
Dent Disease Type 1: Still an Under-Recognized Renal Proximal Tubulopathy: A Case Report


Abstract
1. Introduction
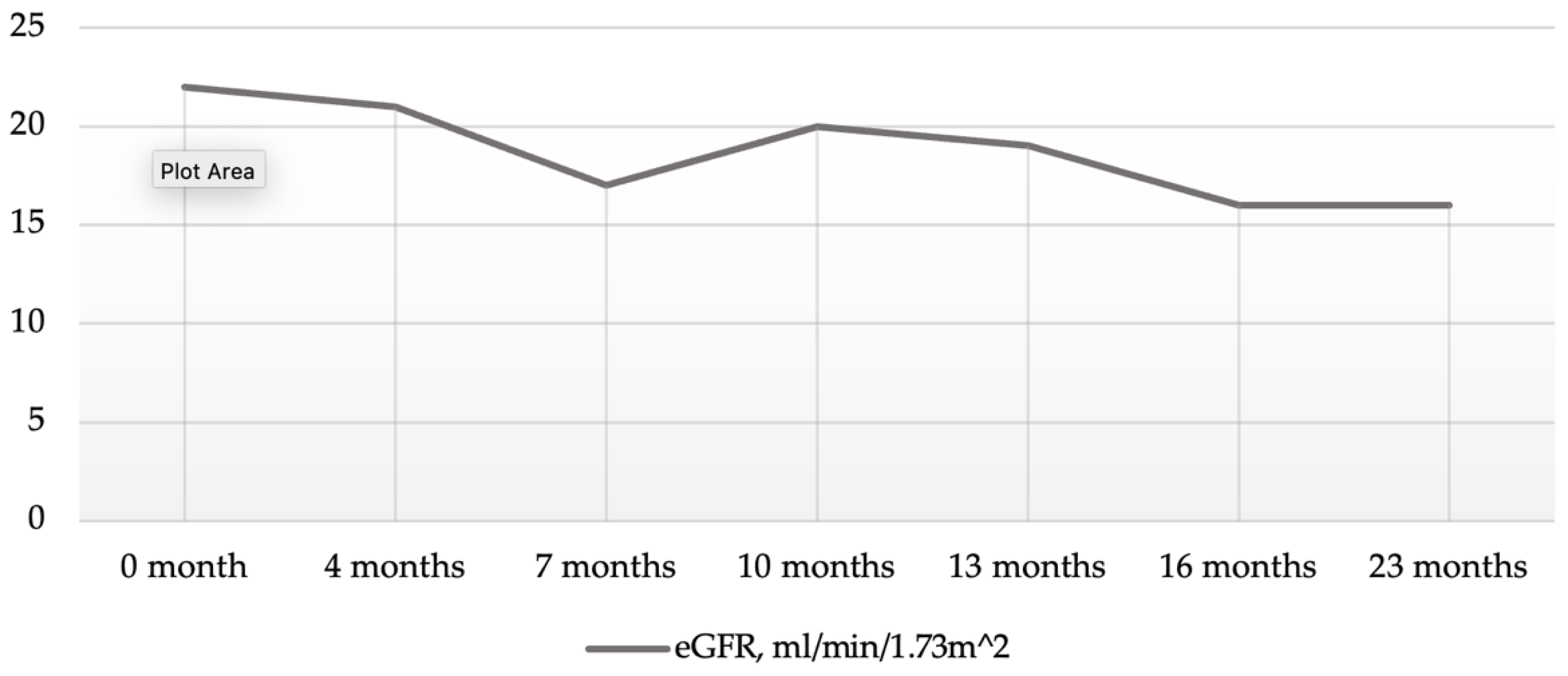
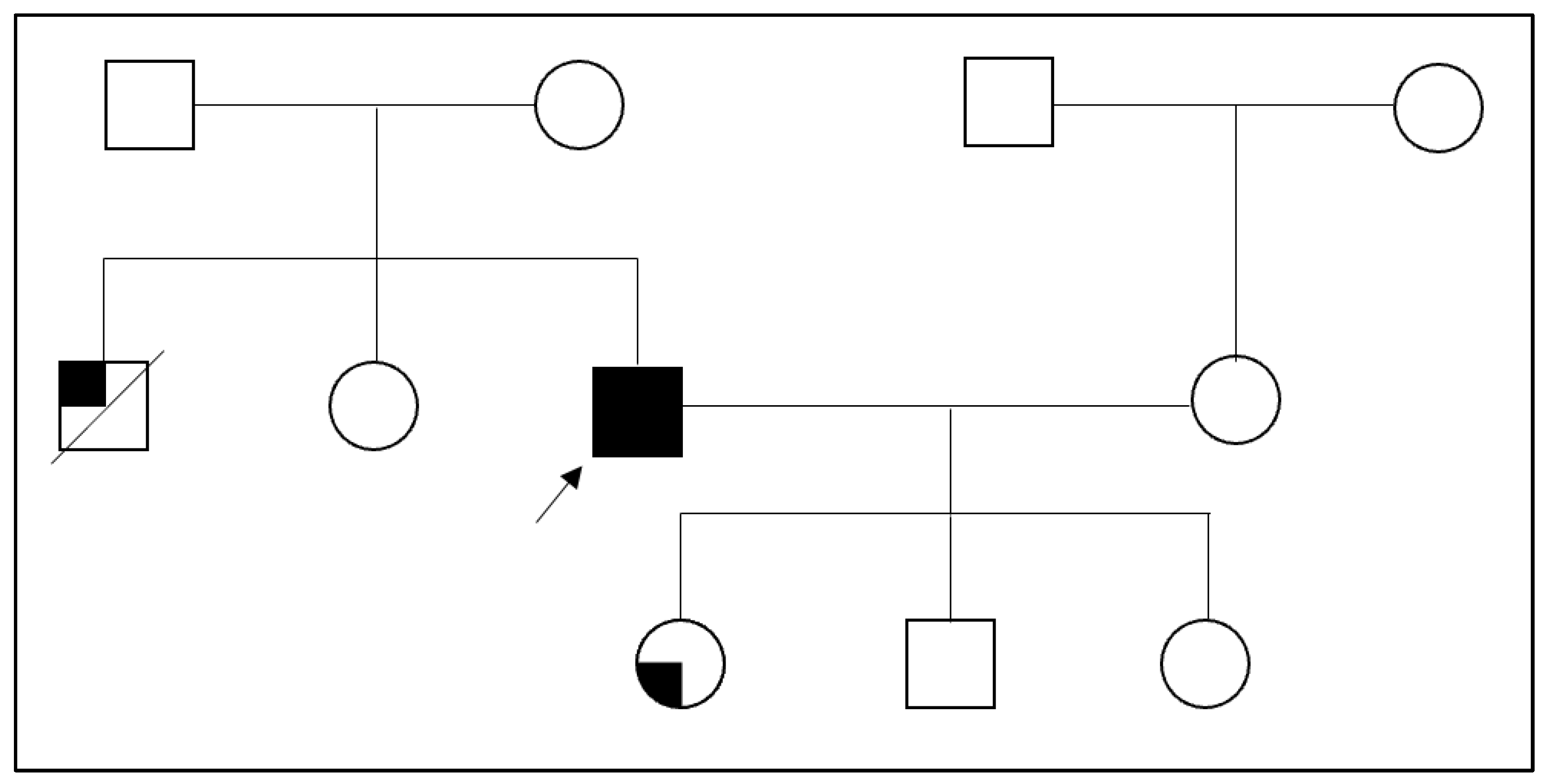
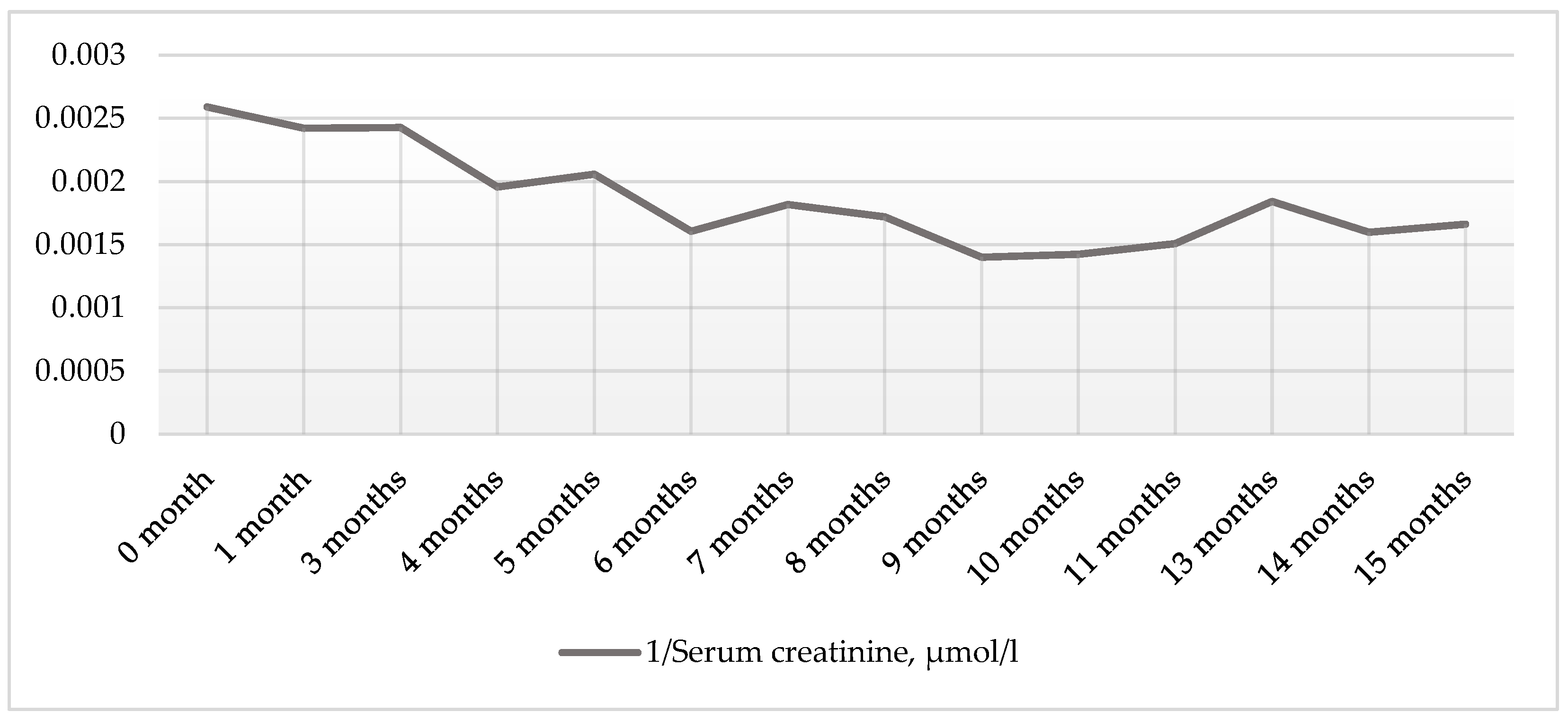
2. Case Presentation Section
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dent, C.E.; Friedman, M. Hypercalcuric Rickets Associated with Renal Tubular Damage. Arch. Dis. Child. 1964, 39, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Solano, A.; Lew, S.Q.; Ing, T.S. Dent–Wrong disease and other rare causes of the Fanconi syndrome. Clin. Kidney J. 2014, 7, 344–347. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Scheinman, S.J. X-linked hypercalciuric nephrolithiasis: Clinical syndromes and chloride channel mutations. Kidney Int. 1998, 53, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Devonald, M.A.J.; Karet, F.E. Renal Epithelial Traffic Jams and One-Way Streets. J. Am. Soc. Nephrol. 2004, 15, 1370–1381. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Devuyst, O.; Thakker, R.V. Dent’s disease. Orphanet J. Rare Dis. 2010, 5, 28. [Google Scholar] [CrossRef]
- Wrong, O.M.; Norden, A.G.; Feest, T.G. Dent’s disease: A familial proximal renal tubular syndrome with low-molecular-weight proteinuria, hypercalciuria, nephrocalcinosis, metabolic bone disease, progressive renal failure and a marked male predominance. QJM Mon. J. Assoc. Physicians 1994, 87, 473–493. [Google Scholar]
- Scheel, O.; Zdebik, A.A.; Lourdel, S.; Jentsch, T.J. Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature 2005, 436, 424–427. [Google Scholar] [CrossRef]
- Hoopes, R.R., Jr.; Shrimpton, A.E.; Knohl, S.J.; Hueber, P.; Hoppe, B.; Matyus, J.; Simckes, A.; Tasic, V.; Toenshoff, B.; Suchy, S.F.; et al. Dent Disease with Mutations in OCRL1. Am. J. Hum. Genet. 2005, 76, 260–267. [Google Scholar] [CrossRef]
- Lloyd, S.E.; Pearce, S.H.; Fisher, S.E.; Steinmeyer, K.; Schwappach, B.; Scheinman, S.J.; Harding, B.; Bolino, A.; Devoto, M.; Goodyer, P.; et al. A common molecular basis for three inherited kidney stone diseases. Nature 1996, 379, 445–449. [Google Scholar] [CrossRef]
- Wang, X.; Anglani, F.; Beara-Lasic, L.; Mehta, A.J.; Vaughan, L.E.; Herrera Hernandez, L.; Cogal, A.; Scheinman, S.J.; Ariceta, G.; Isom, R.; et al. Glomerular Pathology in Dent Disease and Its Association with Kidney Function. Clin. J. Am. Soc. Nephrol. CJASN 2016, 11, 2168–2176. [Google Scholar] [CrossRef]
- Ehlayel, A.M.; Copelovitch, L. Update on Dent Disease. Pediatr. Clin. 2019, 66, 169–178. [Google Scholar] [CrossRef]
- Böckenhauer, D.; Bökenkamp, A.; Nuutinen, M.; Unwin, R.; Van’t Hoff, W.; Sirimanna, T.; Vrljicak, K.; Ludwig, M. Novel OCRL mutations in patients with Dent-2 disease. J. Pediatr. Genet. 2012, 1, 15–23. [Google Scholar] [CrossRef]
- Zaniew, M.; Mizerska-Wasiak, M.; Załuska-Leśniewska, I.; Adamczyk, P.; Kiliś-Pstrusińska, K.; Haliński, A.; Zawadzki, J.; Lipska-Ziętkiewicz, B.S.; Pawlaczyk, K.; Sikora, P.; et al. Dent disease in Poland: What we have learned so far? Int. Urol. Nephrol. 2017, 49, 2005–2017. [Google Scholar] [CrossRef]
- Ludwig, M.; Utsch, B.; Monnens, L.A.H. Recent advances in understanding the clinical and genetic heterogeneity of Dent’s disease. Nephrol. Dial. Transplant. 2006, 21, 2708–2717. [Google Scholar] [CrossRef][Green Version]
- Beara-Lasic, L.; Cogal, A.; Mara, K.; Enders, F.; Mehta, R.A.; Haskic, Z.; Furth, S.L.; Trachtman, H.; Scheinman, S.J.; Milliner, D.S.; et al. Prevalence of low molecular weight proteinuria and Dent disease 1 CLCN5 mutations in proteinuric cohorts. Pediatr. Nephrol. 2020, 35, 633–640. [Google Scholar] [CrossRef]
- Gianesello, L.; Del Prete, D.; Anglani, F.; Calò, L.A. Genetics and phenotypic heterogeneity of Dent disease: The dark side of the moon. Hum. Genet. 2021, 140, 401–421. [Google Scholar] [CrossRef]
- Bhardwaj, S.; Thergaonkar, R.; Sinha, A.; Hari, P.; Hi, C.; Bagga, A. Phenotype of dent disease in a cohort of Indian children. Indian Pediatr. 2016, 53, 977–982. [Google Scholar] [CrossRef]
- Lloyd, S.E.; Pearce, S.H.; Günther, W.; Kawaguchi, H.; Igarashi, T.; Jentsch, T.J.; Thakker, R.V. Idiopathic low molecular weight proteinuria associated with hypercalciuric nephrocalcinosis in Japanese children is due to mutations of the renal chloride channel (CLCN5). J. Clin. Investig. 1997, 99, 967–974. [Google Scholar] [CrossRef]
- Zaniew, M.; Bökenkamp, A.; Kolbuc, M.; La Scola, C.; Baronio, F.; Niemirska, A.; Szczepanska, M.; Bürger, J.; La Manna, A.; Miklaszewska, M.; et al. Long-term renal outcome in children with OCRL mutations: Retrospective analysis of a large international cohort. Nephrol. Dial. Transplant. 2018, 33, 85–94. [Google Scholar]
- Blanchard, A.; Curis, E.; Guyon-Roger, T.; Kahila, D.; Treard, C.; Baudouin, V.; Bérard, E.; Champion, G.; Cochat, P.; Dubourg, J.; et al. Observations of a large Dent disease cohort. Kidney Int. 2016, 90, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Picollo, A.; Pusch, M. Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature 2005, 436, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Claverie-Martín, F.; Ramos-Trujillo, E.; García-Nieto, V. Dent’s disease: Clinical features and molecular basis. Pediatr. Nephrol. 2011, 26, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Poroca, D.R.; Pelis, R.M.; Chappe, V.M. ClC Channels and Transporters: Structure, Physiological Functions, and Implications in Human Chloride Channelopathies. Front. Pharmacol. 2017, 8, 151. [Google Scholar] [CrossRef]
- Jin, Y.Y.; Huang, L.M.; Quan, X.F.; Mao, J.H. Dent disease: Classification, heterogeneity and diagnosis. World J. Pediatr. 2021, 17, 52–57. [Google Scholar] [CrossRef]
- Gianesello, L.; Del Prete, D.; Ceol, M.; Priante, G.; Calò, L.A.; Anglani, F. From protein uptake to Dent disease: An overview of the CLCN5 gene. Gene 2020, 747, 144662. [Google Scholar] [CrossRef]
- Mansour-Hendili, L.; Blanchard, A.; Le Pottier, N.; Roncelin, I.; Lourdel, S.; Treard, C.; González, W.; Vergara-Jaque, A.; Morin, G.; Colin, E.; et al. Mutation Update of the CLCN5 Gene Responsible for Dent Disease 1. Hum. Mutat. 2015, 36, 743–752. [Google Scholar] [CrossRef]
- Günther, W.; Piwon, N.; Jentsch, T.J. The ClC-5 chloride channel knock-out mouse—An animal model for Dent’s disease. Pflüg. Arch. 2003, 445, 456–462. [Google Scholar] [CrossRef]
- Soares, R.B.; Bhat, N. Dent Disease Type 1: A Diagnostic Dilemma and Review. Cureus 2022, 14, e23910. [Google Scholar] [CrossRef]
- Sekine, T.; Komoda, F.; Miura, K.; Takita, J.; Shimadzu, M.; Matsuyama, T.; Ashida, A.; Igarashi, T. Japanese Dent disease has a wider clinical spectrum than Dent disease in Europe/USA: Genetic and clinical studies of 86 unrelated patients with low-molecular-weight proteinuria. Nephrol. Dial. Transplant. 2014, 29, 376–384. [Google Scholar] [CrossRef]
- Akuta, N.; Lloyd, S.E.; Igarashi, T.; Shiraga, H.; Matsuyama, T.; Yokoro, S.; Cox, J.P.; Thakker, R.V. Mutations of CLCN5 in Japanese children with idiopathic low molecular weight proteinuria, hypercalciuria and nephrocalcinosis. Kidney Int. 1997, 52, 911–916. [Google Scholar] [CrossRef]
- Scheinman, S.J. Nephrolithiasis. Semin. Nephrol. 1999, 19, 381–388. [Google Scholar]
- Frishberg, Y.; Dinour, D.; Belostotsky, R.; Becker-Cohen, R.; Rinat, C.; Feinstein, S.; Navon-Elkan, P.; Ben-Shalom, E. Dent’s disease manifesting as focal glomerulosclerosis: Is it the tip of the iceberg? Pediatr. Nephrol. 2009, 24, 2369–2373. [Google Scholar] [CrossRef]
- Van Berkel, Y.; Ludwig, M.; van Wijk, J.A.E.; Bökenkamp, A. Proteinuria in Dent disease: A review of the literature. Pediatr. Nephrol. 2017, 32, 1851–1859. [Google Scholar] [CrossRef]
- Copelovitch, L.; Nash, M.A.; Kaplan, B.S. Hypothesis: Dent Disease Is an Underrecognized Cause of Focal Glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2007, 2, 914–918. [Google Scholar] [CrossRef]
- Fervenza, F.C. A Patient with Nephrotic-Range Proteinuria and Focal Global Glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2013, 8, 1979–1987. [Google Scholar] [CrossRef]
- Yanagida, H.; Ikeoka, M.; Kuwajima, H.; Wada, N.; Tabata, N.; Sugimoto, K.; Okada, M.; Takemura, T. A boy with Japanese Dent’s disease exhibiting abnormal calcium metabolism and osseous disorder of the spine: Defective megalin expression at the brushborder of renal proximal tubules. Clin. Nephrol. 2004, 62, 306–312. [Google Scholar] [CrossRef]
- Raja, K.A.; Schurman, S.; D’mello, R.G.; Blowey, D.; Goodyer, P.; Van Why, S.; Ploutz-Snyder, R.J.; Asplin, J.; Scheinman, S.J. Responsiveness of Hypercalciuria to Thiazide in Dent’s Disease. J. Am. Soc. Nephrol. 2002, 13, 2938–2944. [Google Scholar] [CrossRef]
- Güngör, T.; Eroğlu, F.K.; Yazılıtaş, F.; Gür, G.; Çakıcı, E.K.; Ludwig, M.; Bülbül, M. A case of Type 1 Dent disease presenting with isolated persistent proteinuria. Turk. Arch. Pediatr. Pediatri Arş. 2020, 55, 72–75. [Google Scholar]
- Blanchard, A.; Vargas-Poussou, R.; Peyrard, S.; Mogenet, A.; Baudouin, V.; Boudailliez, B.; Charbit, M.; Deschesnes, G.; Ezzhair, N.; Loirat, C.; et al. Effect of hydrochlorothiazide on urinary calcium excretion in dent disease: An uncontrolled trial. Am. J. Kidney Dis. 2008, 52, 1084–1095. [Google Scholar] [CrossRef]
- Deng, H.; Zhang, Y.; Xiao, H.; Yao, Y.; Zhang, H.; Liu, X.; Su, B.; Guan, N.; Zhong, X.; Wang, S.; et al. Phenotypic spectrum and antialbuminuric response to angiotensin converting enzyme inhibitor and angiotensin receptor blocker therapy in pediatric Dent disease. Mol. Genet. Genom. Med. 2020, 8, e1306. [Google Scholar] [CrossRef]
- Gambaro, G.; Naticchia, A.; Ferraro, P.M.; Spagnoletti, G.; Romagnoli, J.; Salerno, M.P.; Citterio, F. Living Kidney Donation in a Type 1 Dent’s Disease Patient from His Mother. Kidney Blood Press. Res. 2019, 44, 1306–1312. [Google Scholar] [CrossRef]
- Edvardsson, V.O.; Goldfarb, D.S.; Lieske, J.C.; Beara-Lasic, L.; Anglani, F.; Milliner, D.S.; Palsson, R. Hereditary causes of kidney stones and chronic kidney disease. Pediatr. Nephrol. 2013, 28, 1923–1942. [Google Scholar] [CrossRef]
- Alhasan, K.; D’Alessandri-Silva, C.; Mongia, A.; Topaloglu, R.; Tasic, V.; Filler, G. Young Adults with Hereditary Tubular Diseases: Practical Aspects for Adult-Focused Colleagues. Adv. Chronic Kidney Dis. 2022, 29, 292–307. [Google Scholar] [CrossRef]
- Cebotaru, V.; Kaul, S.; Devuyst, O.; Cai, H.; Racusen, L.; Guggino, W.B.; Guggino, S.E. High citrate diet delays progression of renal insufficiency in the ClC-5 knockout mouse model of Dent’s disease. Kidney Int. 2005, 68, 642–652. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Value | Reference Range | |
|---|---|---|
| Complete blood count | ||
| White blood cell, ×109/L | 5.99 | 4.0–9.8 |
| Hemoglobin, g/L | 143 | 128–160 |
| Platelet, ×109/L | 213 | 140–450 |
| Arterial blood gas | ||
| pH | 7.386 | 7.35–7.45 |
| pCO2, mmHg | 37.4 | 35–48 |
| pO2, mmHg | 74.8 | 83–108 |
| HCO3−, mmol/L | 21.9 | 21–28 |
| Actual base excess, mmol/L | −2.1 | −2–+3 |
| Biochemistry | ||
| Serum creatinine, μmol/L | 366 | 64–104 |
| Urea, mmol/L | 9.8 | 2.5–7.5 |
| Uric acid, μmol/L | 304 | 208–428 |
| Total protein, g/L | 67.7 | 66–83 |
| Albumin, g/L | 41.4 | 36–52 |
| C-reactive protein, mg/L | 1.68 | ≤5 |
| Potassium, mmol/L | 4.7 | 3.8–5.3 |
| Sodium, mmol/L | 145 | 134–148 |
| Calcium, mmol/L | 2.18 | 2.10–2.55 |
| Phosphorus, mmol/L | 0.85 | 0.74–1.52 |
| Magnesium, mmol/L | 0.82 | 0.65–1.05 |
| Parathyroid hormone | 31.05 | 1.6–7.3 |
| 25-hydroxyvitamin D, nmol/L | 27.3 | 75–100 |
| Cholesterol, mmol/L | 9.62 | <5.2 |
| Triglycerides, mmol/L | 4.98 | ≤1.8 |
| High-density lipoprotein cholesterol, mmol/L | 1.13 | >0.91 |
| Low-density lipoprotein cholesterol, mmol/L | 5.46 | 2.6–3.5 |
| Value | Reference Range | |
|---|---|---|
| Urinalysis | ||
| pH | 6.5 | 4.0–8.0 |
| Urine-specific gravity | 1.004 | 1.010–1.030 |
| Urine protein, g/L | 0.7 (1+) | ≤0.15 |
| Urine glucose, mmol/L | 0 | ≤2.8 |
| Urine blood, mg/L | 0.6 (1+) | ≤0.6 |
| Urine white blood cells, /μL | 0 | ≤25 |
| Urine chemistries | ||
| Urine total protein, g/L | 0.793 | <0.11 |
| 24 h urine total protein, g/24 h | 6.106 | <0.15 |
| Urine albumin, mg/L | 456 | <20 |
| 24 h urine albumin, mg/24 h | 3511.2 | <30 |
| Albumin–creatinine ratio, mg/mmol | 233.85 | <2.5 |
| Urine potassium, mmol/L | 13.4 | |
| 24 h urine potassium, mmol/24 h | 103.18 | 25–125 |
| Urine sodium, mmol/L | 44 | |
| 24 h urine sodium, mmol/24 h | 338.8 | 40–220 |
| Urine calcium, mmol/L | 0.54 | |
| 24 h urine calcium, mmol/24 h | 4.158 | 2.5–7.5 |
| Urine phosphorus, mmol/L | 4.44 | |
| 24 h urine phosphorus, mmol/L | 34.188 | 16.5–48.5 |
| Urine magnesium, mmol/L | 0.83 | |
| 24 h urine magnesium, mmol/24 h | 6.391 | 3.0–5.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vitkauskaitė, M.; Čerkauskaitė, A.; Miglinas, M. Dent Disease Type 1: Still an Under-Recognized Renal Proximal Tubulopathy: A Case Report. Reports 2022, 5, 50. https://doi.org/10.3390/reports5040050
Vitkauskaitė M, Čerkauskaitė A, Miglinas M. Dent Disease Type 1: Still an Under-Recognized Renal Proximal Tubulopathy: A Case Report. Reports. 2022; 5(4):50. https://doi.org/10.3390/reports5040050
Chicago/Turabian StyleVitkauskaitė, Monika, Agnė Čerkauskaitė, and Marius Miglinas. 2022. "Dent Disease Type 1: Still an Under-Recognized Renal Proximal Tubulopathy: A Case Report" Reports 5, no. 4: 50. https://doi.org/10.3390/reports5040050
APA StyleVitkauskaitė, M., Čerkauskaitė, A., & Miglinas, M. (2022). Dent Disease Type 1: Still an Under-Recognized Renal Proximal Tubulopathy: A Case Report. Reports, 5(4), 50. https://doi.org/10.3390/reports5040050