2. Case Presentation
2.1. Case 1
A 10-year-old girl was first admitted to our department in June 2022 for scleral jaundice, abdominal pain, mostly left-sided, and poor appetite (a proper history of the disease was difficult to obtain because of illiterate relatives). Symptoms began 4 months prior, and she had never been evaluated by a medical team before. Both her parents were apparently in good health, but her 6-year-old brother was known since 2018 with cholestatic hepatitis of undetermined cause.
On admission, the patient presented with failure to thrive: weight = 23 kg (z-score = −2.03), height = 122 cm (z-score = −2.48), BMI = 15.5 kg/m2 (z-score = −0.6), generalized jaundice with scratching lesions, facial angiomas, enlarged liver and spleen. She had dark-brown urine and acholic stools.
Investigations revealed bicytopenia (hemoglobin level = 10.5 g/dL; low platelet count = 134.000/mm3), significant liver cytolysis (transaminases x7-10 the upper limit of normal (ULN)), and cholestasis (total bilirubin = 10 mg/dL, predominantly conjugated, high GGT, significantly increased serum bile acids). Infectious causes of acute or chronic hepatitis were promptly ruled out: hepatitis B virus (HBV), hepatitis C virus (HCV), human immunodeficiency virus (HIV), reactivation of Epstein Barr (EBV), and Cytomegalovirus (CMV). Autoimmune liver disorders were highly unlikely (normal ESR, immunoglobulin G and gamma globulins levels, negative anti-LKM1, pANCA, ANA, and ASMA antibodies). Wilson disease and cystic fibrosis were excluded due to normal serum ceruloplasmin, urinary copper, and normal sweat test, respectively. Metabolic storage diseases (Gaucher and Niemann Pick) were also ruled out through genetic testing on dryspot.
Due to family history (brother with chronic cholestatic hepatitis and particular facial features), Alagille syndrome and Progressive familial intrahepatic cholestasis (PFIC) were taken into consideration. Thoracic spine X-ray showed no vertebrae anomalies; cardiac ultrasonography showed no specific malformations, and eye check-up showed no posterior embriotoxon.
Genetic testing (sequence analysis and deletion/duplication testing of the 136 genes listed in the Genes Analyzed section) came out positive for ABCB4 homozygous mutation (c.2534G>T (p.Gly845Val)); the patient is also homozygous for UGT1A1 gene mutation (c.-41-40dup (non-coding)) and heterozygous for NOTCH2 (c.6178C>T (p.Arg2060Cys)), but considering clinical presentation, PFIC type 3 diagnosis was set.
The father was tested with a dry spot for Gaucher and Niemann Pick disease with negative results. Genetic sequencing, WES or otherwise, of the parents was not financially accessible to this family.
Abdominal ultrasound found ascites, an enlarged liver with an irregular outline, and multiple nodular elements–hepatic structure alterations compatible with cirrhosis (
Figure 2a) and a thick-walled gallbladder (
Figure 2b).
Figure 2.
Imagistic findings in case 1. (a) Abdominal US—cirrhotic liver nodules. (b) Abdominal US—thick-walled gallbladder. (c) Abdominal MRI—enlarged liver and spleen.
Figure 2.
Imagistic findings in case 1. (a) Abdominal US—cirrhotic liver nodules. (b) Abdominal US—thick-walled gallbladder. (c) Abdominal MRI—enlarged liver and spleen.
Abdominal magnetic resonance described the same cirrhotic aspect of the liver, thick-walled gallbladder with inhomogeneous content, and enlarged spleen (
Figure 2c) Upper endoscopy showed esophageal varices with no signs of active bleeding.
PELD score was 18.5 points, meaning 65.5% 1-year waiting list survival and 89.4% 1-year posttransplant survival.
In the next 2 months, her general condition and jaundice progressively worsened; she developed significant abdominal distension due to rapidly progressing hepatomegaly. Encephalopathy was becoming apparent (altered consciousness, aggressive behavior, reversal of sleep rhythm). Laboratory markers showed progressive thrombocytopenia, severe cholestasis (total bilirubin value of 14 mg/dL, rapidly increasing to 36 mg/dL, predominantly conjugated (
Figure 3), significantly altered liver functions (INR = 2.55, hypoproteinemia). Abdominal ultrasound described a medium amount of ascites, and pulmonary X-ray found bilateral pleural effusions.
Figure 3.
Dynamics of bilirubin levels (mg/dL) in case 1 prior to transplantation.
Figure 3.
Dynamics of bilirubin levels (mg/dL) in case 1 prior to transplantation.
Partial parenteral nutrition and immediate antibiotic treatment with meropenem were initiated; also, supportive liver medication was given (arginine, plasma infusions, fat-soluble vitamins, beta-blockers, and diuretics), with no improvement of clinical or biological status. Two courses of urgent plasmapheresis did not manage to lower the bilirubin levels.
She rapidly developed liver failure and received life-saving liver transplantation from a related donor (mother), with a good outcome.
2.2. Case 2
A 6-year-old boy (brother to case 1), previously diagnosed with cholestatic hepatitis of unknown cause back in 2018 (temporarily lost of follow-up), first presented to our department in June 2022 for generalized jaundice and severe pruritus with generalized scratching lesions. Symptoms had progressively developed since the first year of life. Both his parents were seemingly in good health (disease history was difficult to draw because of illiterate family members).
On admission, the patient presented with stunting: weight = 19 kg (z-score = −0.6), height = 105 cm (z-score = −2), BMI = 17.27 kg/m2 (z-score = 1.2), peculiar facial features (triangle-shaped face, large forehead, deep-set eyes, straight nose), xanthomas on hands and feet, generalized jaundice with severe pruritus, and multiple scratching lesions. A heart murmur was discovered during examination, alongside a distended abdomen due to hepatosplenomegaly.
Investigations determined mild anemia, low platelet number, liver enzyme abnormality (transaminases × 2 ULN), cholestasis with a total bilirubin of 3.09 mg/dL predominantly conjugated and high GGT level, and extremely high levels of serum bile acids (×20 times ULN). Infectious causes, autoimmune liver, Wilson disease, cystic fibrosis, and metabolic diseases were ruled out.
Abdominal ultrasound described normal liver size with irregular structure, various nodular elements (
Figure 4a), a thick-walled gallbladder (
Figure 4b), and an enlarged spleen (
Figure 4c).
Figure 4.
Imagistic findings in case 2. (a) Abdominal US—liver nodules. (b) Abdominal US—thick-walled, double-layered gallbladder. (c) Abdominal US—enlarged spleen. (d) Abdominal MRI—enlarged liver and spleen. (e) Abdominal MRI—thick-walled, double-layered gallbladder.
Figure 4.
Imagistic findings in case 2. (a) Abdominal US—liver nodules. (b) Abdominal US—thick-walled, double-layered gallbladder. (c) Abdominal US—enlarged spleen. (d) Abdominal MRI—enlarged liver and spleen. (e) Abdominal MRI—thick-walled, double-layered gallbladder.
Abdominal MRI found an enlarged liver with an irregular outline, two nodular structures (probably regenerative type), no bile duct enlargements, and an otherwise normal aspect (
Figure 4d), but encountered
a gallbladder with double-layered walls (
Figure 4e). Upper endoscopy showed three esophageal variceal cords with no signs of active bleeding.
Keeping in mind his family history, two main differential diagnosis hypotheses were investigated–Alagille syndrome and progressive familial hepatic cholestasis. Thoracic X-ray showed no vertebrae anomalies; cardiac sonography described normal heart structure, and eye check-up did not reveal posterior embryotoxon. Genetic testing for other inherited diseases associated with intrahepatic cholestasis was not financially available, but considering the older sister had now been labeled as homozygous for ABCB4 mutation, liver biopsy was no longer compulsory for diagnosis, more so because the patient embodied phenotype of PFIC type 3. He was immediately listed for liver transplantation because the father was not compatible as a donor, and the mother had donated the left liver lobe to his sister.
PELD score was 5.6 points, meaning 86.8% 1-year waiting list survival and 93.6% 1-year posttransplant survival.
The patient remained under surveillance at home with medical treatment—ursodeoxycholic acid (UCDA)—to slow down disease progression, fat-soluble vitamin supplementation, hepatocyte preserving medication, and specific alleviating tincture for skin lesions.
In April 2023, the patient underwent surgery for a split adult cadaveric organ grafting with the left liver lobe being transplanted. Regarding the vascular anastomosis, the triangulation method was preferred. The arterial anastomosis was performed via a microscopic approach. An intraoperative ultrasound was performed, and it showed good portal flow and a patent arterial anastomosis with an IR = 0.74 (resistive index). The left biliary duct to the common bile duct anastomosis was performed with biliary stent placement. The stent was removed 1 month after surgery. While the patient’s initial postoperative course was favorable, on postoperative day 3, the control ultrasound could not identify blood flow in the hepatic artery. The patient was taken to the operating room, and an exploratory laparotomy was performed. The liver presented with ischemic areas in segments III and IV-a. Intraoperative ultrasound showed no blood flow in the hepatic artery. A revision of the arterial anastomosis was performed with a good resistive index. The patient’s postoperative outcome was favorable, with an IR of 0.8 in the postoperative ultrasounds.
Pathology aspects of the diseased liver are exhibited in
Figure 1 ((1)—literature images, (2–4) histology from case 2).
2.3. Case 3
A 3-year-5-month-old boy was first admitted to our Pediatric Department in April 2023 for failure to thrive, one-year generalized jaundice with pruritus, and significant splenomegaly. Both parents and older brother declared no health issues, but we discovered that the patient had two young uncles, who earlier that year, had received liver grafts because of advanced cirrhosis due to progressive familial hepatic cholestasis (both diagnosed in our department—cases 1 and 2).
Upon evaluation, the boy exhibited failure to thrive: weight = 11.5 kg (z-score = −2.6), height = 87 cm (z-score = −2.9), BMI = 15.19 kg/m2 (z-score = −0.6). He had generalized jaundice with intense pruritus, second-degree heart murmur, and enlarged liver and spleen. No stool or urine anomalies were noted.
Laboratory investigations showed mild anemia and thrombocytopenia. Liver cytolysis was found (transaminases x2 ULN) alongside cholestasis (total bilirubin 2 mg/dL predominantly conjugated, high GGT) and highly elevated levels of serum bile acids. The coagulation panel was normal.
Abdominal ultrasound described an enlarged liver with normal structure and severely enlarged homogeneous spleen. No aspects compatible with liver cirrhosis were encountered, and no alterations of the hepatobiliary tree were found (
Figure 5a).
Figure 5.
Imaging findings in case 3. (a) Abdominal US—hepatosplenomegaly. (b) Abdominal MRI—hepatosplenomegaly.
Figure 5.
Imaging findings in case 3. (a) Abdominal US—hepatosplenomegaly. (b) Abdominal MRI—hepatosplenomegaly.
Abdominal MRI described hepatosplenomegaly (right hepatic lobe 110 mm, bipolar spleen axis 114 mm), no expansions of intra or extrahepatic biliary ducts, gallbladder with normal structure, and no ascites (
Figure 5b).
Cardiac sonography described normal function of ventricular cavities, no pulmonary hypertension, and intact heart walls.
No esophageal varices were described on upper endoscopy.
Because of the family history (two young uncles diagnosed previously with PFIC type 3), longtime jaundice with pruritus, and severely enlarged spleen with hepatomegaly and cholestasis, based on high conjugated bilirubin values and high level of serum bile acids, we had a reasonable suspicion of PFIC associated with HBV infection.
Genetic testing (sequence analysis using the Blueprint Genetics (BpG) Cholestasis Panel) identified a homozygous missense ABCB4 mutation (c.2534G>T, p(Gly845Val)), as well as his girl cousin who had already been transplanted, so PFIC 3 diagnosis was set.
Parents were tested with dry spots for Gaucher and Niemann Pick disease with negative results. Genetic sequencing, WES or otherwise, was not financially accessible to this family.
He received medical treatment with UCDA, which slightly enhanced the clinical and laboratory statuses (improved jaundice and pruritus, lowered liver cytolysis and cholestasis). Imaging also showed a slightly decreasing size of the liver and spleen. PELD score was 2.2 points, meaning 86.8% for 1-year waiting list survival and 93.6% for 1-year posttransplant survival.
He remains under surveillance and will return for regular laboratory and imaging check-ups. Meanwhile, he will continue medical treatment with UDAC, which, so far, has kept the progression of the liver disease under control. Having a steady liver function, he was not yet listed for liver transplantation.
3. Discussion
Genetic analysis is the gold standard for PFIC diagnosis [
5,
11,
23]. Gene analysis requires DNA sequencing of the 27 coding exons and their splice junctions [
3]. Nowadays, a resequencing chip that looks for genetic syndromes of cholestasis has been developed and may facilitate diagnosis [
24].
PFIC 1 occurs due to mutations within the ATP8B1 gene, which is responsible for producing a protein known as FIC1 (which maintains the amino-phospholipid plasma membrane). This protein is located on the canalicular membranes, cholangiocytes, and cells of the small intestine, which explains its involvement in a variety of functions. That is why a broad spectrum of symptoms exists amongst these mutations [
1,
3,
22,
24].
PFIC 2 is caused by mutations in the ABCB11 gene, located on chromosome 2, which encodes for a bile salt export pump (BSEP), a protein responsible for the export of bile salts into the bilious fluid. Mutations led to a buildup of bile salts within the hepatocytes [
1,
3,
6].
PFIC 3 occurs due to a defect in MDR3 (class III multi-drug resistance p-glycoprotein) or a genetic variation in ATP binding cassette subfamily B member 4 gene (ABCB4). Homozygous or compound heterozygous mutations in the ABCB4 gene are considered characteristic variants in PFIC 3 patients. Heterozygous ABCB4 variants result in less severe clinical patterns and partially preserved MDR3 protein function [
5]. ABCB4 gene is located on chromosome 7q21 and encodes a translocator protein involved in bile and lipid excretion [
2,
3,
5,
8,
9,
18,
21,
23,
25].
The mechanism of action of PFIC (all types) is represented in
Figure 6.
Figure 6.
Mechanism of action in PFIC 1, PFIC 2, and PFIC 3.
Figure 6.
Mechanism of action in PFIC 1, PFIC 2, and PFIC 3.
Regarding molecular diagnosis, genetic analysis is the gold standard for PFIC 3 diagnosis [
6]. Gene analysis requires DNA sequencing of the 27 coding exons and their splice junctions [
26]. Nowadays, a resequencing chip that looks for genetic syndromes of cholestasis has been developed and may facilitate diagnosis [
27]. In subtype 3, the ABCB4 gene located on chromosome 7 has been confirmed responsible, and approximately 50 mutations have been linked to this form of PFIC [
6,
28]. ABCB4 protein (
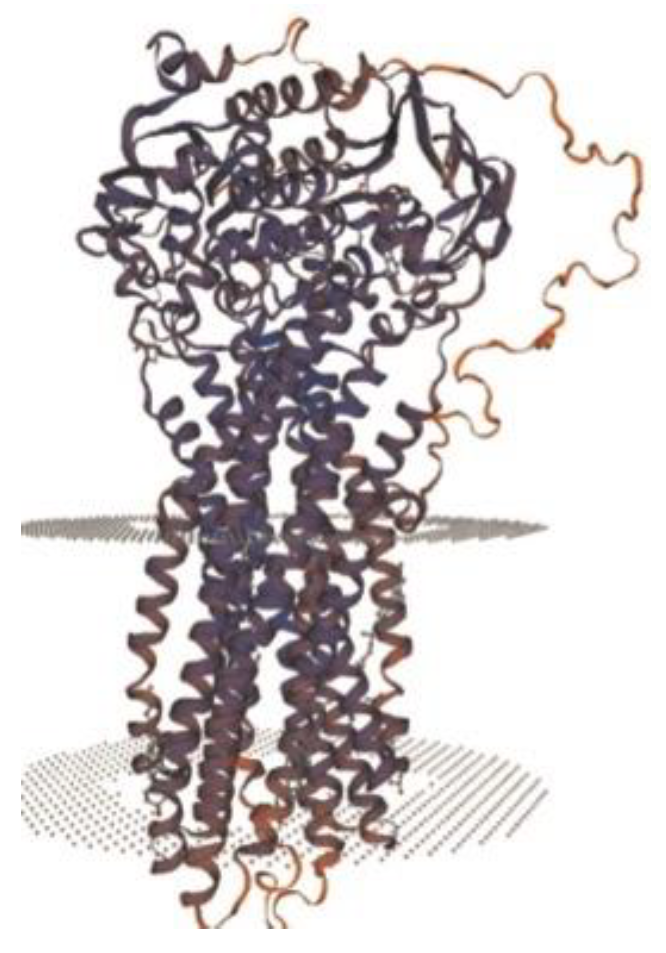
Figure 7) is made of two transmembrane domains (TMDs) and two highly conserved nucleotide-binding domains (NBDs). TMDs are in charge of substrate specificity, and NBDs bind and hydrolyze ATP [
6]. This protein is actually a phosphatidylcholine (PC) enzyme that transforms PC into bile and emulsified bile salts, thus avoiding injury of the epithelial bile duct cells. Blood samples should be obtained from the child and both parents if possible, more so if they desire a future pregnancy [
13].
Figure 7.
Structure of ABCB4 novel pathogenic variant [
6].
Figure 7.
Structure of ABCB4 novel pathogenic variant [
6].
In our series of three cases, two of the patients (cases 1 and 3) exhibited ABCB4 gene pathogenic mutation. For case 2, complete genetic testing was not financially available (he was only tested for Alagille syndrome, with negative results), but considering the familial aggregation, the patient’s clinical phenotype, and the microscopic aspects of liver biopsy (after transplantation), we may fairly state PFIC 3 diagnosis.
Sequence analysis identified the same homozygous missense variant ABCB4 c.2534G>T, p.(Gly845Val) in both cases genetically tested. This variant is absent in gnomAD, a large reference population database (n > 120,000 exomes and >15,000 genomes) that aims to exclude individuals with severe pediatric disease. This variant affects a moderately conserved amino acid within the ABC transporter, the transmembrane domain of the protein. This variant has been submitted to ClinVar by a clinical testing laboratory (variation in ID 2100150) but has, to the best of our knowledge, not been reported in the medical literature.
The onset of symptoms for PFIC 1 and PFIC 2 is in infancy or early childhood, whereas PFIC 3 can present at any age from infancy to adolescence. The main feature in all types of PFIC is hepatic cholestasis, either in the form of pruritus, jaundice, or both [
1,
4,
6]. Subtype 1 usually debuts with extrahepatic symptoms and is characterized by recurrent episodes of jaundice. In the case of PFIC 2, initial presentation and evolution are more severe; liver failure develops in the first years of life, and hepatocellular carcinoma may complicate the course of this disease. On the other hand, because clinical manifestations of PFIC 3 usually develop later in childhood, patients may present with gastrointestinal bleeding due to cirrhosis and portal hypertension, with or without jaundice. Evolution leads to liver failure, with 50% of patients requiring liver transplantation at a mean age of 7.5 years. No malignancies have been associated with subtype 3 [
1,
5,
6,
7,
8,
9].
Similar phenotypes in these related patients were obvious on clinical examination in terms of particular facial features (
Figure 8) and hepatosplenomegaly.
Figure 8.
Similar phenotypes with particular facial features. (a) Case 1—large forehead, deep-set eyes. (b) Case 2—triangle-shaped face, large forehead, deep-set eyes, straight nose. (c) Case 3—big forehead, straight nose, twisted ear lobe.
Figure 8.
Similar phenotypes with particular facial features. (a) Case 1—large forehead, deep-set eyes. (b) Case 2—triangle-shaped face, large forehead, deep-set eyes, straight nose. (c) Case 3—big forehead, straight nose, twisted ear lobe.
A liver biopsy is a required procedure in children with suspected PFIC, as it allows for liver histology and immunostaining to be performed. Liver immunostaining is possible due to MDR3 and BSEP antibodies. Mild immunostaining or absent bile ducts favors a gene defect, but normal staining does not necessarily pair with a genetic mutation (a genetic defect may induce function loss with normal synthesis and expression) [
3,
29].
In PFIC 1, histological findings include hepatocellular and canalicular cholestasis with pseudo-acinar transformation (
Figure 1(1a)); giant cell formation and ballooned hepatocytes translate cellular injury. Progression to lobular and portal fibrosis starts early, by the age of two. On electronic microscopy, Byler bile (coarsely granular bile) may be observed in the canalicular spaces [
1,
3,
14,
18].
PFIC 2 patients exhibit mainly the same histological traits, but their liver architecture is more damaged, with severe lobular and portal fibrosis, as well as inflammation. Necrotizing hepatocytes and giant cells are more perceivable than in subtype 1 (
Figure 1(1b)), thus emphasizing that cell injury is more significant in PFIC 2 [
1,
3,
14,
18].
In PFIC 3, histological examination reveals a distinctive pattern closely resembling that of extrahepatic biliary obstruction, characterized by a florid ductular reaction. However, this entity is defined by a patent biliary tree, and these microscopic features align with the MDR3 deficiency, a canalicular transport defect leading to the onset of cholangiopathy [
27,
30]. Liver biopsy shows true ductular proliferation (
Figure 1(1c2)) with mixed inflammatory infiltrate and portal fibrosis; bile ducts are plugged with bile (
Figure 1(1c1)). Minimal giant transformation of hepatocytes can be noticed [
1,
3,
14,
18,
29]. At the onset of PFIC 3, liver histology exhibits portal fibrosis and evident bile ductular proliferation. Most portal tracts showcase interlobular bile ducts, variable degrees of inflammatory infiltrate, and some ducts containing bile plugs and fibrosis. Intralobularly, there is hepatocellular and canalicular cholestasis [
31]. Subsequently, there is a progression to marked portal fibrosis and biliary cirrhosis with a micronodular pattern, sometimes accompanied by intraductal cholelithiasis. Notably absent are periductal fibrosis and biliary epithelial injury [
32]. These typical changes are found in patient 2’s microscopic pathology, confirming PFIC 3 diagnosis, although genetic testing was not performed.
Differential diagnosis of PFIC includes a large variety of pathologies, from metabolic to endocrine disorders, each one harboring signs and symptoms out of the liver-like area. Clinicians should primarily search for cholestasis evidence, but they must also take into consideration growth pattern, bone status, and hormonal balances because sometimes, the right diagnosis falls somewhere in-between. In order to emphasize the main differential diagnoses, we sorted them alphabetically: A (Aagenaes syndrome, Alagille syndrome, alpha1antitripsin deficiency, arthrogryposis, autoimmune hepatitis), B (benign recurrent intrahepatic cholestasis, bile acid synthetic disorder, biliary atresia), C (choledochal cysts, cystic fibrosis, chole-lithiasis), F (familial Amish hypercholanemia), G (galactosemia), H (hypopituitarism, hypothyroidism), N (North American Indian childhood cholestasis), S (sclerosing cholangitis, sepsis), T (TORCH syndrome, toxic ingestion, tyrosinemia), W (Williams syndrome) [
14].
In terms of differential diagnosis, Spraul et al. propose a rather helpful diagram that enables clinicians to have a panoramic view regarding child cholestasis origins [
1].
In a patient with a clinical history of cholestasis, as was the case for each of our three patients, after ruling out other main causes such as Alagille syndrome, biliary atresia, cystic fibrosis, sclerosing cholangitis, an alpha1antitripsine deficiency, and extrahepatic bile duct obstruction, PFIC is to be taken into consideration [
1,
14].
Natural history and complications of subtypes 1, 2, and 3 include portal hypertension, cirrhosis, liver failure, hepatocellular carcinoma, and extrahepatic manifestations. More so, PFIC 1–3 children are at higher risk of developing cholelithiasis and drug-induced cholestasis further in the course of the disease [
1,
3]. Also, female patients with PFIC who reach adulthood with native liver must continue taking ursodeoxycholic acid (UCDA) during pregnancy to reduce the risk of developing intrahepatic cholestasis of pregnancy (ICP) [
1,
3]. Children with BSEP deficiency are at high risk of developing hepatobiliary malignancies like hepatocellular carcinoma or cholangiocarcinoma; thus, regular monitoring every 6 months of alpha-fetoprotein and annual abdominal sonography are required for these patients [
1,
3,
33].
Our series of cases clearly illustrates the natural course of a genetic disorder with severe hepatic lesions: three patients with the same disease timely caught in different stages as their respective PELD scores described: 18.5 in case 1 (10-year-old girl), 5.6 in case 2 (her 6-year-old brother), and 2.2 in case 3 (their second-degree 3-year-5-month-old cousin).
Table 1 summarizes the clinical, biochemical, histological, and genetic features of our three cases. The last column contains comparisons between each of our cases and similar ones in the literature.
Agarwal et al. developed a great tool for addressing children with PFIC, starting from dietary and medical treatment options all the way down to liver transplantation [
4].
First in line is dietary treatment because poor bile flow means that patients with PFIC are not able to absorb fats and fat-soluble vitamins [
12]. Medium-chain triglycerides (MCT) are a special category of easily absorbable fats and represent a solid form of energy for these children. Common foods do not usually contain MCT, but there are some milk formulas and supplements enriched with medium-chain triglycerides. Oral or intravenous supplementation with fat-soluble vitamins (A, D, K, E) is also relevant. Vitamin K 10 mg/week, Vitamin D 3000–5000 UI/day, Vitamin A 2500 UI/day, Vitamin E 50–400 UI/day [
4]. Growth impairment, if present, should be approached by setting a nasogastric tube for overnight feedings, thus increasing the protein (3–4 g/kg/day) and energy (180–200 calories/kg/day) intake [
4,
12].
“Frontline treatment” is considered a secondary bile acid called UCDA at a dosage of 20–30 mg/kg/day. UCDA improves bile flow out of the liver and has numerous benefits, such as reducing bile accumulation in the hepatocellular tissue and reversal of hepatotoxicity, increasing mitochondrial integrity, and decreasing the degree of cholestasis. This medication is particularly useful in MDR3 deficiency [
4,
12,
13,
14].
All three of our patients received UDCA with temporary efficacy in case 1 and fairly good results in cases 2 and 3.
Cholestyramine is a medication that binds to bile acids in the gut and prevents their absorption into the bloodstream. A dosage of 200–300 mg/kg/day may reduce itching, although patient feedback varies from case to case. Cholestyramine should not be given at the same time as fat-soluble vitamins because it can stop proper absorption [
4,
12].
Pruritus had a negative effect on the quality of life of all three patients, but cholestyramine is not available in Romania, so none of them received it.
If, after 4 weeks of therapy with UCDA and Cholestyramine, there is no improvement, alternative medication must be provided. Rifampicin, at a dosage of 5–10 mg/kg/day, is a liver stimulant that upregulates specific detoxification enzymes and may reduce itching in some patients. It has its pitfalls because it can alter liver function tests (must be used with caution in severe liver disease) and can cause urine to turn pink or red [
4,
12,
13,
14]. After 8–12 weeks of classical therapy and unresponsive pruritus, other medications may be tried. Phenobarbital, due to its ability to induce CYP enzymes, has long been used to treat newborn hyperbilirubinemia. Naltrexone, at a dosage of 0.25–0.5 mg/kg/day, and Ondansetron, at a dosage of 0.1–0.2 mg/kg/day, can be used for symptomatic relief [
4,
12,
13]. None of our three patients received this medication.
If the severity of pruritus and nutritional status do not improve after 3 months of medical therapy, surgical intervention must be taken into consideration. There are a number of procedures that can be performed in an attempt to reduce the effects of PFIC, but they must be adapted to each patient, and results vary from case to case. The commonest types are internal or external biliary diversion procedures, which prevent enterohepatic recirculation of bile acid salts, thus decreasing serum levels and alleviating itching. External diversion is possible in children who have not yet developed cirrhosis and supposes a conduit connecting the round end of the gallbladder and abdominal skin, creating a permanent communication. Sometimes, a short section of a child’s bowel is used to connect the gallbladder to the surface of the abdomen. Up to 50% of bile flow can be drawn away from enterohepatic recirculation. In addition, other benefits of external diversion are improved hepatocyte function, reduced or reversed progression of disease, increased time until liver transplantation, and prolonged life expectancy. A common complication of this procedure is the collapse of external biliary drainage. Other drawbacks include worsening malabsorption and episodes of cholangitis [
12,
13,
14,
20,
22,
34]. Partially internal biliary drainage is a newer surgical procedure that creates a jejunal connection between the gallbladder and the large intestine, bypassing the ileal reabsorption of bile acids. There is little evidence to back its safety and efficacy, but patients seem to perceive an improvement in their itching status. Effects on hepatocellular function and disease progression should be similar to those induced after external diversion. It can cause diarrhea because more bile enters the colon [
12,
13,
14,
22]. An alternative surgical procedure is internal ileal exclusion or ileal bypass, in which the last 15% of the small bowel is bypassed by connecting the proximal ileum to the cecum. This way, the largest site of enterohepatic recirculation of bile acid salts is avoided, providing immediate relief. Downfalls include malabsorption and recurrent cholestasis within the first year after the procedure is conducted [
12,
13,
14].
A great number of patients do not respond to either medical or surgical therapy, developing end-stage liver disease, which will require liver transplantation. Liver grafting has proven its efficacy in children with all types of PFIC and remains, until today, the best treatment option, especially for subtype 3 (MDR3 deficiency). In many centers, it is considered a first-line therapy choice, even in patients with no evidence of end-stage liver disease [
12,
13,
14,
22,
34].
Cases 1 and 2 progressed to end-stage liver disease and received liver transplantation; case 3 is currently in a steady state in terms of liver function. The timing of liver transplantation was different for case 2 in comparison to his sister because, as there was no family member available as a donor, he received a graft when a compatible deceased donor was found. He presented complications after liver transplantation, including portal thrombosis, which imposed surgical reintervention for de-obstruction. Subsequently, he developed chronic ischemia due to a stenosis of the hepatic artery, highlighted on CT angiography, after identifying persistently high levels of transaminases during follow-up. The evolution after the liver transplant was surprisingly better in case 1, although she received the draft while in critical condition from a possible mutation carrier (the mother) by comparison to case 2, who received a scheduled unrelated deceased donor graft before hepatic failure.
Vascular complications following liver transplant have been reported in up to 22% of cases, more frequently in cases where the graft originated from a living donor and, more often, in younger recipients due to a smaller graft area. These complications can occur in the early period (most related to the hepatic artery and portal vein) or later during evolution. Hepatic artery stenosis has been reported in 4.1% of cases after pediatric liver transplant, less often than hepatic artery thrombosis. Endovascular revascularization is reported to have an 85.7% success rate, in comparison to surgical revascularization, which reaches 100% success rate. The spectrum of clinical symptoms includes minor elevations of liver function tests to acute fulminant failure [
35,
36,
37].
When looking at our two transplanted cases (cases 1 and 2), vascular complications occurred for the younger patient who received the graft from a deceased donor, which is not typical. Portal thrombosis was found shortly after the transplant and was surgically resolved. However, hepatic artery stenosis developed later during follow-up and was identified because of persistently elevated transaminases, even though it is usually reported to be an early-period vascular complication.


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}