
An Ultra-Rare Disorder: Case Report on Cerebrotendinous Xanthomatosis

 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
Abstract
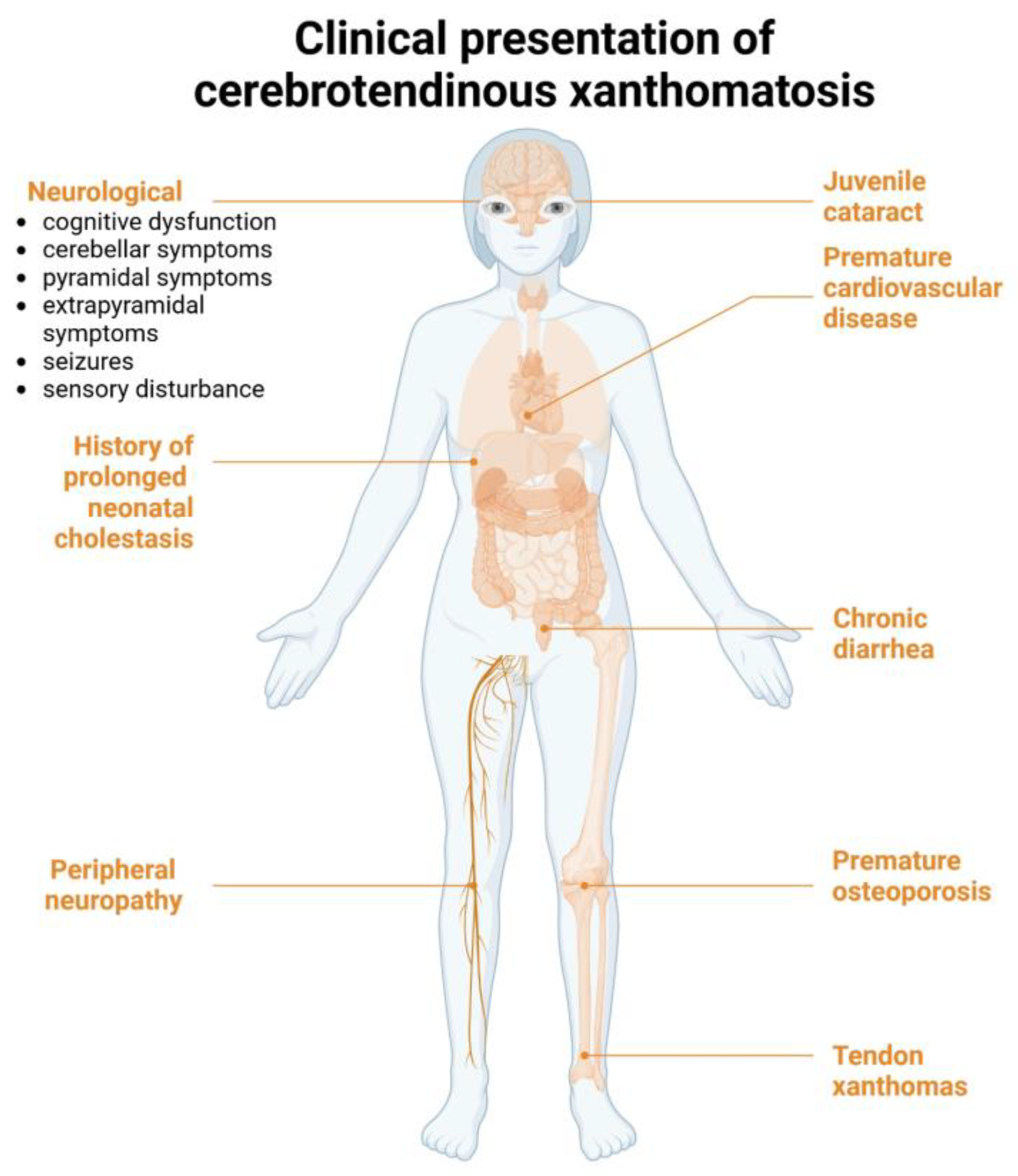
1. Introduction and Clinical Significance
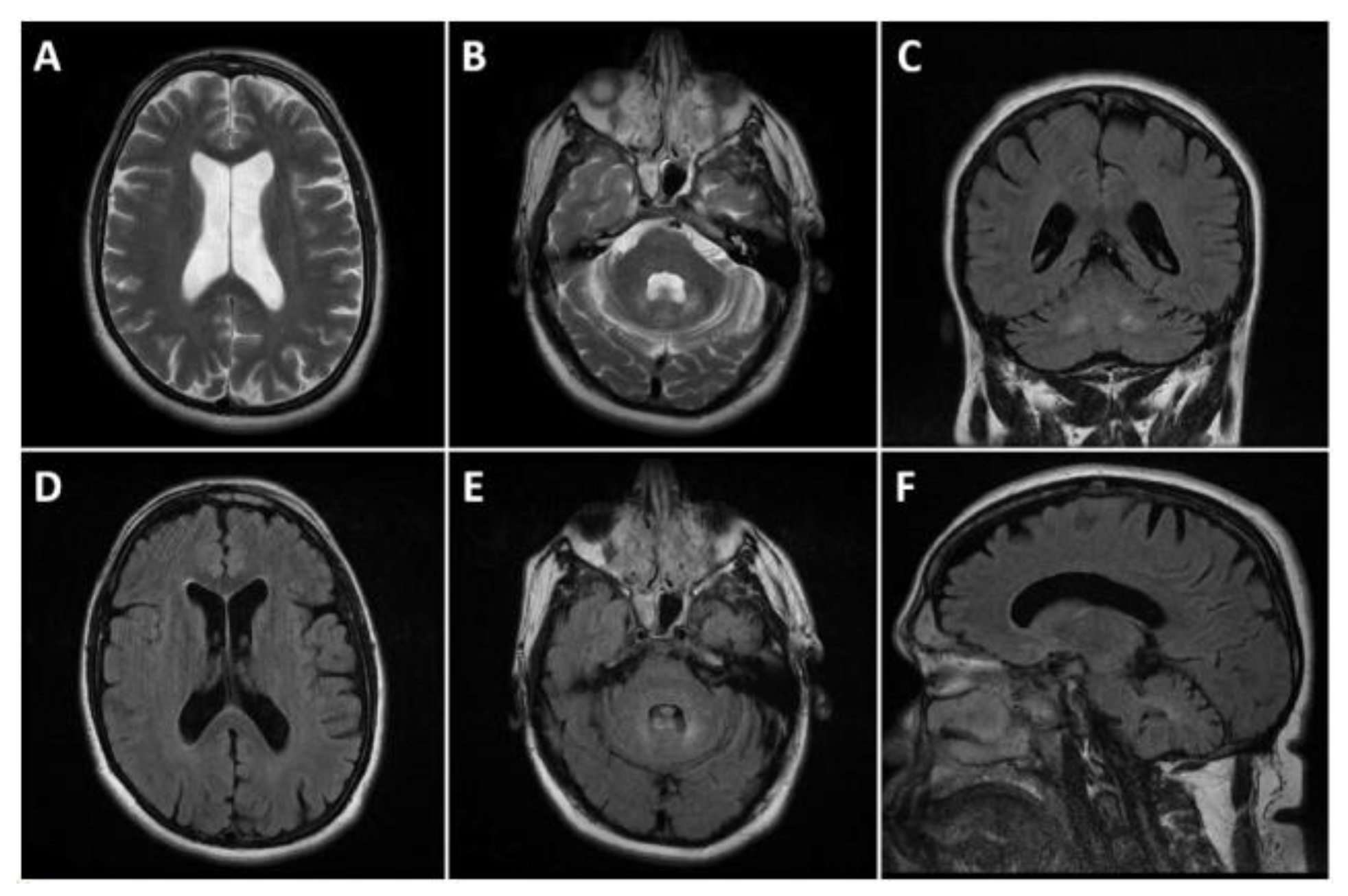
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CTX | Cerebrotendinous xanthomatosis |
| CYP27A1 | enzyme 27-hydroxylase |
| MRI | magnetic resonance imaging |
| EEG | electroencephalogram |
| SWDs | single spike–wave discharges |
| MMSE | Mini-Mental State Examination |
| IADL | Instrumental Activities of Daily Living Scale |
| CDCA | chenodeoxycholic acid |
| CNV | copy number variant |
References
- Luo, F.; Ding, Y.; Zhang, S.; Diao, J.; Yuan, B. Frontier and hotspot evolution in cerebrotendinous xanthomatosis: A bibliometric analysis from 1993 to 2023. Front. Neurol. 2024, 15, 1371375. [Google Scholar] [CrossRef] [PubMed]
- Zubarioglu, T.; Kıykım, E.; Köse, E.; Eminoğlu, F.T.; Kısa, P.T.; Balcı, M.C.; Özer, I.; İnci, A.; Çilesiz, K.; Canda, E.; et al. Clinical, biochemical, and molecular insights into Cerebrotendinous Xanthomatosis: A nationwide study of 100 Turkish individuals. Mol. Genet. Metab. 2024, 142, 108493. [Google Scholar] [CrossRef] [PubMed]
- Behari, M. A Rare Symptomatic Case of Heterozygous Cerebro-Tendinous Xanthomatosis (CTX) Treated with Urso-Deoxycholic Acid (UDCA): With Mini Review. J. Neurosci. Neurol. Disord. 2024, 8, 57–63. [Google Scholar]
- Pavisich, K.; Jones, H.; Baynam, G. The Diagnostic Odyssey for Children Living with a Rare Disease–Caregiver and Patient Perspectives: A Narrative Review with Recommendations. Rare 2024, 2, 100022. [Google Scholar] [CrossRef]
- Guay, S.-P.; Paquette, M.; Poulin, V.; Levtova, A.; Baass, A.; Bernard, S. The 20-Year diagnostic odyssey of a milder form of cerebrotendinous xanthomatosis. JCEM Case Rep. 2024, 2, luae004. [Google Scholar] [CrossRef] [PubMed]
- Franklin by Genoox. Available online: https://franklin.genoox.com (accessed on 30 October 2024).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Garuti, R.; Lelli, N.; Barozzini, M.; Tiozzo, R.; Dotti, M.; Federico, A.; Ottomano, A.; Croce, A.; Bertolini, S.; Calandra, S. Cerebrotendinous xanthomatosis caused by two new mutations of the sterol-27-hydroxylase gene that disrupt mRNA splicing. J. Lipid Res. 1996, 37, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Bartholdi, D.; Zumsteg, D.; Verrips, A.; Wevers, R.; Sistermans, E.; Hess, K.; Jung, H. Spinal phenotype of cerebrotendinous xanthomatosis. J. Neurol. 2004, 251, 105–107. [Google Scholar] [CrossRef] [PubMed]
- Ginanneschi, F.; Mignarri, A.; Mondelli, M.; Gallus, G.; Del Puppo, M.; Giorgi, S.; Federico, A.; Rossi, A.; Dotti, M. Polyneuropathy in cerebrotendinous xanthomatosis and response to treatment with chenodeoxycholic acid. J. Neurol. 2013, 260, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Sekijima, Y.; Koyama, S.; Yoshinaga, T.; Koinuma, M.; Inaba, Y. Nationwide survey on cerebrotendinous xanthomatosis in Japan. J. Hum. Genet. 2018, 63, 271–280. [Google Scholar] [CrossRef] [PubMed]
- DeBarber, A.E.; Schaefer, E.J.; Do, J.; Ray, J.W.; Larson, A.; Redder, S.; Fowler, M.; Duell, P.B. Genetically and clinically confirmed atypical cerebrotendinous xanthomatosis with normal cholestanol and marked elevations of bile acid precursors and bile alcohols. J. Clin. Lipidol. 2024, 18, e465–e476. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.S.; Sannegowda, R.B.; Agrawal, A.; Hemrajani, D.; Jain, R.; Mathur, T. ‘Hot cross bun’sign in a case of cerebrotendinous xanthomatosis: A rare neuroimaging observation. Case Rep. 2013, 2013, bcr2012006641. [Google Scholar]
- Fussiger, H.; Lima, P.L.G.S.B.; Souza, P.V.S.; Freua, F.; Husny, A.S.E.; Leão, E.K.E.A.; Braga-Neto, P.; Kok, F.; Lynch, D.S.; Saute, J.A.M.; et al. Clinicogenetic characterization of cerebrotendinous xanthomatosis in Brazil. Clin. Genet. 2024, 106, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Salardaine, Q.; Shor, N.; Villain, N.; Bozon, F.; Amador, M.D.M.; Duchon, C.; Mélé, N.; Schiff, M.; Brassier, A.; Nadjar, Y. Cognitive impairment in children and adults with cerebrotendinous xanthomatosis: A French cohort study. J. Inherit. Metab. Dis. 2024, 47, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Han, J.; Tao, D.; Zheng, H. Cerebrotendinous xanthomatosis with tremor as the main manifestation: A case report. Medicine 2024, 103, e37976. [Google Scholar] [CrossRef] [PubMed]
- Chun, M.Y.; Heo, N.J.; Seo, S.W.; Jang, H.; Suh, Y.-L.; Jang, J.-H.; Kim, Y.-E.; Kim, E.-J.; Moon, S.Y.; Jung, N.-Y.; et al. Case report: Cerebrotendinous xanthomatosis with a novel mutation in the CYP27A1 gene mimicking behavioral variant frontotemporal dementia. Front. Neurol. 2023, 14, 1131888. [Google Scholar] [CrossRef] [PubMed]
- Min, J.H.; Kim, Y.S.; Son, M.J.; Joo, I.S. A double CYP27A1 gene mutation in spinal cerebrotendinous xanthomatosis in a patient presenting with spastic gait: A case report. J. Med. Case Rep. 2024, 18, 334. [Google Scholar] [CrossRef]
- Jenzer, H.; Groesser, S.; Miljković, N. Availability of Medicines. In Practical Pharmaceutics; Springer: Berlin/Heidelberg, Germany, 2023; pp. 23–55. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levkova, M.; Hachmeriyan, M.; Grudkova, M.; Tsalta-Mladenov, M.; Kaprelyan, A. An Ultra-Rare Disorder: Case Report on Cerebrotendinous Xanthomatosis. Reports 2025, 8, 77. https://doi.org/10.3390/reports8020077
Levkova M, Hachmeriyan M, Grudkova M, Tsalta-Mladenov M, Kaprelyan A. An Ultra-Rare Disorder: Case Report on Cerebrotendinous Xanthomatosis. Reports. 2025; 8(2):77. https://doi.org/10.3390/reports8020077
Chicago/Turabian StyleLevkova, Mariya, Mari Hachmeriyan, Margarita Grudkova, Mihael Tsalta-Mladenov, and Ara Kaprelyan. 2025. "An Ultra-Rare Disorder: Case Report on Cerebrotendinous Xanthomatosis" Reports 8, no. 2: 77. https://doi.org/10.3390/reports8020077
APA StyleLevkova, M., Hachmeriyan, M., Grudkova, M., Tsalta-Mladenov, M., & Kaprelyan, A. (2025). An Ultra-Rare Disorder: Case Report on Cerebrotendinous Xanthomatosis. Reports, 8(2), 77. https://doi.org/10.3390/reports8020077