, S.; Bochicchio, C.; Barber, K.; et al. A database and synthesis of northern peatland soil properties and Holocene carbon and nitrogen accumulation. Holocene 2014, 24, 1028–1042. [Google Scholar] [CrossRef]
- Christensen, T.R.; Ekberg, A.; Ström, L.; Mastepanov, M.; Panikov, N.; Öquist, M.; Svensson, B.H.; Nykanen, H.; Martikainen, P.J.; Oskarsson, H. Factors controlling large scale variations in methane emissions from wetlands. Geophys. Res. Lett. 2003, 30. [Google Scholar] [CrossRef]
- McCalley, C.K.; Woodcroft, B.J.; Hodgkins, S.B.; Wehr, R.A.; Kim, E.H.; Mondav, R.; Crill, P.M.; Chanton, J.P.; Rich, V.I.; Tyson, G.W.; et al. Methane dynamics regulated by microbial community response to permafrost thaw. Nature 2014, 514, 478–481. [Google Scholar] [CrossRef] [PubMed]
- Granberg, G.; Mikkela, C.; Sundh, I.; Svensson, B.H.; Nilsson, M. Sources of spatial variation in methane emission from mires in northern Sweden: A mechanistic approach in statistical modeling. Glob. Biogeochem. Cycle 1997, 11, 135–150. [Google Scholar] [CrossRef]
- Wu, Y.Q.; Verseghy, D.L.; Melton, J.R. Integrating peatlands into the coupled Canadian Land Surface Scheme (CLASS) v3.6 and the Canadian Terrestrial Ecosystem Model (CTEM) v2.0. Geosci. Model Dev. 2016, 9, 2639–2663. [Google Scholar] [CrossRef]
- Wu, J.H.; Roulet, N.T.; Nilsson, M.; Lafleur, P.; Humphreys, E. Simulating the Carbon Cycling of Northern Peat lands Using a Land Surface Scheme Coupled to a Wetland Carbon Model (CLASS3W-MWM). Atmos.-Ocean 2012, 50, 487–506. [Google Scholar] [CrossRef]
- Abdalla, M.; Hastings, A.; Bell, M.J.; Smith, J.U.; Richards, M.; Nilsson, M.B.; Peichl, M.; Lofvenius, M.O.; Lund, M.; Helfter, C.; et al. Simulation of CO2 and Attribution Analysis at Six European Peatland Sites Using the ECOSSE Model. Water Air Soil Poll. 2014, 225, 14. [Google Scholar] [CrossRef]
- Qiu, C.; Zhu, D.; Ciais, P.; Guenet, B.; Krinner, G.; Peng, S.; Aurela, M.; Bernhofer, C.; Brümmer, C.; Bret-Harte, S.; et al. ORCHIDEE-PEAT (revision 4596), a model for northern peatland CO2, water and energy fluxes on daily to annual scales. Geosci. Mod. Dev. Discuss. 2017. [Google Scholar] [CrossRef]
- Metzger, C.; Nilsson, M.B.; Peichl, M.; Jansson, P.E. Parameter interactions and sensitivity analysis for modelling carbon heat and water fluxes in a natural peatland, using CoupModel v5. Geosci. Model Dev. 2016, 9, 4313–4338. [Google Scholar] [CrossRef]
- Gerard, G.; Chanton, J. Quantification of methane oxidation in the rhizosphere of emergent aquatic macrophytes: Defining upper limits. Biogeochemistry 1993, 23, 79–97. [Google Scholar] [CrossRef]
- Kankaala, P.; Bergström, I. Emission and oxidation of methane in Equisetum fluviatile stands growing on organic sediment and sand bottoms. Biogeochemistry 2004, 67, 21–37. [Google Scholar] [CrossRef]
- Sundh, I.; Mikkela, C.; Nilsson, M.; Svensson, B.H. Potential Aerobic Methane Oxidation in a Sphagnum-Dominated Peatland—Controlling Factors and Relation to Methane Emission. Soil Biol. Biochem. 1995, 27, 829–837. [Google Scholar] [CrossRef]
- Ding, W.X.; Cai, Z.C.; Tsuruta, H. Factors affecting seasonal variation of methane concentration in water in a freshwater marsh vegetated with Carex lasiocarpa. Biol. Fert. Soils 2005, 41, 1–8. [Google Scholar] [CrossRef]
- Lombardi, J.E.; Epp, M.A.; Chanton, J.P. Investigation of the methyl fluoride technique for determining rhizospheric methane oxidation. Biogeochemistry 1997, 36, 153–172. [Google Scholar] [CrossRef]
- van der Nat, F.; Middelburg, J.J. Seasonal variation in methane oxidation by the rhizosphere of Phragmites australis and Scirpus lacustris. Aquat. Bot. 1998, 61, 95–110. [Google Scholar] [CrossRef]
- Popp, T.J.; Chanton, J.P.; Whiting, G.J.; Grant, N. Methane stable isotope distribution at a Carex dominated fen in north central Alberta. Glob. Biogeochem. Cycle 1999, 13, 1063–1077. [Google Scholar] [CrossRef]
- Groot, T.T.; Bodegom, P.M.V.; Harren, F.J.M.; Meijer, H.A.J. Quantification of methane oxidation in the rice rhizosphere using 13C-labelled methane. Biogeochemistry 2003, 64, 355–372. [Google Scholar] [CrossRef]
- Riveros-Iregui, D.A.; King, J.Y. Isotopic Evidence of Methane Oxidation across the Surface Water-Ground Water Interface. Wetlands 2008, 28, 928–937. [Google Scholar] [CrossRef]
- Cadieux, S.B.; White, J.R.; Sauer, P.E.; Peng, Y.B.; Goldman, A.E.; Pratt, L.M. Large fractionations of C and H isotopes related to methane oxidation in Arctic lakes. Geochim. Cosmochim. Acta 2016, 187, 141–155. [Google Scholar] [CrossRef]
- Fechner, E.J.; Hemond, H.F. Methane transport and oxidation in the unsaturated zone of a Sphagnum peatland. Glob. Biogeochem. Cycles 1992, 6, 33–44. [Google Scholar] [CrossRef]
- Urmann, K.; Gonzalez-Gil, G.; Schroth, M.H.; Zeyer, J. Quantification of Microbial Methane Oxidation in an Alpine Peat Bog. Vadose Zone J. 2007, 6, 705–712. [Google Scholar] [CrossRef]
- King, G.M. In situ analyses of methane oxidation associated with the roots and rhizomes of a bur reed, Sparganium eurycarpum, in a Maine wetland. Appl. Environ. Microbiol. 1996, 62, 4548–4555. [Google Scholar] [PubMed]
- Popp, T.J.; Chanton, J.P.; Whiting, G.J.; Grant, N. Evaluation of methane oxidation in the rhizosphere of a Carex dominated fen in north central Alberta, Canada. Biogeochemistry 2000, 51, 259–281. [Google Scholar] [CrossRef]
- Preuss, I.; Knoblauch, C.; Gebert, J.; Pfeiffer, E.M. Improved quantification of microbial CH4 oxidation efficiency in arctic wetland soils using carbon isotope fractionation. Biogeosciences 2013, 10, 2539–2552. [Google Scholar] [CrossRef]
- Alexandersson, H.; Karlström, C.; Larsson-Mccann, S. Temperature and Precipitation in Sweden during 1961–1990. Reference Normals; SMHI Meteorologi: Norrköping, Sweden, 1991; Volume 81. [Google Scholar]
- Peichl, M.; Öquist, M.; Löfvenius, M.O.; Ilstedt, U.; Sagerfors, J.; Grelle, A.; Lindroth, A.; Nilsson, M.B. A 12-year record reveals pre-growing season temperature and water table level threshold effects on the net carbon dioxide exchange in a boreal fen. Environ. Res. Lett. 2014, 9, 11. [Google Scholar] [CrossRef]
- Eriksson, T.; Öquist, M.G.; Nilsson, M.B. Effects of decadal deposition of nitrogen and sulfur, and increased temperature, on methane emissions from a boreal peatland. J. Geophys. Res.-Biogeosci. 2010, 115, 13. [Google Scholar] [CrossRef]
- Nilsson, M.; Sagerfors, J.; Buffam, I.; Laudon, H.; Eriksson, T.; Grelle, A.; Klemedtsson, L.; Weslien, P.; Lindroth, A. Contemporary carbon accumulation in a boreal oligotrophic minerogenic mire—A significant sink after accounting for all C-fluxes. Glob. Chang. Biol. 2008, 14, 2317–2332. [Google Scholar] [CrossRef]
- Laine, A.M.; Bubier, J.; Riutta, T.; Nilsson, M.B.; Moore, T.R.; Vasander, H.; Tuittila, E.-S. Abundance and composition of plant biomass as potential controls for mire net ecosytem CO2 exchange. Botany 2011, 90, 63–74. [Google Scholar] [CrossRef]
- Järveoja, J.; Nilsson, M.B.; Gažovič, M.; Crill, P.M.; Peichl, M. Partitioning of the net CO2 exchange using an automated chamber system reveals plant phenology as key control of production and respiration fluxes in a boreal peatland. Glob. Chang. Biol. 2018, 24, 3436–3451. [Google Scholar] [CrossRef]
- Eriksson, T.; Öquist, M.G.; Nilsson, M.B. Production and oxidation of methane in a boreal mire after a decade of increased temperature and nitrogen and sulfur deposition. Glob. Change Biol. 2010, 16, 2130–2144. [Google Scholar] [CrossRef]
- Keeling, C.D. The Concentration and Isotopic Abundances of Atmospheric Carbon Dioxide in Rural Areas. Geochim. Cosmochim. Acta 1958, 13, 322–334. [Google Scholar] [CrossRef]
- Happell, J.D.; Chanton, J.P.; Showers, W.S. The Influence of Methane Oxidation on the Stable Isotopic Composition of Methane Emitted from Florida Swamp Forests. Geochim. Cosmochim. Acta 1994, 58, 4377–4388. [Google Scholar] [CrossRef]
- Coleman, D.D.; Risatti, J.B.; Schoell, M. Fractionation of Carbon and Hydrogen Isotopes by Methane-Oxidizing Bacteria. Geochim. Cosmochim. Acta 1981, 45, 1033–1037. [Google Scholar] [CrossRef]
- Fry, B. Stable Isotope Ecology; Springer: New York, NY, USA, 2006; p. 316. [Google Scholar]
- Schipper, L.A.; Reddy, K.R. Determination of methane oxidation in the rhizosphere of Sagittaria lancifolia using methyl fluoride. Soil Sci. Soc. Am. J. 1996, 60, 611–616. [Google Scholar] [CrossRef]
- Kankaala, P.; Taipale, S.; Nykanen, H.; Jones, R.I. Oxidation, efflux, and isotopic fractionation of methane during autumnal turnover in a polyhumic, boreal lake. J. Geophys. Res.-Biogeosci. 2007, 112. [Google Scholar] [CrossRef]
- De Visscher, A.; De Pourcq, I.; Chanton, J. Isotope fractionation effects by diffusion and methane oxidation in landfill cover soils. J. Geophys. Res.-Atmos. 2004, 109, 8. [Google Scholar] [CrossRef]
- Reeburgh, W.S.; Hirsch, A.I.; Sansone, F.J.; Popp, B.N.; Rust, T.M. Carbon kinetic isotope effect accompanying microbial oxidation of methane in boreal forest soils. Geochim. Cosmochim. Acta 1997, 61, 4761–4767. [Google Scholar] [CrossRef]
- Mikkelä, C.; Sundh, I.; Svensson, B.H.; Nilsson, M. Diurnal variation in methane emission in relation to the water table, soil temperature, climate and vegetation cover in a Swedish acid mire. Biogeochemistry 1995, 28, 93–114. [Google Scholar] [CrossRef]
- Moore, T.R.; Dalva, M. The influence of temperature and water table position on carbon dioxide and methane emissions from laboratory columns of peatland soils. J. Soil Sci. 1993, 44, 651–664. [Google Scholar] [CrossRef]
- van Asperen, H.; Warneke, T.; Sabbatini, S.; Höpker, M.; Nicolini, G.; Chiti, T.; Papale, D.; Böhm, M.; Notholt, J. Diel variation in isotopic composition of soil respiratory CO2 fluxes: The role of non-steady state conditions. Agric. For. Meteorol. 2017, 234–235, 95–105. [Google Scholar] [CrossRef]
- Braendholt, A.; Larsen, K.S.; Ibrom, A.; Pilegaard, K. Overestimation of closed-chamber soil CO2 effluxes at low atmospheric turbulence. Biogeosciences 2017, 14, 1603–1616. [Google Scholar] [CrossRef]
- Lasslop, G.; Reichstein, M.; Papale, D.; Richardson, A.D.; Arneth, A.; Barr, A.; Stoy, P.; Wohlfahrt, G. Separation of net ecosystem exchange into assimilation and respiration using a light response curve approach: Critical issues and global evaluation. Glob. Chang. Biol. 2010, 16, 187–208. [Google Scholar] [CrossRef]
- Reichstein, M.; Falge, E.; Baldocchi, D.; Papale, D.; Aubinet, M.; Berbigier, P.; Bernhofer, C.; Buchmann, N.; Gilmanov, T.; Granier, A.; et al. On the separation of net ecosystem exchange into assimilation and ecosystem respiration: Review and improved algorithm. Glob. Chang. Biol. 2005, 11, 1424–1439. [Google Scholar] [CrossRef]
- Roslev, P.; King, G.M. Regulation of methane oxidation in a freshwater wetland by water table changes and anoxia. FEMS Microbiol. Ecol. 1996, 19, 105–115. [Google Scholar] [CrossRef]
- Lai, D.Y.F. Methane Dynamics in Northern Peatlands: A Review. Pedosphere 2009, 19, 409–421. [Google Scholar] [CrossRef]
- Redeker, K.R.; Baird, A.J.; Teh, Y.A. Quantifying wind and pressure effects on trace gas fluxes across the soil–atmosphere interface. Biogeosciences 2015, 12, 7423–7434. [Google Scholar] [CrossRef]
- Smemo, K.A.; Yavitt, J.B. Anaerobic oxidation of methane: An underappreciated aspect of methane cycling in peatland ecosystems? Biogeosciences 2011, 8, 779–793. [Google Scholar] [CrossRef]
- Smemo, K.A.; Yavitt, J.B. Evidence for Anaerobic CH4 Oxidation in Freshwater Peatlands. Geomicrobiol. J. 2007, 24, 583–597. [Google Scholar] [CrossRef]
- Corbett, J.E.; Tfaily, M.M.; Burdige, D.J.; Cooper, W.T.; Glaser, P.H.; Chanton, J.P. Partitioning pathways of CO2 production in peatlands with stable carbon isotopes. Biogeochemistry 2013, 114, 327–340. [Google Scholar] [CrossRef]
- Bond-Lamberty, B.; Bronson, D.; Bladyka, E.; Gower, S.T. A comparison of trenched plot techniques for partitioning soil respiration. Soil Biol. Biochem. 2011, 43, 2108–2114. [Google Scholar] [CrossRef]
- Larmola, T.; Tuittila, E.S.; Tiirola, M.; Nykanen, H.; Martikainen, P.J.; Yrjala, K.; Tuomivirta, T.; Fritze, H. The role of Sphagnum mosses in the methane cycling of a boreal mire. Ecology 2010, 91, 2356–2365. [Google Scholar] [CrossRef] [PubMed]
- Teh, Y.A.; Silver, W.L.; Conrad, M.E.; Borglin, S.E.; Carlson, C.M. Carbon isotope fractionation by methane-oxidizing bacteria in tropical rain forest soils. J. Geophys. Res.-Biogeosci. 2006, 111. [Google Scholar] [CrossRef]
- Templeton, A.S.; Chu, K.-H.; Alvarez-Cohen, L.; Conrad, M.E. Variable carbon isotope fractionation expressed by aerobic CH4-oxidizing bacteria. Geochim. Cosmochim. Acta 2006, 70, 1739–1752. [Google Scholar] [CrossRef]
- Dorodnikov, M.; Marushchak, M.; Biasi, C.; Wilmking, M. Effect of microtopography on isotopic composition of methane in porewater and efflux at a boreal peatland. Boreal Environ. Res. 2013, 18, 269–279. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
| Term | Abbreviation |
|---|---|
| Concentration of headspace CH4 at time 0 in the incubation experiment for the fractionation factor | [CH4]0 |
| Concentration of headspace CH4 at time t in the incubation experiment for the fractionation factor | [CH4]t |
| Ecosystem respiration | ER |
| Fractional contribution of CH4 oxidation to heterotrophic respiration | fCH4 |
| Kinetic fractionation factor | α |
| Heterotrophic respiration | Rh |
| Isotopic 13C signature | δ13C |
| Isotopic 13C signature of ecosystem respiration | δ13CER |
| Isotopic 13C signature of headspace CH4 at times 0 in the incubation experiment for the fractionation factor | δ13CCH4,0 |
| Isotopic 13C signature of headspace CH4 at times t in the incubation experiment for the fractionation factor | δ13CCH4,t |
| Isotopic 13C signature of heterotrophic respiration | δ13CRh |
| Isotopic 13C signature of pore water CH4 | δ13CCH4,pw |
| Isotopic 13C signature of organic matter | δ13COM |
| Net ecosystem exchange | NEE |
| Organic matter | OM |
| Permil fractionation factor | Δ |
| Relative CH4 oxidation % | %CH4oxi |
| Soil organic matter | SOM |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nielsen, C.S.; Hasselquist, N.J.; Nilsson, M.B.; Öquist, M.; Järveoja, J.; Peichl, M. A Novel Approach for High-Frequency in-situ Quantification of Methane Oxidation in Peatlands. Soil Syst. 2019, 3, 4. https://doi.org/10.3390/soilsystems3010004
Nielsen CS, Hasselquist NJ, Nilsson MB, Öquist M, Järveoja J, Peichl M. A Novel Approach for High-Frequency in-situ Quantification of Methane Oxidation in Peatlands. Soil Systems. 2019; 3(1):4. https://doi.org/10.3390/soilsystems3010004
Chicago/Turabian StyleNielsen, Cecilie Skov, Niles J. Hasselquist, Mats B. Nilsson, Mats Öquist, Järvi Järveoja, and Matthias Peichl. 2019. "A Novel Approach for High-Frequency in-situ Quantification of Methane Oxidation in Peatlands" Soil Systems 3, no. 1: 4. https://doi.org/10.3390/soilsystems3010004
APA StyleNielsen, C. S., Hasselquist, N. J., Nilsson, M. B., Öquist, M., Järveoja, J., & Peichl, M. (2019). A Novel Approach for High-Frequency in-situ Quantification of Methane Oxidation in Peatlands. Soil Systems, 3(1), 4. https://doi.org/10.3390/soilsystems3010004