Effects of Ionic Strength on Arsenate Adsorption at Aluminum Hydroxide–Water Interfaces


Abstract
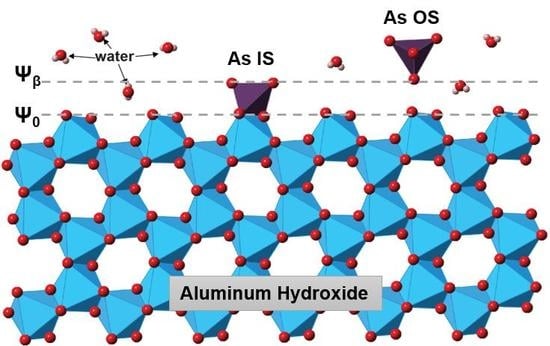
:
1. Introduction
2. Materials and Methods
2.1. Mineral and Reagent Preparation
2.2. Macroscopic Arsenate Adsorption Isotherms
2.3. ζ-Potential Analysis
2.4. EXAFS Spectroscopic Measurements
3. Results
3.1. Properties of Synthesized Materials
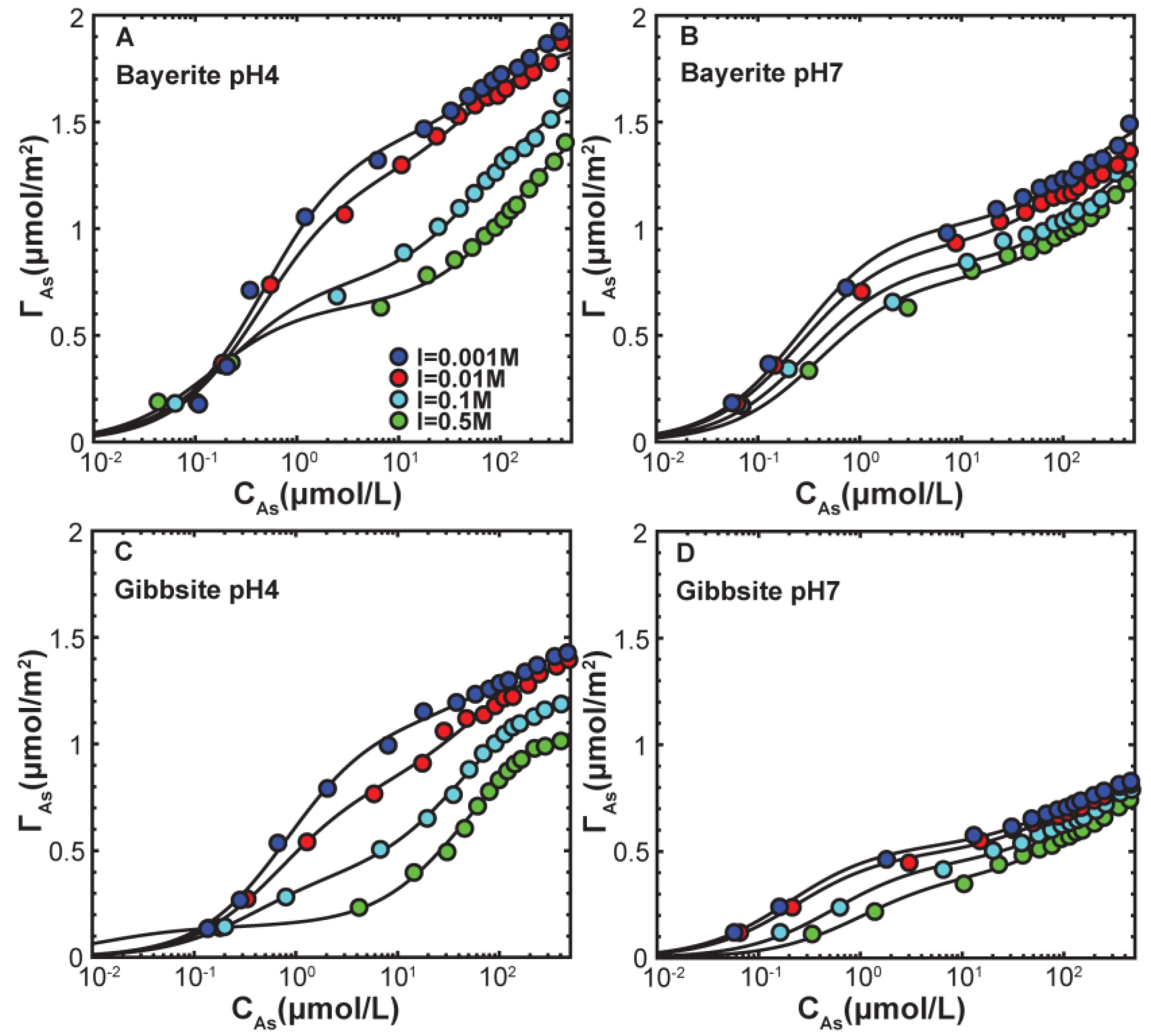
3.2. Macroscopic Arsenate Adsorption
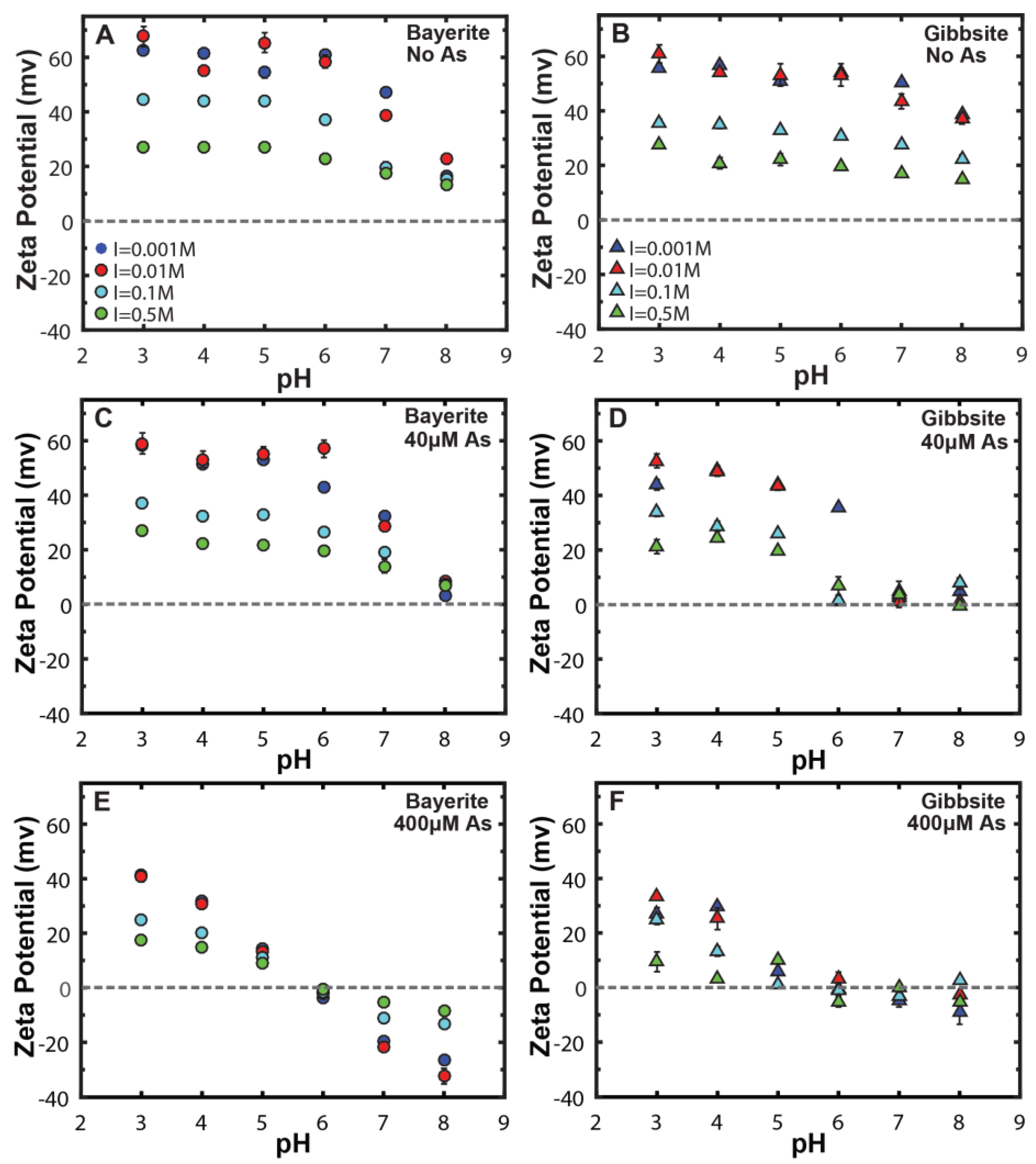
3.3. Ionic Strength Effect on Surface Charge Properties
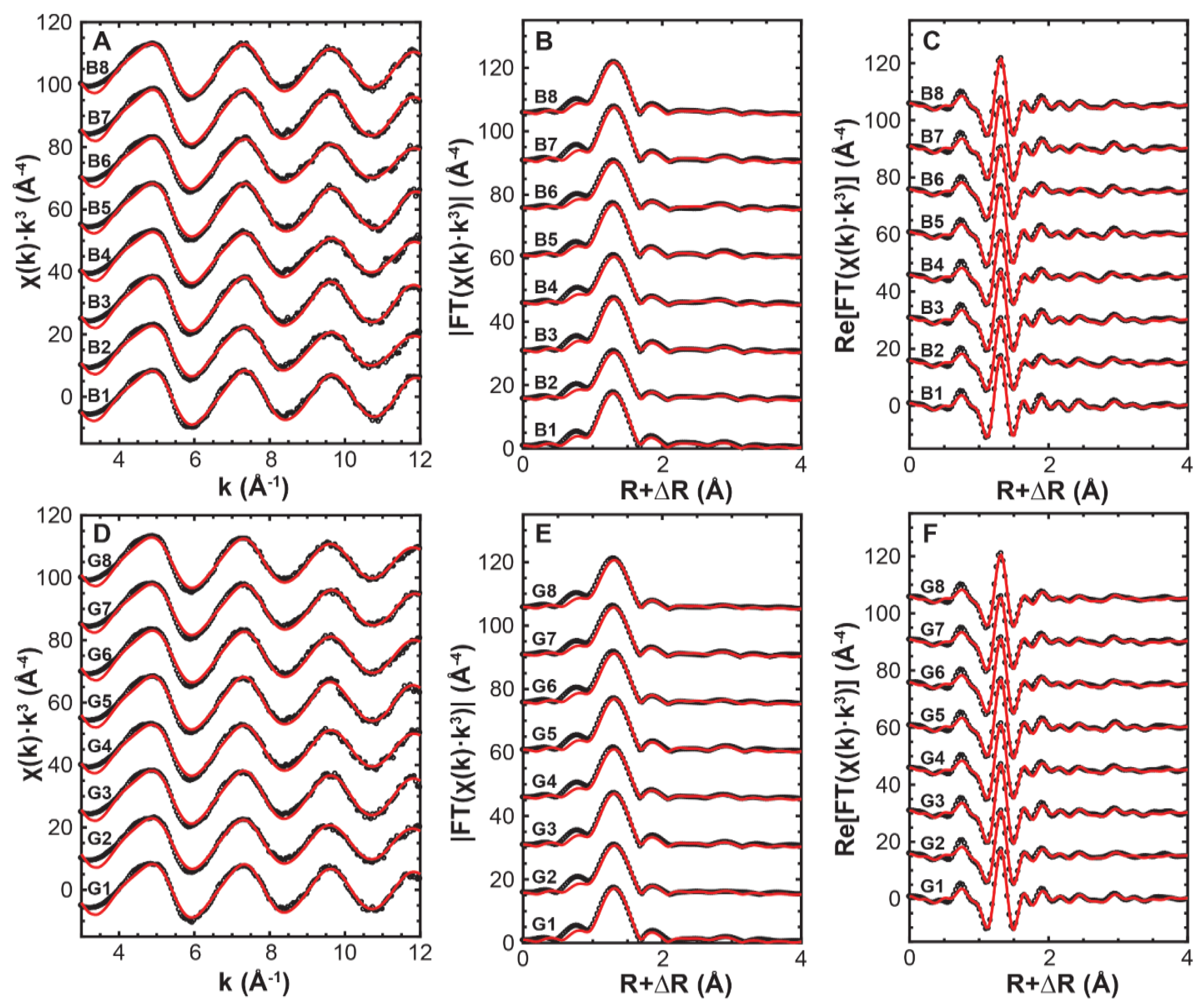
3.4. Arsenate Adsorption Mechanisms
4. Discussion
4.1. Implications to Arsenate Adsorption Mechanisms
4.2. Implications to Arsenate in the Environment
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Foster, A.L.; Brown, G.E.; Tingle, T.N.; Park, G.A. Quantitative arsenic speciation in mine tailings using X-ray absorption spectroscopy. Am. Miner. 1998, 83, 553–568. [Google Scholar] [CrossRef]
- Jain, C.K.; Ali, I. Arsenic: Occurrence, toxicity and speciation techniques. Water Res. 2000, 34, 4304–4312. [Google Scholar] [CrossRef]
- Mandal, B.K.; Suzuku, K.T. Arsenic around the world: A review. Talanta 2003, 58, 201–235. [Google Scholar] [CrossRef]
- Arai, Y.; Lanzirotti, A.; Sutton, S.; Davis, J.A.; Sparks, D.L. Arsenic speciation and reactivity in poultry litter. Environ. Sci. Technol. 2003, 37, 4083–4090. [Google Scholar] [CrossRef] [PubMed]
- Smedley, P.L.; Kinniburgh, D.G. A review of the source, behavior and distribution of arsenic in natural waters. Appl. Geochem. 2002, 17, 517–568. [Google Scholar] [CrossRef]
- Chwirka, J.D.; Thomson, B.M.; Stomp, J.M. Removing arsenic from groundwater. J. Am. Water Works Assoc. 2000, 92, 79–88. [Google Scholar]
- Garelick, H.; Dybowska, A.; Valsami-Jones, E.; Priest, N.D. Remediation technologies for arsenic contaminated drinking waters. J. Soils Sediments 2005, 5, 182–190. [Google Scholar] [CrossRef]
- Arai, Y.; Elzinga, E.J.; Sparks, D.L. X-ray absorption spectroscopic investigation of arsenite and arsenate adsorption at the aluminum oxide-water interface. J. Colloid Interface Sci. 2001, 235, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, S.; Johnston, C.T. Mechanisms of arsenic adsorption on amorphous oxides evaluated using macroscopic measurements, vibrational spectroscopy, and surface complexation modeling. J. Colloid Interface Sci. 2001, 234, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Antelo, J.; Avena, M.; Fiol, S.; Lopez, R.; Arce, F. Effects of pH and ionic strength on the adsorption of phosphate and arsenate at the goethite-water interface. J. Colloid Interface Sci. 2005, 285, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Hayes, K.F.; Papelis, C.; Leckie, J.O. Modeling ionic strength effects of anion adsorption at hydrous oxide/solution interface. J. Colloid Interface Sci. 1988, 125, 717–726. [Google Scholar] [CrossRef]
- Manceau, A. The mechanism of anion adsorption on iron oxides: Evidence for the bonding of arsenate tetrahedra on free Fe(O,OH)6 edges. Geochim. Cosmochim. Acta 1995, 59, 3647–3653. [Google Scholar] [CrossRef]
- Fendorf, S.; Eick, M.J.; Grossl, P.; Sparks, D.L. Arsenate and chromate retention mechanisms on goethite. 1. Surface structure. Environ. Sci. Technol. 1997, 31, 315–320. [Google Scholar] [CrossRef]
- Lumsdon, D.G.; Fraser, A.R.; Russell, J.D.; Livesey, N.T. New infrared band assignments for the arsenate ion adsorbed on synthetic goethite (α-FeOOH). J. Soil Sci. 1984, 35, 381–386. [Google Scholar] [CrossRef]
- Sun, X.; Doner, H.E. An investigation of arsenate and arsenite bonding structures on goethite by FTIR. Soil Sci. 1996, 161, 865–872. [Google Scholar] [CrossRef]
- Myneni, S.C.B.; Traina, S.J.; Waychunas, G.A.; Logan, T.J. Experimental and theoretical vibrational spectroscopic evaluation of arsenate coordination in aqueous solutions, solids, and at mineral-water interfaces. Geochim. Cosmochim. Acta 1998, 62, 3285–3300. [Google Scholar] [CrossRef]
- Waychunas, G.A.; Rea, B.A.; Fuller, C.C.; Davis, J.A. Surface chemistry of ferrihydrite: Part 1. EXAFS studies of the geometry of coprecipitated and adsorbed arsenate. Geochim. Cosmochim. Acta 1993, 57, 2251–2269. [Google Scholar] [CrossRef]
- Goldberg, S. Chemical modeling of arsenate adsorption on aluminum and iron oxide minerals. Soil Sci. Soc. Am. J. 1986, 50, 1154–1157. [Google Scholar] [CrossRef]
- Hiemstra, T.; Van Riemsdijk, W.H. Surface structural ion adsorption modeling of competitive binding of oxyanions by metal (hydr)oxides. J. Colloid Interface Sci. 1999, 210, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Dixit, S.; Hering, J.G. Comparison of arsenic(V) and arsenic(III) sorption onto iron oxide minerals: Implications for arsenic mobility. Environ. Sci. Technol. 2003, 37, 4182–4189. [Google Scholar] [CrossRef] [PubMed]
- Catalano, J.G.; Park, C.; Fenter, P.; Zhang, Z. Simultaneous inner- and outer-sphere arsenate complexation on corundum and hematite. Geochim. Cosmochim. Acta 2008, 72, 1986–2004. [Google Scholar] [CrossRef]
- Xu, T.; Catalano, J.G. Impacts of surface site coordination on arsenate adsorption: Macroscopic uptake and binding mechanisms on aluminum hydroxide surfaces. Langmuir 2016, 32, 13261–13269. [Google Scholar] [CrossRef] [PubMed]
- Hiemstra, T.; Yong, H.; van Riemsdijk, W.H. Interfacial charging phenomena of aluminum (hydr)oxides. Langmuir 1999, 15, 5942–5955. [Google Scholar]
- Shen, S.; Chow, P.S.; Chen, F.; Feng, S.; Tan, R.B.H. Synthesis of submicron gibbsite platelets by organic-free hydrothermal crystallization process. J. Cryst. Growth 2006, 292, 136–142. [Google Scholar] [CrossRef]
- Lefevre, G.; Fedoroff, M. Synthesis of bayerite [β-Al(OH)3] microrods by neutralization of alyminate ions at constant pH. Mater. Lett. 2002, 56, 978–983. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Newville, M. IFEFFIT: Interactive EXAFS analysis and FEFF fitting. J. Synchrotron Radiat. 2001, 8, 322–324. [Google Scholar] [CrossRef] [PubMed]
- Webb, S.M. SixPack: A graphical user interface for XAS analysis using IFEFFIT. Phys. Scr. 2005, 115, 1011–1014. [Google Scholar] [CrossRef]
- Harrison, W.T.A. Synthetic mansfieldite, AlAsO4·2H2O. Acta Cryst. 2000, 56, 421. [Google Scholar] [CrossRef]
- Ankudinov, A.L.; Rehr, J.J. Relativistic calculations of independent X-ray absorption spectra. Phys. Rev. B 1997, 56, 1712–1715. [Google Scholar] [CrossRef]
- Mikutta, C.; Frommer, J.; Voegelin, A.; Kaegi, R.; Kretzschmar, R. Effect of citrate on the local Fe coordination in ferrihydrite, arsenate binding, and ternary arsenate complex formation. Geochim. Cosmochim. Acta 2010, 74, 5574–5592. [Google Scholar] [CrossRef]
- Rosenqvist, J.; Persson, P.; Sjoberg, S. Protonation and charging of nanosized gibbsite [α-Al(OH)3] particles in aqueous suspension. Langmuir 2002, 18, 4598–4604. [Google Scholar] [CrossRef]
- Gan, Y.; Franks, G.V. Charging behavior of the gibbsite (001) surface in NaCl solution investigated by AFM colloidal probe technique. Langmuir 2006, 22, 6087−6092. [Google Scholar] [CrossRef] [PubMed]
- Adekola, F.; Fedoroff, M.; Geckeis, H.; Kupcik, T.; Lefevre, G.; Lutzenkirchen, J.; Plaschke, M.; Preocanin, T.; Rabung, T.; Schild, D. Characterization of acid-base properties of two gibbsite samples in the context of literature results. J. Colloid Interface Sci. 2011, 354, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.A.; Goldberg, S. Modeling Competitive Adsorption of arsenate with phosphate and molybdate on oxide minerals. Soil Sci. Soc. Am. J. 1996, 60, 121–131. [Google Scholar] [CrossRef]
- Ladeira, A.C.Q.; Ciminelli, V.S.T.; Duarte, H.A.; Alives, M.C.M.; Ramos, A.Y. Mechanism of anion retention from EXAFS and density functional calculations: Arsenic(V) adsorbed on gibbsite. Geochim. Cosmochim. Acta 2001, 65, 1211–1217. [Google Scholar] [CrossRef]
- Kappen, P.; Webb, J. An EXAFS study of arsenic bonding on amorphous aluminum hydroxide. Appl. Geochem. 2013, 31, 79–83. [Google Scholar] [CrossRef]
- Kelly, S.D.; Hesterberg, D.; Ravel, B. Analysis of soils and minerals using X-ray absorption spectroscopy. In Methods of Soil Analysis, Part 5—Mineralogical Methods; Ulery, A.L., Drees, L.R., Eds.; Soil Science Society of American: Madison, WI, USA, 2008; pp. 387–463. [Google Scholar]
- Fukushi, K.; Sverjensky, D.A. A predictive model (ETLM) for arsenate adsorption and surface speciation on oxides consistent with spectroscopic and theoretical molecular evidence. Geochim. Cosmochim. Acta 2007, 71, 3717–3745. [Google Scholar] [CrossRef]
- Lee, S.S.; Fenter, P.; Nagy, K.L.; Sturchio, N.C. Real-time observation of cation exchange kinetics and dynamics at the muscovite-water interface. Nat. Commun. 2017, 8, 15826. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, S.E.; Strawn, D.G.; Sparks, D.L. Residence time effects on arsenate adsorption/desorption mechanisms on goethite. Soil Sci. Soc. Am. J. 2001, 65, 67–77. [Google Scholar] [CrossRef]
- Quaghebeur, M.; Rate, A.; Rengel, Z.; Hinz, C. Desorption kinetics of arsenate from kaolinite as influenced by pH. J. Environ. Qual. 2005, 34, 479–486. [Google Scholar] [PubMed]
- Arai, Y.; Spark, D.L. Residence time effects on arsenate surface speciation at the aluminum oxide-water interface. Soil Sci. 2002, 167, 303–314. [Google Scholar] [CrossRef]
- Pigna, M.; Krishnamurti, G.S.R.; Violante, A. Kinetics of arsenate sorption–desorption from metal oxides: Effect of residence time. Soil Sci. Soc. Am. J. 2006, 70, 2017–2027. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Mineral | pH | Desired I (mol/L NaNO3) | [AsO43]init (μmol/L) | [AsO43]final (μmol/L) | ΓAs (μmol/m2) |
|---|---|---|---|---|---|---|
| G1 | 4 g/L gibbsite | 7.0 | 0.5 | 40 | 10 | 0.35 |
| G2 | 4 g/L gibbsite | 7.0 | 0.5 | 400 | 338 | 0.71 |
| G3 | 4 g/L gibbsite | 7.0 | 0.001 | 40 | 2 | 0.44 |
| G4 | 4 g/L gibbsite | 7.0 | 0.001 | 400 | 329 | 0.82 |
| G5 | 4 g/L gibbsite | 4.0 | 0.5 | 40 | 9 | 0.36 |
| G6 | 4 g/L gibbsite | 4.0 | 0.5 | 400 | 312 | 1.01 |
| G7 | 4 g/L gibbsite | 4.0 | 0.001 | 40 | 1 | 0.45 |
| G8 | 4 g/L gibbsite | 4.0 | 0.001 | 400 | 278 | 1.41 |
| B1 | 4 g/L bayerite | 7.0 | 0.5 | 40 | 3 | 0.63 |
| B2 | 4 g/L bayerite | 7.0 | 0.5 | 400 | 333 | 1.16 |
| B3 | 4 g/L bayerite | 7.0 | 0.001 | 40 | 2 | 0.65 |
| B4 | 4 g/L bayerite | 7.0 | 0.001 | 400 | 319 | 1.39 |
| B5 | 4 g/L bayerite | 4.0 | 0.5 | 40 | 7 | 0.63 |
| B6 | 4 g/L bayerite | 4.0 | 0.5 | 400 | 329 | 1.31 |
| B7 | 4 g/L bayerite | 4.0 | 0.001 | 40 | 1 | 0.71 |
| B8 | 4 g/L bayerite | 4.0 | 0.001 | 400 | 101 | 1.92 |
| Mineral | pH | I (M) | Γmax,1 (μmol/m2) | K1 (L/μmol) | Γmax,2 (μmol/m2) | K2 (L/μmol) | R-Factor a |
|---|---|---|---|---|---|---|---|
| Bayerite | 4 | 0.5 | 0.64 ± 0.03 | 7.23 ± 1.37 | 0.98 ± 0.05 | 0.007 ± 0.001 | 0.020 |
| 0.1 | 0.75 ± 0.04 | 4.83 ± 0.89 | 0.94 ± 0.04 | 0.014 ± 0.003 | 0.017 | ||
| 0.01 | 1.24 ± 0.06 | 2.25 ± 0.32 | 0.65 ± 0.06 | 0.02 ± 0.007 | 0.019 | ||
| 0.001 | 1.43 ± 0.07 | 2.11 ± 0.32 | 0.62 ± 0.09 | 0.0092 ± 0.006 | 0.022 | ||
| Bayerite | 7 | 0.5 | 0.77 ± 0.03 | 2.54 ± 0.48 | 0.62 ± 0.08 | 0.0054 ± 0.002 | 0.021 |
| 0.1 | 0.83 ± 0.04 | 3.17 ± 0.65 | 0.59 ± 0.13 | 0.004 ± 0.002 | 0.026 | ||
| 0.01 | 0.92 ± 0.30 | 3.86 ± 0.47 | 0.51 ± 0.04 | 0.01 ± 0.003 | 0.016 | ||
| 0.001 | 1.02 ± 0.03 | 3.87 ± 0.50 | 0.60 ± 0.07 | 0.0056 ± 0.002 | 0.020 | ||
| Gibbsite | 4 | 0.5 | 0.15 ± 0.03 | 78.38 ± 9.01 | 0.99 ± 0.03 | 0.02 ± 0.003 | 0.022 |
| 0.1 | 0.39 ± 0.03 | 2.74 ± 0.60 | 0.90 ± 0.03 | 0.023 ± 0.002 | 0.012 | ||
| 0.01 | 0.81 ± 0.05 | 1.45 ± 0.26 | 0.63 ± 0.05 | 0.018 ± 0.005 | 0.018 | ||
| 0.001 | 1.11 ± 0.04 | 1.21 ± 0.11 | 0.39 ± 0.04 | 0.0089 ± 0.004 | 0.013 | ||
| Gibbsite | 7 | 0.5 | 0.38 ± 0.02 | 1.02 ± 0.21 | 0.47 ± 0.03 | 0.0064 ± 0.002 | 0.020 |
| 0.1 | 0.45 ± 0.02 | 1.85 ± 0.26 | 0.44 ± 0.03 | 0.0064 ± 0.002 | 0.018 | ||
| 0.01 | 0.49 ± 0.02 | 4.39 ± 0.62 | 0.38 ± 0.02 | 0.011 ± 0.003 | 0.019 | ||
| 0.001 | 0.53 ± 0.01 | 5.02 ± 0.40 | 0.36 ± 0.01 | 0.010 ± 0.002 | 0.010 |
| Sample | Path | CN a | R (Å) b | σ2 (Å2) c | ∆E0 (eV) d | R-Factor e | χv2 e |
|---|---|---|---|---|---|---|---|
| G1 | As-O | 4 | 1.696 (4) | 0.0019 (2) | 7 (1) | 0.008 | 37.70 |
| As-Al | 0.9 (5) f | 3.19 (3) | 0.006 | ||||
| G2 | As-O | 4 | 1.701 (4) | 0.0027 (2) | 8 (1) | 0.008 | 26.50 |
| As-Al | 0.6 (4) | 3.21 (5) | 0.006 | ||||
| G3 | As-O | 4 | 1.697 (4) | 0.0020 (2) | 7 (1) | 0.008 | 41.28 |
| As-Al | 0.9 (5) | 3.19 (3) | 0.006 | ||||
| G4 | As-O | 4 | 1.694 (4) | 0.0024 (2) | 6 (1) | 0.011 | 49.88 |
| As-Al | 0.8 (5) | 3.19 (4) | 0.006 | ||||
| G5 | As-O | 4 | 1.697 (3) | 0.0021 (3) | 8 (1) | 0.007 | 20.25 |
| As-Al | 1.1 (4) | 3.22 (3) | 0.006 | ||||
| G6 | As-O | 4 | 1.696 (5) | 0.0024 (5) | 7 (1) | 0.013 | 58.38 |
| As-Al | 0.8 (6) | 3.19 (5) | 0.006 | ||||
| G7 | As-O | 4 | 1.690 (5) | 0.0026 (2) | 6 (1) | 0.012 | 62.78 |
| As-Al | 1.0 (5) | 3.18 (4) | 0.006 | ||||
| G8 | As-O | 4 | 1.698 (5) | 0.0027 (2) | 7 (1) | 0.012 | 32.16 |
| As-Al | 0.7 (5) | 3.21 (5) | 0.006 | ||||
| B1 | As-O | 4 | 1.686 (4) | 0.0018 (2) | 5 (1) | 0.009 | 42.92 |
| As-Al | 1.2 (5) | 3.18 (3) | 0.006 | ||||
| B2 | As-O | 4 | 1.697 (5) | 0.0028 (5) | 7 (1) | 0.013 | 27.54 |
| As-Al | 1.1 (5) | 3.17 (3) | 0.006 | ||||
| B3 | As-O | 4 | 1.696 (4) | 0.0019 (2) | 7 (1) | 0.007 | 34.56 |
| As-Al | 1.2 (4) | 3.20 (3) | 0.006 | ||||
| B4 | As-O | 4 | 1.696 (5) | 0.0027 (3) | 7 (1) | 0.013 | 73.71 |
| As-Al | 1.2 (5) | 3.17 (3) | 0.006 | ||||
| B5 | As-O | 4 | 1.686 (5) | 0.0020 (2) | 5 (1) | 0.011 | 56.60 |
| As-Al | 1.2 (6) | 3.19 (3) | 0.006 | ||||
| B6 | As-O | 4 | 1.695 (5) | 0.0028 (2) | 7 (1) | 0.012 | 75.51 |
| As-Al | 1.4 (5) | 3.18 (3) | 0.006 | ||||
| B7 | As-O | 4 | 1.697 (4) | 0.0018 (2) | 7 (1) | 0.007 | 37.21 |
| As-Al | 1.2 (5) | 3.19 (3) | 0.006 | ||||
| B8 | As-O | 4 | 1.694 (3) | 0.0021 (2) | 6 (1) | 0.006 | 20.21 |
| As-Al | 1.1 (4) | 3.18 (3) | 0.006 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, T.; Catalano, J.G. Effects of Ionic Strength on Arsenate Adsorption at Aluminum Hydroxide–Water Interfaces. Soil Syst. 2018, 2, 1. https://doi.org/10.3390/soils2010001
Xu T, Catalano JG. Effects of Ionic Strength on Arsenate Adsorption at Aluminum Hydroxide–Water Interfaces. Soil Systems. 2018; 2(1):1. https://doi.org/10.3390/soils2010001
Chicago/Turabian StyleXu, Tingying, and Jeffrey G. Catalano. 2018. "Effects of Ionic Strength on Arsenate Adsorption at Aluminum Hydroxide–Water Interfaces" Soil Systems 2, no. 1: 1. https://doi.org/10.3390/soils2010001
APA StyleXu, T., & Catalano, J. G. (2018). Effects of Ionic Strength on Arsenate Adsorption at Aluminum Hydroxide–Water Interfaces. Soil Systems, 2(1), 1. https://doi.org/10.3390/soils2010001