
Long QT Interval Syndrome and Female Sex—Review and Case Report

{kind=link}
Abstract
1. Introduction and Clinical Significance
- Pathophysiology
- LQT1
- LQT2
- LQT3
- Acquired QT interval
1.1. Genotype–Phenotype Correlation
- Epilepsy and long QT syndrome
- Women and long QT interval syndrome
1.2. Pregnancy and Long QT Interval Syndrome
1.3. Therapeutic Approach in Long QT Interval Syndrome
1.4. Gene Therapy
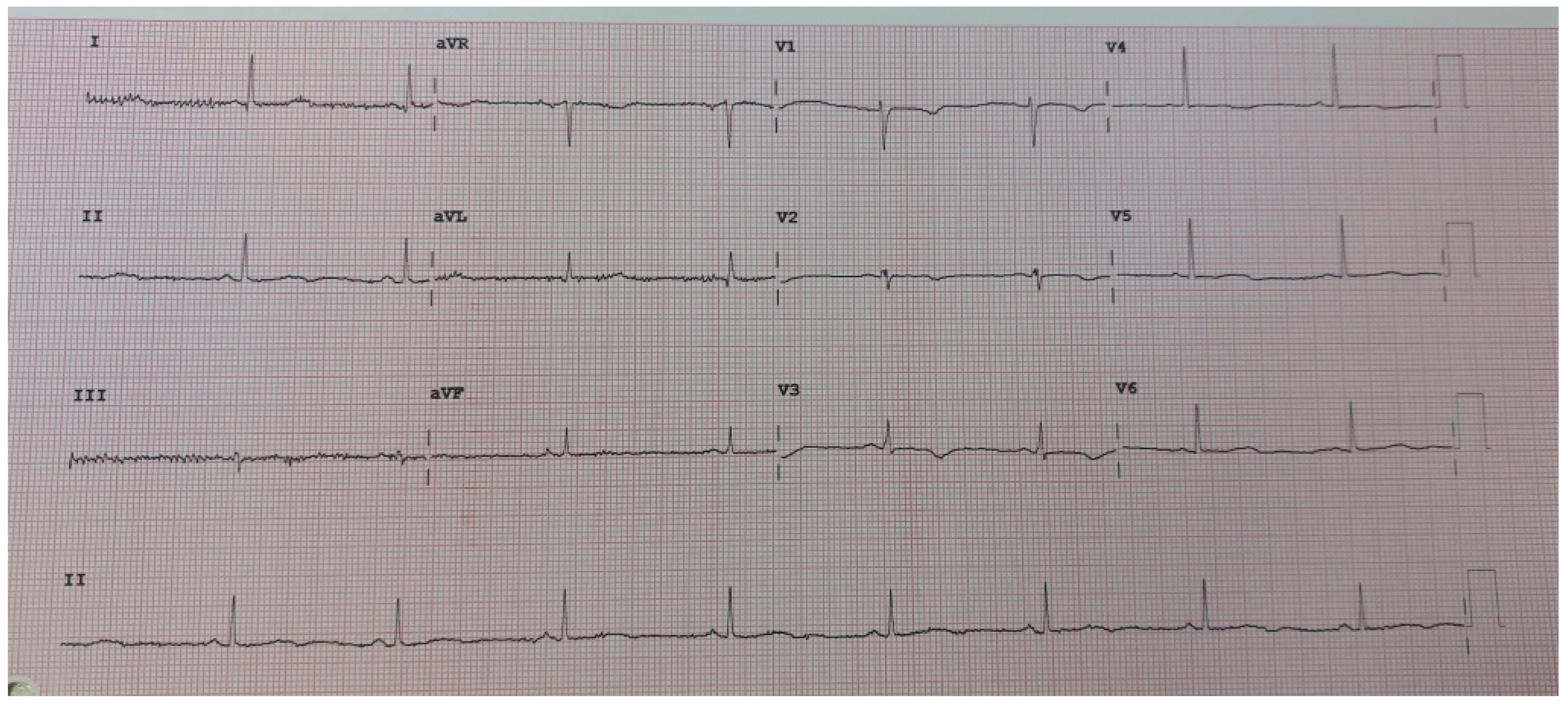
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| LQTS | long QT syndrome |
| ICD | implantable cardioverter defibrillator |
| LCSD | left cardiac sympathetic denervation |
References
- Schwartz, P.J.; Moss, A.J.; Vincent, G.M.; Crampton, R.S. Diagnostic criteria for the long QT syndrome. An update. Circulation 1993, 88, 782–784. [Google Scholar] [CrossRef]
- Ponce-Balbuena, D.; Deschênes, I. Long QT syndrome–Bench to bedside. Heart Rhythm O2 2021, 2, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Jervell, A.; Lange-Nielsen, F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am. Heart J. 1957, 54, 59–68. [Google Scholar] [CrossRef]
- Romano, C.; Gemme, G.; Ponginglione, R. Accesi sincopali per fibrillazione ventricolare paossistica. (Presentazione del primo caso della letteratura pediatrica Italiana) Rare Ccardiac arrytmias of the pediatric age II. Syncopal attacks due to paryoxysmla ventricular fibrillation. (Presentation of of 1st case in Italian pediatric literature). Clin. Pediatr. 1963, 45, 656–683. [Google Scholar]
- Ward, O.C. A new familial cardiac syndrome in children. J. Ir. Med. Assoc. 1964, 54, 103–106. [Google Scholar]
- Napolitano, C.; Priori, S.G.; Schwartz, P.J.; Bloise, R.; Ronchetti, E.; Nastoli, J.; Bottelli, G.; Cerrone, M.; Leonardi, S. Genetic testing in the long QT syndrome: Development and validation of an efficient approach to genotyping in clinical practice. JAMA 2005, 294, 2975–2980. [Google Scholar] [CrossRef] [PubMed]
- Splawski, I.; Shen, J.; Timothy, K.W.; Lehmann, M.H.; Priori, S.; Robinson, J.L.; Moss, A.J.; Schwartz, P.J.; Towbin, J.A.; Vincent, G.M.; et al. Spectrum of mutations in long-QT syndrome genes: KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 2000, 102, 1178–1185. [Google Scholar] [CrossRef]
- Tester, D.J.; Will, M.L.; Haglund, C.M.; Ackerman, M.J. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005, 2, 507–517. [Google Scholar] [CrossRef]
- Landstrom, A.P.; Shah, S.H. Rare Things Being Common: Implications for Common Genetic Variants in Rare Diseases Like Long-QT Syndrome. Circulation 2020, 142, 339–341. [Google Scholar] [CrossRef]
- Al-Akchar, M.; Siddique, M.S. Long QT Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: https://www.ncbi.nlm.nih.gov/books/NBK441860/ (accessed on 26 December 2022).
- Wilde, A.A.; Jongbloed, R.J.; Doevendans, P.A.; Düren, D.R.; Hauer, R.N.; van Langen, I.M.; van Tintelen, J.P.; Smeets, H.J.; Meyer, H.; Geelen, J.L. Auditory stimuli as a trigger for arrhythmic events differentiate HERG-related (LQTS2) patients from KVLQT1-related patients (LQTS1). J. Am. Coll. Cardiol. 1999, 33, 327–332. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Priori, S.G.; Spazzolini, C.; Moss, A.J.; Vincent, G.M.; Napolitano, C.; Denjoy, I.; Guicheney, P.; Breithardt, G.; Keating, M.T.; et al. Genotype-phenotype correlation in the long-QT syndrome: Gene-specific triggers for life-threatening arrhythmias. Circulation 2001, 103, 89–95. [Google Scholar] [CrossRef]
- Tan, H.L.; Bardai, A.; Shimizu, W.; Moss, A.J.; Schulze-Bahr, E.; Noda, T.; Wilde, A.A. Genotype-specific onset of arrhythmias in congenital long-QT syndrome: Possible therapy implications. Circulation 2006, 114, 2096–2103. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Wang, Y.; Tseng, G.N. Adult Ventricular Myocytes Segregate KCNQ1 and KCNE1 to Keep the IKs Amplitude in Check Until When Larger IKs Is Needed. Circ. Arrhythm. Electrophysiol. 2017, 10, e005084. [Google Scholar] [CrossRef]
- Dotzler, S.M.; Kim, C.S.J.; Gendron, W.A.C.; Zhou, W.; Ye, D.; Bos, J.M.; Tester, D.J.; Barry, M.A.; Ackerman, M.J. Suppression-Replacement KCNQ1 Gene Therapy for Type 1 Long QT Syndrome. Circulation 2021, 143, 1411–1425. [Google Scholar] [CrossRef]
- Warmke, J.W.; Ganetzky, B. A family of potassium channel genes related to eag in Drosophila and mammals. Proc. Natl. Acad. Sci. USA 1994, 91, 3438–3442. [Google Scholar] [CrossRef] [PubMed]
- London, B.; Trudeau, M.C.; Newton, K.P.; Beyer, A.K.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Satler, C.A.; Robertson, G.A. Two isoforms of the mouse ether-a-go-go-related gene coassemble to form channels with properties similar to the rapidly activating component of the cardiac delayed rectifier K+ current. Circ. Res. 1997, 81, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.K.; Liu, F.; Vaidyanathan, R.; Eckhardt, L.L.; Trudeau, M.C.; Robertson, G.A. hERG 1b is critical for human cardiac repolarization. Proc. Natl. Acad. Sci. USA 2014, 111, 18073–18077. [Google Scholar] [CrossRef]
- Jones, E.M.; Roti Roti, E.C.; Wang, J.; Delfosse, S.A.; Robertson, G.A. Cardiac IKr channels minimally comprise hERG 1a and 1b subunits. J. Biol. Chem. 2004, 279, 44690–44694. [Google Scholar] [CrossRef]
- Lees-Miller, J.P.; Kondo, C.; Wang, L.; Duff, H.J. Electrophysiological characterization of an alternatively processed ERG K+ channel in mouse and human hearts. Circ. Res. 1997, 81, 719–726. [Google Scholar] [CrossRef]
- Sanguinetti, M.C.; Jiang, C.; Curran, M.E.; Keating, M.T. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 1995, 81, 299–307. [Google Scholar] [CrossRef]
- Spector, P.S.; Curran, M.E.; Zou, A.; Keating, M.T.; Sanguinetti, M.C. Fast incativation causes rectification of th eIkr channel. J. Gen. Physiol. 1996, 107, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Schönherr, R.; Heinemann, S.H. Molecular determinants for activation and inactivation of HERG, a human inward rectifier potassium channel. J. Physiol. 1996, 493, 635–642. [Google Scholar] [CrossRef]
- Wang, Q.; Shen, J.; Splawski, I.; Atkinson, D.; Li, Z.; Robinson, J.L.; Moss, A.J.; Towbin, J.A.; Keating, M.T. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 1995, 80, 805–811. [Google Scholar] [CrossRef]
- Remme, C.A. Cardiac sodium channelopathy associated with SCN5A mutations: Electrophysiological, molecular and genetic aspects. J. Physiol. 2013, 591, 4099–4116. [Google Scholar] [CrossRef]
- Sakaguchi, T.; Shimizu, W.; Itoh, H.; Noda, T.; Miyamoto, Y.; Nagaoka, I.; Oka, Y.; Ashihara, T.; Ito, M.; Tsuji, K.; et al. Age-and genotype-specific triggers for life-threatening arrhythmia in the genotyped long QT syndrome. J. Cardiovasc. Electrophysiol. 2008, 19, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, I.; Thottathil, P.; Lopes, C.M.; Twobin, J.A.; Vincet, M.; Barshesshet, A. Trigger-specific ion-channel mechanisms, risk factors, and response to therapy in type 1 long QT syndrome. Heart Rhythm 2012, 9, 49–56. [Google Scholar] [CrossRef]
- Shimizu, W.; Horie, M.; Ohno, S.; Takenaka, K.; Yamaguchi, M.; Shimizu, M.; Washizuka, T.; Aizawa, Y.; Nakamura, K.; Ohe, T.; et al. Mutation site-specific differences in arrhythmic risk and sensitivity to sympathetic stimulation in the LQT1 form of congenital long QT syndrome: Multicenter study in Japan. J. Am. Coll. Cardiol. 2004, 44, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Moss, A.J.; Shimizu, W.; Wilde, A.A.; Towbin, J.A.; Zareba, W.; Robinson, J.L.; Qi, M.; Vincent, G.M.; Ackerman, M.J.; Kaufman, E.S.; et al. Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation 2007, 115, 2481–2489. [Google Scholar] [CrossRef]
- Kim, J.A.; Lopes, C.M.; Moss, A.J.; McNitt, S.; Barsheshet, A.; Robinson, J.L.; Zareba, W.; Ackerman, M.J.; Kaufman, E.S.; Towbin, J.A.; et al. Trigger-specific risk factors and response to therapy in long QT syndrome type 2. Heart Rhythm 2010, 7, 1797–1805. [Google Scholar] [CrossRef]
- Shimizu, W.; Moss, A.J.; Wilde, A.A.; Towbin, J.A.; Ackerman, M.J.; January, C.T.; Tester, D.J.; Zareba, W.; Robinson, J.L.; Qi, M.; et al. Genotype-phenotype aspects of type 2 long QT syndrome. J. Am. Coll. Cardiol. 2009, 54, 2052–2062. [Google Scholar] [CrossRef]
- Moss, A.J.; Zareba, W.; Kaufman, E.S.; Gartman, E.; Peterson, D.R.; Benhorin, J.; Towbin, J.A.; Keating, M.T.; Priori, S.G.; Schwartz, P.J.; et al. Increased risk of arrhythmic events in long-QT syndrome with mutations in the pore region of the human ether-a-go-go-related gene potassium channel. Circulation 2002, 105, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hao, N.; Sander, J.W.; Lin, X.; Xiong, W.; Zhou, D. KCNH2 variants in a family with epilepsy and long QT syndrome: A case report and literature review. Epileptic Disord. 2023, 4, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Díez-Escuté, N.; Arbelo, E.; Martínez-Barrios, E.; Cerralbo, P.; Cesar, S.; Cruzalegui, J.; Chipa, F.; Fiol, V.; Zschaeck, I.; Hernández, C.; et al. Sex differences in long QT syndrome. Front. Cardiovasc. Med. 2023, 10, 1164028. [Google Scholar] [CrossRef] [PubMed]
- Okin, P.M.; Kligfield, P.; Prineas, R.J.; Grandits, G.; Rautaharju, P.M.; Cohen, J.D.; Crow, R.S.; Roman, M.J.; Schwartz, J.E.; Pickering, T.G.; et al. Gender-specific criteria and performance of the exercise electrocardiogram. Circulation 1995, 92, 1209–1216. [Google Scholar] [CrossRef]
- Stramba-Badiale, M.; Spagnolo, D.; Bosi, G.; Schwartz, P.J. Are gender differences in QTc present at birth? MISNES Investigators. Multicenter Italian Study on Neonatal Electrocardiography and Sudden Infant Death Syndrome. Am. J. Cardiol. 1995, 75, 1277–1278. [Google Scholar] [CrossRef]
- Vink, A.S.; Clur, S.B.; Wilde, A.A.M.; Blom, N.A. Effect of age and gender on the QTc-interval in healthy individuals and patients with long-QT syndrome. Trends Cardiovasc. Med. 2018, 28, 64–75. [Google Scholar] [CrossRef]
- Odening, K.E.; Choi, B.R.; Liu, G.X.; Hartmann, K.; Ziv, O.; Chaves, L.; Schofield, L.; Centracchio, J.; Zehender, M.; Peng, X.; et al. Estradiol promotes sudden cardiac death in transgenic long QT type 2 rabbits while progesterone is protective. Heart Rhythm 2012, 9, 8223–8232. [Google Scholar] [CrossRef]
- Nakamura, H.; Kurokawa, J.; Bai, C.X.; Asada, K.; Xu, J.; Oren, R.V.; Zhu, Z.I.; Clancy, C.E.; Isobe, M.; Furukawa, T. Progesterone regulates cardiac repolarization through a nongenomic pathway: An in vitro patch-clamp and computational modeling study. Circulation 2007, 116, 2913–2922. [Google Scholar] [CrossRef]
- Song, M.; Helguera, G.; Eghbali, M.; Zhu, N.; Zarei, M.M.; Olcese, R.; Toro, L.; Stefani, E. Remodeling of Kv4.3 potassium channel gene expression under the control of sex hormones. J. Biol. Chem. 2001, 276, 31883–31890. [Google Scholar] [CrossRef]
- Helguera, G.; Olcese, R.; Song, M.; Toro, L.; Stefani, E. Tissue specific regulation of Ca2+ channel protein expression by sex hormones. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2002, 1569, 59–66. [Google Scholar] [CrossRef]
- Moshal, K.S.; Zhang, Z.; Roder, K.; Kim, T.Y.; Cooper, L.; Patedakis Litvinov, B.; Lu, Y.; Reddy, V.; Terentyev, D.; Choi, B.R.; et al. Progesterone modulates SERCA2a expression and function in rabbit cardiomyocytes. Am. J. Physiol. Cell Physiol. 2014, 307, C1050–C1057. [Google Scholar] [CrossRef] [PubMed]
- Buber, J.; Mathew, J.; Moss, A.J.; Hall, W.J.; Barsheshet, A.; McNitt, S.; Robinson, J.L.; Zareba, W.; Ackerman, M.J.; Kaufman, E.S.; et al. Risk of recurrent cardiac events after onset of menopause in women with congenital long-QT syndrome types 1 and 2. Circulation 2011, 123, 2784–2791. [Google Scholar] [CrossRef] [PubMed]
- Odening, K.E.; Koren, G. How do sex hormones modify arrhythmogenesis in long QT syndrome? Sex hormone effects on arrhythmogenic substrate and triggered activity. Heart Rhythm 2014, 11, 2107–2115. [Google Scholar] [CrossRef]
- Rashba, E.J.; Zareba, W.; Moss, A.J.; Hall, W.J.; Robinson, J.; Locati, E.H.; Schwartz, P.J.; Andrews, M. Influence of pregnancy on the risk for cardiac events in patients with hereditary long QT syndrome. LQTS Investigators. Circulation 1998, 97, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Odening, K.E.; Koren, G.; Kirk, M. Normalization of QT interval duration in a long QT syndrome patient during pregnancy and the postpartum period due to sex hormone effects on cardiac repolarization. HeartRhythm Case Rep. 2016, 2, 223–227. [Google Scholar] [CrossRef]
- Cuneo, B.F.; Kaizer, A.M.; Clur, S.A.; Swan, H.; Herberg, U.; Winbo, A.; Rydberg, A.; Haugaa, K.; Etheridge, S.; Ackerman, M.J.; et al. Mothers with long QT syndrome are at increased risk for fetal death: Findings from a multicenter international study. Am. J. Obstet. Gynecol. 2020, 222, 263.e1–263.e11. [Google Scholar] [CrossRef]
- Neves, R.; Bains, S.; Bos, J.M.; MacIntyre, C.; Giudicessi, J.R.; Ackerman, M.J. Precision therapy in congenital long QT syndrome. Trends Cardiovasc. Med. 2024, 34, 39–47. [Google Scholar] [CrossRef]
- Wilde, A.A.M.; Amin, A.S.; Postema, P.G. Diagnosis, management and therapeutic strategies for congenital long QT syndrome. Heart 2022, 108, 332–338. [Google Scholar] [CrossRef]
- Salanti, G.; Ades, A.E.; Ioannidis, J.P. Graphical methods and numerical summaries for presenting results from multiple-treatment meta-analysis: An overview and tutorial. J. Clin. Epidemiol. 2011, 64, 163–171. [Google Scholar] [CrossRef]
- Abu-Zeitone, A.; Peterson, D.R.; Polonsky, B.; McNitt, S.; Moss, A.J. Efficacy of different beta-blockers in the treatment of long QT syndrome. J. Am. Coll. Cardiol. 2014, 64, 1352–1358. [Google Scholar] [CrossRef]
- Han, L.; Liu, F.; Li, Q.; Qing, T.; Zhai, Z.; Xia, Z.; Li, J. The Efficacy of Beta-Blockers in Patients With Long QT Syndrome 1-3 According to Individuals’ Gender, Age, and QTc Intervals: A Network Meta-analysis. Front. Pharmacol. 2020, 11, 579525. [Google Scholar] [CrossRef] [PubMed]
- Chockalingam, P.; Crotti, L.; Girardengo, G.; Johnson, J.N.; Harris, K.M.; van der Heijden, J.F.; Hauer, R.N.; Beckmann, B.M.; Spazzolini, C.; Rordorf, R.; et al. Not all beta-blockers are equal in the management of long QT syndrome types 1 and 2: Higher recurrence of events under metoprolol. J. Am. Coll. Cardiol. 2012, 60, 2092–2099. [Google Scholar] [CrossRef] [PubMed]
- Mazzanti, A.; Maragna, R.; Faragli, A.; Monteforte, N.; Bloise, R.; Memmi, M.; Novelli, V.; Baiardi, P.; Bagnardi, V.; Etheridge, S.P.; et al. Gene-Specific Therapy With Mexiletine Reduces Arrhythmic Events in Patients With Long QT Syndrome Type 3. J. Am. Coll. Cardiol. 2016, 67, 1053–1058. [Google Scholar] [CrossRef]
- Bos, J.M.; Crotti, L.; Rohatgi, R.K.; Castelletti, S.; Dagradi, F.; Schwartz, P.J.; Ackerman, M.J. Mexiletine Shortens the QT Interval in Patients With Potassium Channel-Mediated Type 2 Long QT Syndrome. Circ. Arrhythm. Electrophysiol. 2019, 12, e007280. [Google Scholar] [CrossRef] [PubMed]
- Dagres, N.; Chao, T.F.; Fenelon, G.; Aguinaga, L.; Benhayon, D.; Benjamin, E.J.; Bunch, T.J.; Chen, L.Y.; Chen, S.A.; Dar-rieux, F.; et al. ESC Scientific Document Group. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) expert consensus on arrhythmias and cognitive function: What is the best practice? J. Arrhythm. 2018, 20, 1399–1421. [Google Scholar]
- Cano, J.; Zorio, E.; Mazzanti, A.; Arnau, M.Á.; Trenor, B.; Priori, S.G.; Saiz, J.; Romero, L. Ranolazine as an Alternative Therapy to Flecainide for SCN5A V411M Long QT Syndrome Type 3 Patients. Front. Pharmacol. 2020, 11, 580481. [Google Scholar] [CrossRef]
- Cho, Y. Management of Patients with Long QT Syndrome. Korean Circ. J. 2016, 46, 747–752. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Priori, S.G.; Cerrone, M.; Spazzolini, C.; Odero, A.; Napolitano, C.; Bloise, R.; De Ferrari, G.M.; Klersy, C.; Moss, A.J.; et al. Left cardiac sympathetic denervation in the management of high-risk patients affected by the long-QT syndrome. Circulation 2004, 109, 1826–1833. [Google Scholar] [CrossRef]
- Dubey, N.; Ubhadiya, T.J.; Garg, V.S.; Vadnagara, H.; Sojitra, M.H.; Gandhi, S.K.; Patel, P. Unlocking the Potential of Left Cardiac Sympathetic Denervation: A Scoping Review of a Promising Approach for Long QT Syndrome. Cureus 2023, 15, e47306. [Google Scholar] [CrossRef]
- Zhu, W.; Bian, X.; Lv, J. From genes to clinical management: A comprehensive review of long QT syndrome pathogenesis and treatment. Heart Rhythm O2 2024, 5, 573–586. [Google Scholar] [CrossRef]
- Yu, Y.; Deschenes, I.; Zhao, M.T. Precision medicine for long QT syndrome: Patient-specific iPSCs take the lead. Expert Rev. Mol. Med. 2023, 25, e5. [Google Scholar] [CrossRef] [PubMed]
- Marcinkeviciene, A.; Rinkuniene, D.; Puodziukynas, A. Long QT Syndrome Management during and after Pregnancy. Medicina 2022, 58, 1694. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maričić, L.; Sušić, L.; Mihić, D.; Šego, N. Long QT Interval Syndrome and Female Sex—Review and Case Report. Reports 2025, 8, 32. https://doi.org/10.3390/reports8010032
Maričić L, Sušić L, Mihić D, Šego N. Long QT Interval Syndrome and Female Sex—Review and Case Report. Reports. 2025; 8(1):32. https://doi.org/10.3390/reports8010032
Chicago/Turabian StyleMaričić, Lana, Livija Sušić, Damir Mihić, and Nikolina Šego. 2025. "Long QT Interval Syndrome and Female Sex—Review and Case Report" Reports 8, no. 1: 32. https://doi.org/10.3390/reports8010032
APA StyleMaričić, L., Sušić, L., Mihić, D., & Šego, N. (2025). Long QT Interval Syndrome and Female Sex—Review and Case Report. Reports, 8(1), 32. https://doi.org/10.3390/reports8010032