Schizophrenic Psychosis Symptoms in a Background of Mild-To-Moderate Carnitine Palmitoyltransferase II Deficiency: A Case Report

 ,
, 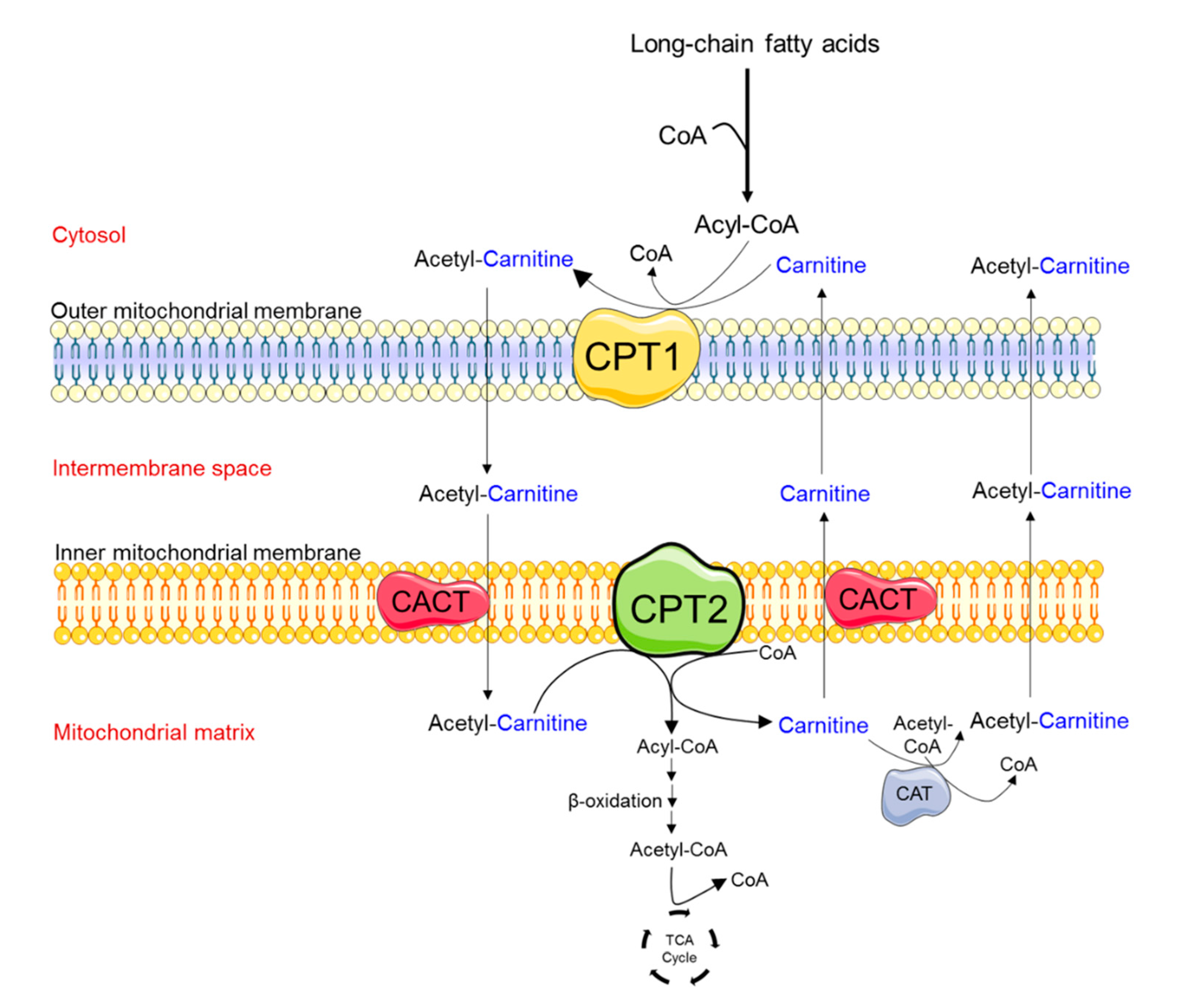{kind=link}
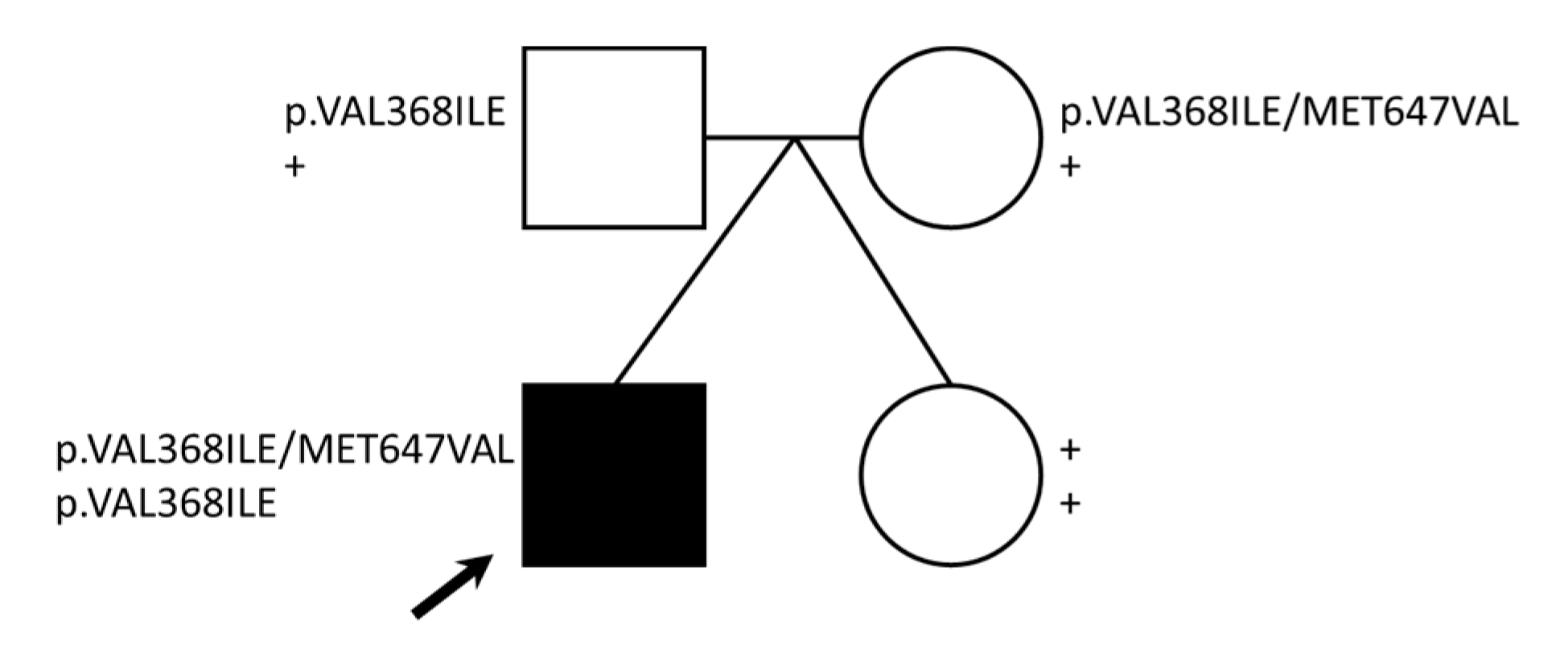{kind=link}
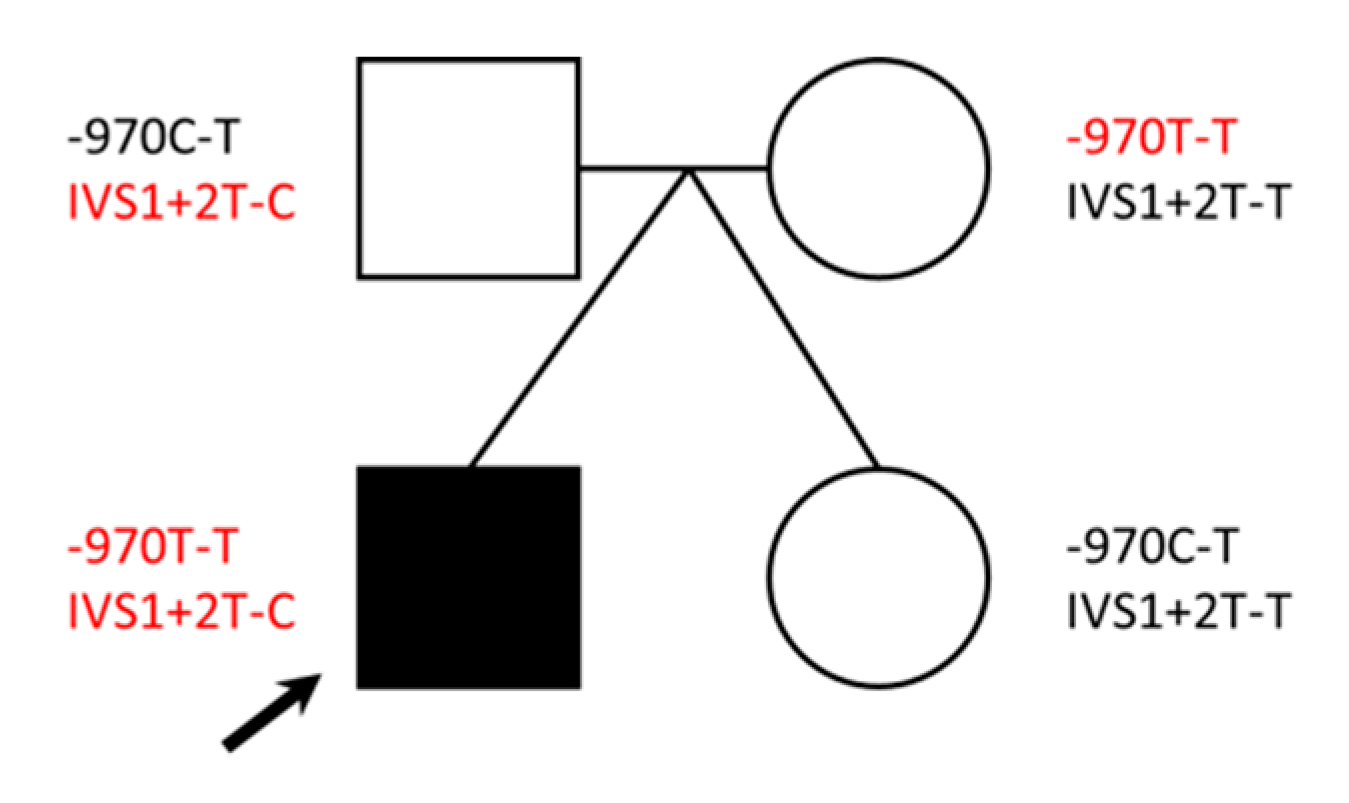{kind=link}
Abstract
1. Introduction
2. Case Presentation
2.1. Clinical Course
2.2. Family-Based Sequencing Identifies Compound CPT2 Variation in the Proband
2.3. Identification of DBH Compound Heterozygous Variants
2.4. Polygenic Risk Likely Underlies Schizophrenia Diagnosis in This Case
2.5. Incidental Findings
3. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Elia, M.; Klepper, J.; Leiendecker, B.; Hartmann, H. Ketogenic Diets in the Treatment of Epilepsy. Curr. Pharm. Des. 2018, 23, 5691–5701. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Prasad, A.N. Inborn Errors of Metabolism and Epilepsy: Current Understanding, Diagnosis, and Treatment Approaches. Int. J. Mol. Sci. 2017, 18, 1384. [Google Scholar] [CrossRef] [PubMed]
- Schönfeld, P.; Reiser, G. Brain Energy Metabolism Spurns Fatty Acids as Fuel Due to Their Inherent Mitotoxicity and Potential Capacity to Unleash Neurodegeneration. Neurochem. Int. 2017, 109, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Romano, A.; Koczwara, J.B.; Gallelli, C.A.; Vergara, D.; Di Bonaventura, M.V.M.; Gaetani, S.; Giudetti, A.M. Fats for Thoughts: An Update on Brain Fatty Acid Metabolism. Int. J. Biochem. Cell Biol. 2017, 84, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Kerner, J.; Hoppel, C.L. Mitochondrial Carnitine Palmitoyltransferase 1a (CPT1a) Is Part of an Outer Membrane Fatty Acid Transfer Complex. J. Biol. Chem. 2011, 286, 25655–25662. [Google Scholar] [CrossRef] [PubMed]
- Longo, N. Primary Carnitine Deficiency and Newborn Screening for Disorders of the Carnitine Cycle. Ann. Nutr. Metab. 2016, 68, 5–9. [Google Scholar] [CrossRef]
- Jones, L.L.; McDonald, D.A.; Borum, P.R. Acylcarnitines: Role in Brain. Prog. Lipid Res. 2010, 49, 61–75. [Google Scholar] [CrossRef]
- Jernberg, J.N.; Bowman, C.E.; Wolfgang, M.J.; Scafidi, S. Developmental Regulation and Localization of Carnitine Palmitoyltransferases (CPTs) in Rat Brain. J. Neurochem. 2017, 142, 407–419. [Google Scholar] [CrossRef]
- Waisbren, S.E.; Landau, Y.; Wilson, J.; Vockley, J. Neuropsychological Outcomes in Fatty Acid Oxidation Disorders: 85 Cases Detected by Newborn Screening. Dev. Disabil. Res. Rev. 2013, 17, 260–268. [Google Scholar] [CrossRef]
- Malik, S.; Paldiwal, A.A.; Korday, C.S.; Jadhav, S.S. Neonatal Carnitine Palmitoyltransferase II Deficiency: A Lethal Entity. J. Clin. Diagn. Res. 2015, 9, SD01–SD02. [Google Scholar] [CrossRef]
- Roe, C.R.; Yang, B.-Z.; Brunengraber, H.; Roe, D.S.; Wallace, M.; Garritson, B.K. Carnitine Palmitoyltransferase II Deficiency: Successful Anaplerotic Diet Therapy. Neurology 2008, 71, 260–264. [Google Scholar] [CrossRef]
- Brown, A.; Crowe, L.M.; Andresen, B.S.; Anderson, V.A.; Boneh, A. Neurodevelopmental profiles of Children with Very Long Chain Acyl-CoA Dehydrogenase Deficiency Diagnosed by Newborn Screening. Mol. Genet. Metab. 2014, 113, 278–282. [Google Scholar] [CrossRef]
- Bozzi, Y.; Provenzano, G.; Casarosa, S. Neurobiological Bases of Autism-Epilepsy Comorbidity: A Focus on Excitation/Inhibition Imbalance. Eur. J. Neurosci. 2018, 47, 534–548. [Google Scholar] [CrossRef]
- Rinaldo, P.; Cowan, T.M.; Matern, D. Acylcarnitine Profile Analysis. Genet. Med. 2008, 10, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Topcu, Y.; Bayram, E.; Karaoglu, P.; Yis, U.; Kurul, S.; Kurul, S.H. Importance of Acylcarnitine Profile Analysis for Disorders of Lipid Metabolism in Adolescent Patients With Recurrent Rhabdomyolysis: Report of Two Cases. Ann. Indian Acad. Neurol. 2014, 17, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Demaugre, F.; Bonnefont, J.-P.; Cépanec, C.; Scholte, J.; Saudubray, J.-M.; Leroux, J.-P. Immunoquantitative Analysis of Human Carnitine Palmitoyltransferase I and II Defects. Pediatr. Res. 1990, 27, 497–500. [Google Scholar] [CrossRef]
- Demaugre, F.; Bonnefont, J.P.; Colonna, M.; Cepanec, C.; Leroux, J.P.; Saudubray, J.M. Infantile Form of Carnitine Palmitoyltransferase II Deficiency with Hepatomuscular Symptoms and Sudden Death. Physiopathological Approach to Carnitine Palmitoyltransferase II Deficiencies. J. Clin. Investig. 1991, 87, 859–864. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhu, Q.; Hu, Q.; Shepherd, L.; Wang, J.; Wei, L.; Morrison, C.D.; Conroy, J.M.; Glenn, S.T.; Davis, W.; Kwan, M.L.; et al. The Impact of DNA Input Amount and DNA Source on the Performance of Whole-Exome Sequencing in Cancer Epidemiology. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1207–1213. [Google Scholar] [CrossRef]
- Ibrahim, O.; Sutherland, H.G.; Haupt, L.M.; Griffiths, L.R. Saliva as a Comparable-Quality Source of DNA for Whole Exome Sequencing on Ion Platforms. Genomics 2020, 112, 1437–1443. [Google Scholar] [CrossRef] [PubMed]
- Feliciano, P.; The SPARK Consortium; Zhou, X.; Astrovskaya, I.; Turner, T.N.; Wang, T.; Brueggeman, L.; Barnard, R.; Hsieh, A.; Snyder, L.G.; et al. Exome Sequencing of 457 Autism Families Recruited Online Provides Evidence for Autism Risk Genes. NPJ Genom. Med. 2019, 4, 1–14. [Google Scholar] [CrossRef]
- Nohesara, S.; Ghadirivasfi, M.; Mostafavi, S.; Eskandari, M.-R.; Ahmadkhaniha, H.; Thiagalingam, S.; Abdolmaleky, H.M. DNA Hypomethylation of MB-COMT Promoter in the DNA Derived from Saliva in Schizophrenia and Bipolar Disorder. J. Psychiatr. Res. 2011, 45, 1432–1438. [Google Scholar] [CrossRef]
- Ghadirivasfi, M.; Nohesara, S.; Ahmadkhaniha, H.; Eskandari, M.-R.; Mostafavi, S.; Thiagalingam, S.; Abdolmaleky, H.M. Hypomethylation of the Serotonin Receptor Type-2A Gene (HTR2A) at T102C Polymorphic Site in DNA Derived From the Saliva of Patients With Schizophrenia and Bipolar Disorder. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2011, 156, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Epi25 Collaborative. Epi25 Collaborative Ultra-Rare Genetic Variation in the Epilepsies: A Whole-Exome Sequencing Study of 17,606 Individuals. Am. J. Hum. Genet. 2019, 105, 267–282. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Mizuguchi, H.; Yamaguchi, M.; Yamada, H.; Chida, J.; Shikata, K.; Kido, H. Thermal Instability of Compound Variants of Carnitine Palmitoyltransferase II and Impaired Mitochondrial Fuel Utilization in Influenza-Associated Encephalopathy. Hum. Mutat. 2008, 29, 718–727. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Saitoh, M.; Takanashi, J.-I.; Yamanouchi, H.; Kubota, M.; Goto, T.; Kikuchi, M.; Shiihara, T.; Yamanaka, G.; Mizuguchi, M. Carnitine Palmitoyl Transferase II Polymorphism Is Associated with Multiple Syndromes of Acute Encephalopathy with Various Infectious Diseases. Brain Dev. 2011, 33, 512–517. [Google Scholar] [CrossRef]
- Mak, C.M.; Lam, C.-W.; Fong, N.-C.; Siu, W.-K.; Lee, H.-C.H.; Siu, T.-S.; Lai, C.-K.; Law, C.-Y.; Tong, S.-F.; Poon, W.-T.; et al. Fatal Viral Infection-Associated Encephalopathy in Two Chinese Boys: A Genetically Determined Risk Factor of Thermolabile Carnitine Palmitoyltransferase II Variants. J. Hum. Genet. 2011, 56, 617–621. [Google Scholar] [CrossRef]
- Chen, Y.; Mizuguchi, H.; Yao, D.; Ide, M.; Kuroda, Y.; Shigematsu, Y.; Yamaguchi, S.; Kinoshita, M.; Kido, H.; Yamaguchi, M. Thermolabile Phenotype of Carnitine Palmitoyltransferase II Variations as a Predisposing Factor for Influenza-Associated Encephalopathy. FEBS Lett. 2005, 579, 2040–2044. [Google Scholar] [CrossRef]
- Brown, N.F.; Mullur, R.S.; Subramanian, I.; Esser, V.; Bennett, M.J.; Saudubray, J.M.; Feigenbaum, A.S.; Kobari, J.A.; MacLeod, P.M.; McGarry, J.D.; et al. Molecular Characterization of L-CPT I Deficiency in Six Patients: Insights Into Function of the Native Enzyme. J. Lipid Res. 2001, 42, 1134–1142. [Google Scholar]
- Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kim, C.-H.; Leung, A.; Huh, Y.H.; Yang, E.; Kim, D.-J.; Leblanc, P.; Ryu, H.; Kim, K.; Kim, D.-W.; Garland, E.M.; et al. Norepinephrine Deficiency Is Caused by Combined Abnormal mRNA Processing and Defective Protein Trafficking of Dopamine β-Hydroxylase. J. Biol. Chem. 2011, 286, 9196–9204. [Google Scholar] [CrossRef]
- Kim, C.-H.; Zabetian, C.P.; Cubells, J.F.; Cho, S.; Biaggioni, I.; Cohen, B.M.; Robertson, D.; Kim, K.-S. Mutations in the Dopamine Beta-Hydroxylase Gene Are Associated with Human Norepinephrine Deficiency. Am. J. Med. Genet. 2002, 108, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Zabetian, C.P.; Romero, R.; Robertson, D.; Sharma, S.; Padbury, J.F.; Kuivaniemi, H.; Kim, K.-S.; Kim, C.-H.; Köhnke, M.D.; Kranzler, H.R.; et al. A Revised Allele Frequency Estimate and Haplotype Analysis of the DBH Deficiency Mutation IVS1+2T → C in African- and European-Americans. Am. J. Med. Genet. Part A 2003, 123, 190–192. [Google Scholar] [CrossRef]
- Deinum, J.; Steenbergen-Spanjers, G.C.H.; Jansen, M.; Boomsma, F.; Lenders, J.W.M.; Van Ittersum, F.J.; Hück, N.; van den Heuvel, L.P.; Wevers, R.A. DBH Gene Variants That Cause Low Plasma Dopamine Beta Hydroxylase with or Without a Severe Orthostatic Syndrome. J. Med. Genet. 2004, 41, e38. [Google Scholar] [CrossRef]
- Jepma, M.; Deinum, J.; Asplund, C.L.; Rombouts, S.A.; Tamsma, J.T.; Tjeerdema, N.; Spapé, M.M.; Garland, E.M.; Robertson, D.; Lenders, J.W.; et al. Neurocognitive Function in Dopamine-β-Hydroxylase Deficiency. Neuropsychopharmacology 2011, 36, 1608–1619. [Google Scholar] [CrossRef] [PubMed]
- Arnold, A.C.; Garland, E.M.; Celedonio, J.E.; Raj, S.R.; Abumrad, N.N.; Biaggioni, I.; Robertson, D.; Luther, J.M.; Shibao, C.A. Hyperinsulinemia and Insulin Resistance in Dopamine β-Hydroxylase Deficiency. J. Clin. Endocrinol. Metab. 2016, 102, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Zabetian, C.P.; Anderson, G.M.; Buxbaum, S.G.; Elston, R.C.; Ichinose, H.; Nagatsu, T.; Kim, K.S.; Kim, C.H.; Malison, R.T.; Gelernter, J.; et al. A Quantitative-Trait Analysis of Human Plasma-Dopamine Beta-Hydroxylase Activity: Evidence for a Major Functional Polymorphism at the DBH Locus. Am. J. Hum. Genet. 2001, 68, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Senard, J.-M.; Rouet, P. Dopamine Beta-Hydroxylase Deficiency. Orphanet J. Rare Dis. 2006, 1, 7. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, X.; Liu, J.; Luo, X.-J.; Yao, Y.-G. SZDB2.0: An Updated Comprehensive Resource for Schizophrenia Research. Hum. Genet. 2020, 139, 1285–1297. [Google Scholar] [CrossRef]
- Howrigan, D.P.; Rose, S.A.; Samocha, K.E.; Fromer, M.; Cerrato, F.; Chen, W.J.; Churchhouse, C.; Chambert, K.; Chandler, S.D.; Daly, M.J.; et al. Exome Sequencing in Schizophrenia-Affected Parent–Offspring Trios Reveals Risk Conferred by Protein-Coding de Novo Mutations. Nat. Neurosci. 2020, 23, 185–193. [Google Scholar] [CrossRef]
- Key Statistics for Colorectal Cancer. Available online: https://www.cancer.org/cancer/colon-rectal-cancer/about/key-statistics.html (accessed on 8 September 2020).
- Pearlman, R.; Frankel, W.L.; Swanson, B.; Zhao, W.; Yilmaz, A.; Miller, K.; Bacher, J.; Bigley, C.; Nelsen, L.; Goodfellow, P.J.; et al. Ohio Colorectal Cancer Prevention Initiative Study Group Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients with Early-Onset Colorectal Cancer. JAMA Oncol. 2017, 3, 464–471. [Google Scholar] [CrossRef]
- Al-Mubarak, B.; Abouelhoda, M.; Omar, A.; AlDhalaan, H.; Aldosari, M.; Nester, M.; Alshamrani, H.A.; El-Kalioby, M.; Goljan, E.; Albar, R.; et al. Whole Exome Sequencing Reveals Inherited and de Novo Variants in Autism Spectrum Disorder: A Trio Study From Saudi Families. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hosseini Bereshneh, A.; Garshasbi, M. Novel in-Frame Deletion in MFSD8 Gene Revealed by Trio Whole Exome Sequencing in an Iranian Affected with Neuronal Ceroid Lipofuscinosis Type 7: A Case Report. J. Med. Sci. Rep. 2018, 12, 281. [Google Scholar] [CrossRef]
- Lee, I.-C.; Chang, T.-M.; Liang, J.-S.; Li, S.-Y. KCNQ2 Mutations in Childhood Nonlesional Epilepsy: Variable Phenotypes and a Novel Mutation in a Case Series. Mol. Genet. Genom. Med. 2019, 7, e00816. [Google Scholar] [CrossRef]
- Wang, D.; Sun, X.; Maziade, M.; Mao, W.; Zhang, C.; Lu, Q.; Cao, B. Characterising Phospholipids and Free Fatty Acids in Patients with Schizophrenia: A Case-Control Study. World J. Biol. Psychiatry 2020, 1–14. [Google Scholar] [CrossRef]
- Sachdev, P. Schizophrenia-Like Psychosis and Epilepsy: The Status of the Association. Am. J. Psychiatry 1998, 155, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Biervert, C.; Schroeder, B.C.; Kubisch, C.; Berkovic, S.F.; Propping, P.; Jentsch, T.J.; Steinlein, O.K. A Potassium Channel Mutation in Neonatal Human Epilepsy. Science 1998, 279, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Castaldo, P.; del Giudice, E.M.; Coppola, G.; Pascotto, A.; Annunziato, L.; Taglialatela, M. Benign Familial Neonatal Convulsions Caused by Altered Gating of KCNQ2/KCNQ3 Potassium Channels. J. Neurosci. 2002, 22, RC199. [Google Scholar] [CrossRef]
- Wang, Q.; Teng, P.; Luan, G. Schizophrenia-Like Psychosis of Epilepsy: From Clinical Characters to Underlying Mechanisms. Neuropsychiatry 2017, 7. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.E.; Cummings, B.B.; et al. The ExAC Browser: Displaying Reference Data Information From Over 60 000 Exomes. Nucleic Acids Res. 2016, 45, D840–D845. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting Amino Acid Changes That Affect Protein Function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7.20.1–7.20.41. [Google Scholar] [CrossRef] [PubMed]
- Olpin, S.E.; Afifi, A.; Clark, S.; Manning, N.J.; Bonham, J.R.; Dalton, A.; Leonard, J.V.; Land, J.M.; Andresen, B.S.; Morris, A.A.; et al. Mutation and Biochemical Analysis in Carnitine Palmitoyltransferase Type II (CPT II) Deficiency. J. Inherit. Metab. Dis. 2003, 26, 543–557. [Google Scholar] [CrossRef]
- Prabakaran, S.; Swatton, J.E.; Ryan, M.M.; Huffaker, S.J.; Huang, J.T.; Griffin, J.L.; Wayland, M.; Freeman, T.; Dudbridge, F.; Lilley, K.S.; et al. Mitochondrial Dysfunction in Schizophrenia: Evidence for Compromised Brain Metabolism and Oxidative Stress. Mol. Psychiatry 2004, 9, 643, 684–697. [Google Scholar] [CrossRef]
- Yang, X.; Sun, L.; Zhao, A.; Hu, X.; Qing, Y.; Jiang, J.; Yang, C.; Xu, T.; Wang, P.; Liu, J.; et al. Serum Fatty Acid Patterns in Patients with Schizophrenia: A Targeted Metabonomics Study. Transl. Psychiatry 2017, 7, e1176. [Google Scholar] [CrossRef]
- Brisch, R.; Esaniotis, A.; Ewolf, R.; Ebielau, H.; Ebernstein, H.-G.; Esteiner, J.; Ebogerts, B.; Braun, A.K.; Ejankowski, Z.; Ekumaratilake, J.; et al. The Role of Dopamine in Schizophrenia from a Neurobiological and Evolutionary Perspective: Old Fashioned, but Still in Vogue. Front. Psychiatry 2014, 5, 47. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wickramasekara, R.N.; Lookian, P.P.; Ngo, J.; Shibata, A.; Stessman, H.A.F. Schizophrenic Psychosis Symptoms in a Background of Mild-To-Moderate Carnitine Palmitoyltransferase II Deficiency: A Case Report. Reports 2020, 3, 31. https://doi.org/10.3390/reports3040031
Wickramasekara RN, Lookian PP, Ngo J, Shibata A, Stessman HAF. Schizophrenic Psychosis Symptoms in a Background of Mild-To-Moderate Carnitine Palmitoyltransferase II Deficiency: A Case Report. Reports. 2020; 3(4):31. https://doi.org/10.3390/reports3040031
Chicago/Turabian StyleWickramasekara, Rochelle N., Pashayar P. Lookian, Jeannie Ngo, Annemarie Shibata, and Holly A. F. Stessman. 2020. "Schizophrenic Psychosis Symptoms in a Background of Mild-To-Moderate Carnitine Palmitoyltransferase II Deficiency: A Case Report" Reports 3, no. 4: 31. https://doi.org/10.3390/reports3040031
APA StyleWickramasekara, R. N., Lookian, P. P., Ngo, J., Shibata, A., & Stessman, H. A. F. (2020). Schizophrenic Psychosis Symptoms in a Background of Mild-To-Moderate Carnitine Palmitoyltransferase II Deficiency: A Case Report. Reports, 3(4), 31. https://doi.org/10.3390/reports3040031