
BMAL1 in Astrocytes: A Protective Role in Alzheimer’s and Parkinson’s Disease

 ,
,  ,
,  ,
,  ,
, {kind=link}
{kind=link}
Abstract
1. Introduction
2. Circadian Rhythms and Astrocytes
3. The Role of Bmal1 in Alzheimer’s and Parkinson’s Disease
3.1. BMAL1 in Alzheimer’s Disease
3.2. Astrocyte BMAL1 in Alzheimer’s Disease
3.3. BMAL1 in Parkinson’s Disease
4. Astrocyte Bmal1 Deletion: A Potential Protective Role Against AD and PD
5. Discussion and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Bass, J.; Lazar, M.A. Circadian time signatures of fitness and disease. Science 2016, 354, 994–999. [Google Scholar] [CrossRef] [PubMed]
- Bass, J.; Takahashi, J.S. Circadian integration of metabolism and energetics. Science 2010, 330, 1349–1354. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.W.; Wei, S.Z.; Huang, G.D.; Liu, L.B.; Gu, C.; Shen, Y.; Wang, X.H.; Xia, S.T.; Xie, A.M.; Hu, L.F.; et al. BMAL1 regulation of microglia-mediated neuroinflammation in MPTP-induced Parkinson’s disease mouse model. FASEB J. 2020, 34, 6570–6581. [Google Scholar] [CrossRef] [PubMed]
- Fifel, K.; De Boer, T. The circadian system in Parkinson’s disease, multiple system atrophy, and progressive supranuclear palsy. Handb. Clin. Neurol. 2021, 179, 301–313. [Google Scholar] [CrossRef]
- Mohawk, J.A.; Green, C.B.; Takahashi, J.S. Central and peripheral circadian clocks in mammals. Annu. Rev. Neurosci. 2012, 35, 445–462. [Google Scholar] [CrossRef]
- Yoo, S.H.; Mohawk, J.A.; Siepka, S.M.; Shan, Y.; Huh, S.K.; Hong, H.K.; Kornblum, I.; Kumar, V.; Koike, N.; Xu, M.; et al. Competing E3 ubiquitin ligases govern circadian periodicity by degradation of CRY in nucleus and cytoplasm. Cell 2013, 152, 1091–1105. [Google Scholar] [CrossRef]
- Hirano, A.; Fu, Y.H.; Ptáček, L.J. The intricate dance of post-translational modifications in the rhythm of life. Nat. Struct. Mol. Biol. 2016, 23, 1053–1060. [Google Scholar] [CrossRef]
- Hastings, M.H.; Smyllie, N.J.; Patton, A.P. Molecular-genetic Manipulation of the Suprachiasmatic Nucleus Circadian Clock. J. Mol. Biol. 2020, 432, 3639–3660. [Google Scholar] [CrossRef]
- Schurhoff, N.; Toborek, M. Circadian rhythms in the blood-brain barrier: Impact on neurological disorders and stress responses. Mol. Brain 2023, 16, 5. [Google Scholar] [CrossRef]
- Yu, E.A.; Weaver, D.R. Disrupting the circadian clock: Gene-specific effects on aging, cancer, and other phenotypes. Aging 2011, 3, 479–493. [Google Scholar] [CrossRef]
- Musiek, E.S.; Holtzman, D.M. Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science 2016, 354, 1004–1008. [Google Scholar] [CrossRef] [PubMed]
- Breen, D.P.; Vuono, R.; Nawarathna, U.; Fisher, K.; Shneerson, J.M.; Reddy, A.B.; Barker, R.A. Sleep and circadian rhythm regulation in early Parkinson disease. JAMA Neurol. 2014, 71, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Moon, M.; Choe, H.K.; Han, D.H.; Jang, C.; Kim, A.; Cho, S.; Kim, K.; Mook-Jung, I. Aβ-induced degradation of BMAL1 and CBP leads to circadian rhythm disruption in Alzheimer’s disease. Mol. Neurodegener. 2015, 10, 13. [Google Scholar] [CrossRef] [PubMed]
- Cronin, P.; McCarthy, M.J.; Lim, A.S.P.; Salmon, D.P.; Galasko, D.; Masliah, E.; Jager, P.L.D.; Bennett, D.A.; Desplats, P. Circadian alterations during early stages of Alzheimer’s disease are associated with aberrant cycles of DNA methylation in BMAL1. Alzheimers Dement. 2017, 13, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Pilleri, M.; Levedianos, G.; Weis, L.; Gasparoli, E.; Facchini, S.; Biundo, R.; Formento-Dojot, P.; Antonini, A. Heart rate circadian profile in the differential diagnosis between Parkinson disease and multiple system atrophy. Park. Relat. Disord. 2014, 20, 217–221. [Google Scholar] [CrossRef]
- Raggi, A.; Neri, W.; Ferri, R. Sleep-related behaviors in Alzheimer’s disease and dementia with Lewy bodies. Rev. Neurosci. 2015, 26, 31–38. [Google Scholar] [CrossRef]
- Vallelonga, F.; Di Stefano, C.; Merola, A.; Romagnolo, A.; Sobrero, G.; Milazzo, V.; Burrello, A.; Burrello, J.; Zibetti, M.; Veglio, F.; et al. Blood pressure circadian rhythm alterations in alpha-synucleinopathies. J. Neurol. 2019, 266, 1141–1152. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef]
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef]
- Chang, C.W.; Shao, E.; Mucke, L. Tau: Enabler of diverse brain disorders and target of rapidly evolving therapeutic strategies. Science 2021, 371, eabb8255. [Google Scholar] [CrossRef]
- Ju, Y.S.; Ooms, S.J.; Sutphen, C.; Macauley, S.L.; Zangrilli, M.A.; Jerome, G.; Fagan, A.M.; Mignot, E.; Zempel, J.M.; Claassen, J.A.H.R.; et al. Slow wave sleep disruption increases cerebrospinal fluid amyloid-β levels. Brain 2017, 140, 2104–2111. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.E.; Lim, M.M.; Bateman, R.J.; Lee, J.J.; Smyth, L.P.; Cirrito, J.R.; Fujiki, N.; Nishino, S.; Holtzman, D.M. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science 2009, 326, 1005–1007. [Google Scholar] [CrossRef] [PubMed]
- Sprecher, K.E.; Koscik, R.L.; Carlsson, C.M.; Zetterberg, H.; Blennow, K.; Okonkwo, O.C.; Sager, M.A.; Asthana, S.; Johnson, S.C.; Benca, R.M.; et al. Poor sleep is associated with CSF biomarkers of amyloid pathology in cognitively normal adults. Neurology 2017, 89, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Holth, J.K.; Fritschi, S.K.; Wang, C.; Pedersen, N.P.; Cirrito, J.R.; Mahan, T.E.; Finn, M.B.; Manis, M.; Geerling, J.C.; Fuller, P.M.; et al. The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science 2019, 363, 880–884. [Google Scholar] [CrossRef]
- Bendor, J.T.; Logan, T.P.; Edwards, R.H. The function of α-synuclein. Neuron 2013, 79, 1044–1066. [Google Scholar] [CrossRef]
- Du, X.Y.; Xie, X.X.; Liu, R.T. The Role of α-Synuclein Oligomers in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 8645. [Google Scholar] [CrossRef]
- Brash-Arias, D.; García, L.I.; Pérez-Estudillo, C.A.; Rojas-Durán, F.; Aranda-Abreu, G.E.; Herrera-Covarrubias, D.; Chi-Castañeda, D. The Role of Astrocytes and Alpha-Synuclein in Parkinson’s Disease: A Review. NeuroSci 2024, 5, 71–86. [Google Scholar] [CrossRef]
- Gros, P.; Videnovic, A. Overview of Sleep and Circadian Rhythm Disorders in Parkinson Disease. Clin. Geriatr. Med. 2020, 36, 119–130. [Google Scholar] [CrossRef]
- Maetzler, W.; Liepelt, I.; Berg, D. Progression of Parkinson’s disease in the clinical phase: Potential markers. Lancet Neurol. 2009, 8, 1158–1171. [Google Scholar] [CrossRef]
- Torres-Pasillas, G.; Chi-Castañeda, D.; Carrillo-Castilla, P.; Marín, G.; Hernández-Aguilar, M.E.; Aranda-Abreu, G.E.; Manzo, J.; García, L.I. Olfactory Dysfunction in Parkinson’s Disease, Its Functional and Neuroanatomical Correlates. NeuroSci 2023, 4, 134–151. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Hayashi, S.; Yoshimoto, M.; Kudo, H.; Takahashi, H. NACP/alpha-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta Neuropathol. 2000, 99, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Sastre, M.; Del Tredici, K. Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol. 2007, 114, 231–241. [Google Scholar] [CrossRef]
- Acioglu, C.; Li, L.; Elkabes, S. Contribution of astrocytes to neuropathology of neurodegenerative diseases. Brain Res. 2021, 1758, 147291. [Google Scholar] [CrossRef]
- Prolo, L.M.; Takahashi, J.S.; Herzog, E.D. Circadian rhythm generation and entrainment in astrocytes. J. Neurosci. 2005, 25, 404–408. [Google Scholar] [CrossRef]
- Tso, C.F.; Simon, T.; Greenlaw, A.C.; Puri, T.; Mieda, M.; Herzog, E.D. Astrocytes Regulate Daily Rhythms in the Suprachiasmatic Nucleus and Behavior. Curr. Biol. 2017, 27, 1055–1061. [Google Scholar] [CrossRef]
- Brancaccio, M.; Edwards, M.D.; Patton, A.P.; Smyllie, N.J.; Chesham, J.E.; Maywood, E.S.; Hastings, M.H. Cell-autonomous clock of astrocytes drives circadian behavior in mammals. Science 2019, 363, 187–192. [Google Scholar] [CrossRef]
- Marpegan, L.; Swanstrom, A.E.; Chung, K.; Simon, T.; Haydon, P.G.; Khan, S.K.; Liu, A.C.; Herzog, E.D.; Beaulé, C. Circadian regulation of ATP release in astrocytes. J. Neurosci. 2011, 31, 8342–8350. [Google Scholar] [CrossRef]
- Rath, M.F.; Rohde, K.; Fahrenkrug, J.; Møller, M. Circadian clock components in the rat neocortex: Daily dynamics, localization and regulation. Brain Struct. Funct. 2013, 218, 551–562. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Astrogliosis. Cold Spring Harb. Perspect. Biol. 2014, 7, a020420. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, K.K. Glial fibrillary acidic protein: From intermediate filament assembly and gliosis to neurobiomarker. Trends Neurosci. 2015, 38, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Lananna, B.V.; Nadarajah, C.J.; Izumo, M.; Cedeño, M.R.; Xiong, D.D.; Dimitry, J.; Tso, C.F.; McKee, C.A.; Griffin, P.; Sheehan, P.W.; et al. Cell-Autonomous Regulation of Astrocyte Activation by the Circadian Clock Protein BMAL1. Cell Rep. 2018, 25, 1–9.e5. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, P.W.; Nadarajah, C.J.; Kanan, M.F.; Patterson, J.N.; Novotny, B.; Lawrence, J.H.; King, M.W.; Brase, L.; Inman, C.E.; Yuede, C.M.; et al. An astrocyte BMAL1-BAG3 axis protects against alpha-synuclein and tau pathology. Neuron 2023, 111, 2383–2398.e7. [Google Scholar] [CrossRef] [PubMed]
- Servière, J.; Lavialle, M. Astrocytes in the mammalian circadian clock: Putative roles. Prog. Brain Res. 1996, 111, 57–73. [Google Scholar] [CrossRef]
- Womac, A.D.; Burkeen, J.F.; Neuendorff, N.; Earnest, D.J.; Zoran, M.J. Circadian rhythms of extracellular ATP accumulation in suprachiasmatic nucleus cells and cultured astrocytes. Eur. J. Neurosci. 2009, 30, 869–876. [Google Scholar] [CrossRef]
- Burkeen, J.F.; Womac, A.D.; Earnest, D.J.; Zoran, M.J. Mitochondrial calcium signaling mediates rhythmic extracellular ATP accumulation in suprachiasmatic nucleus astrocytes. J. Neurosci. 2011, 31, 8432–8440. [Google Scholar] [CrossRef]
- Koyanagi, S.; Kusunose, N.; Taniguchi, M.; Akamine, T.; Kanado, Y.; Ozono, Y.; Masuda, T.; Kohro, Y.; Matsunaga, N.; Tsuda, M.; et al. Glucocorticoid regulation of ATP release from spinal astrocytes underlies diurnal exacerbation of neuropathic mechanical allodynia. Nat. Commun. 2016, 7, 13102. [Google Scholar] [CrossRef]
- Leone, M.J.; Beaule, C.; Marpegan, L.; Simon, T.; Herzog, E.D.; Golombek, D.A. Glial and light-dependent glutamate metabolism in the suprachiasmatic nuclei. Chronobiol. Int. 2015, 32, 573–578. [Google Scholar] [CrossRef]
- McKee, C.A.; Lananna, B.V.; Musiek, E.S. Circadian regulation of astrocyte function: Implications for Alzheimer’s disease. Cell Mol. Life Sci. 2020, 77, 1049–1058. [Google Scholar] [CrossRef]
- Hassler, C.; Burnier, M. Circadian variations in blood pressure: Implications for chronotherapeutics. Am. J. Cardiovasc. Drugs 2005, 5, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Manfredini, R.; Boari, B.; Smolensky, M.H.; Salmi, R.; Cecilia, O.I.; Malagoni, A.M.; Haus, E.; Manfredini, F. Circadian variation in stroke onset: Identical temporal pattern in ischemic and hemorrhagic events. Chronobiol. Int. 2005, 22, 417–453. [Google Scholar] [CrossRef] [PubMed]
- Karmarkar, S.W.; Tischkau, S.A. Influences of the circadian clock on neuronal susceptibility to excitotoxicity. Front. Physiol. 2013, 4, 313. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- You, S.; Fulga, T.A.; Van Vactor, D.; Jackson, F.R. Regulation of Circadian Behavior by Astroglial MicroRNAs in Drosophila. Genetics 2018, 208, 1195–1207. [Google Scholar] [CrossRef]
- Maywood, E.S.; Elliott, T.S.; Patton, A.P.; Krogager, T.P.; Chesham, J.E.; Ernst, R.J.; Beránek, V.; Brancaccio, M.; Chin, J.W.; Hastings, M.H. Translational switching of Cry1 protein expression confers reversible control of circadian behavior in arrhythmic Cry-deficient mice. Proc. Natl. Acad. Sci. USA 2018, 115, E12388–E12397. [Google Scholar] [CrossRef]
- Patton, A.P.; Smyllie, N.J.; Chesham, J.E.; Hastings, M.H. Astrocytes Sustain Circadian Oscillation and Bidirectionally Determine Circadian Period, but Do Not Regulate Circadian Phase in the Suprachiasmatic Nucleus. J. Neurosci. 2022, 42, 5522–5537. [Google Scholar] [CrossRef]
- Hastings, M.H.; Brancaccio, M.; Gonzalez-Aponte, M.F.; Herzog, E.D. Circadian Rhythms and Astrocytes: The Good, the Bad, and the Ugly. Annu. Rev. Neurosci. 2023, 46, 123–143. [Google Scholar] [CrossRef]
- Anashkina, A.A.; Poluektov, Y.M.; Dmitriev, V.A.; Kuznetsov, E.N.; Mitkevich, V.A.; Makarov, A.A.; Petrushanko, I.Y. A novel approach for predicting protein S-glutathionylation. BMC Bioinform. 2020, 21 (Suppl. S11), 282. [Google Scholar] [CrossRef]
- Bunger, M.K.; Wilsbacher, L.D.; Moran, S.M.; Clendenin, C.; Radcliffe, L.A.; Hogenesch, J.B.; Simon, M.C.; Takahashi, J.S.; Bradfield, C.A. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell 2000, 103, 1009–1017. [Google Scholar] [CrossRef]
- Welz, P.S.; Zinna, V.M.; Symeonidi, A.; Koronowski, K.B.; Kinouchi, K.; Smith, J.G.; Guillén, I.M.; Castellanos, A.; Furrow, S.; Aragón, F.; et al. BMAL1-Driven Tissue Clocks Respond Independently to Light to Maintain Homeostasis. Cell 2019, 177, 1436–1447.e12. [Google Scholar] [CrossRef] [PubMed]
- Kondratov, R.V.; Kondratova, A.A.; Gorbacheva, V.Y.; Vykhovanets, O.V.; Antoch, M.P. Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev. 2006, 20, 1868–1873. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.; Peng, X.; Xie, L.; Dong, K.; Ma, D.; Xu, W.; Shi, X.; Zhang, S.; Chen, J.; Yu, X.; et al. Importance of Bmal1 in Alzheimer’s disease and associated aging-related diseases: Mechanisms and interventions. Aging Cell 2022, 21, e13704. [Google Scholar] [CrossRef] [PubMed]
- Chhunchha, B.; Kubo, E.; Singh, D.P. Clock Protein Bmal1 and Nrf2 Cooperatively Control Aging or Oxidative Response and Redox Homeostasis by Regulating Rhythmic Expression of Prdx6. Cells 2020, 9, 1861. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Tang, Q.; Nie, J.; Zhang, C.; Zhou, X.; Yu, S.; Sun, J.; Cheng, X.; Dong, N.; Hu, Y.; et al. BMAL1-Downregulation Aggravates Porphyromonas gingivalis-Induced Atherosclerosis by Encouraging Oxidative Stress. Circ. Res. 2020, 126, e15–e29. [Google Scholar] [CrossRef]
- Shi, S.Q.; Ansari, T.S.; McGuinness, O.P.; Wasserman, D.H.; Johnson, C.H. Circadian disruption leads to insulin resistance and obesity. Curr. Biol. 2013, 23, 372–381. [Google Scholar] [CrossRef]
- Li, E.; Li, X.; Huang, J.; Xu, C.; Liang, Q.; Ren, K.; Bai, A.; Lu, C.; Qian, R.; Sun, N. BMAL1 regulates mitochondrial fission and mitophagy through mitochondrial protein BNIP3 and is critical in the development of dilated cardiomyopathy. Protein Cell 2020, 11, 661–679. [Google Scholar] [CrossRef]
- Ray, S.; Valekunja, U.K.; Stangherlin, A.; Howell, S.A.; Snijders, A.P.; Damodaran, G.; Reddy, A.B. Circadian rhythms in the absence of the clock gene Bmal1. Science 2020, 367, 800–806. [Google Scholar] [CrossRef]
- Mattis, J.; Sehgal, A. Circadian Rhythms, Sleep, and Disorders of Aging. Trends Endocrinol. Metab. 2016, 27, 192–203. [Google Scholar] [CrossRef]
- Niu, L.; Zhang, F.; Xu, X.; Yang, Y.; Li, S.; Liu, H.; Le, W. Chronic sleep deprivation altered the expression of circadian clock genes and aggravated Alzheimer’s disease neuropathology. Brain Pathol. 2022, 32, e13028. [Google Scholar] [CrossRef]
- Wang, Y.; Lv, D.; Liu, W.; Li, S.; Chen, J.; Shen, Y.; Wang, F.; Hu, L.F.; Liu, C.F. Disruption of the Circadian Clock Alters Antioxidative Defense via the SIRT1-BMAL1 Pathway in 6-OHDA-Induced Models of Parkinson’s Disease. Oxid. Med. Cell Longev. 2018, 2018, 4854732. [Google Scholar] [CrossRef] [PubMed]
- Kress, G.J.; Liao, F.; Dimitry, J.; Cedeno, M.R.; FitzGerald, G.A.; Holtzman, D.M.; Musiek, E.S. Regulation of amyloid-β dynamics and pathology by the circadian clock. J. Exp. Med. 2018, 215, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- McKee, C.A.; Lee, J.; Cai, Y.; Saito, T.; Saido, T.; Musiek, E.S. Astrocytes deficient in circadian clock gene Bmal1 show enhanced activation responses to amyloid-beta pathology without changing plaque burden. Sci. Rep. 2022, 12, 1796. [Google Scholar] [CrossRef] [PubMed]
- Akladious, A.; Azzam, S.; Hu, Y.; Feng, P. Bmal1 knockdown suppresses wake and increases immobility without altering orexin A, corticotrophin-releasing hormone, or glutamate decarboxylase. CNS Neurosci. Ther. 2018, 24, 549–563. [Google Scholar] [CrossRef]
- Qiu, P.; Jiang, J.; Liu, Z.; Cai, Y.; Huang, T.; Wang, Y.; Liu, Q.; Nie, Y.; Liu, F.; Cheng, J.; et al. BMAL1 knockout macaque monkeys display reduced sleep and psychiatric disorders. Natl. Sci. Rev. 2019, 6, 87–100. [Google Scholar] [CrossRef]
- Ettcheto, M.; Olloquequi, J.; Sánchez-López, E.; Busquets, O.; Cano, A.; Manzine, P.R.; Beas-Zarate, C.; Castro-Torres, R.D.; García, M.L.; Bulló, M.; et al. Benzodiazepines and Related Drugs as a Risk Factor in Alzheimer’s Disease Dementia. Front. Aging Neurosci. 2020, 11, 344. [Google Scholar] [CrossRef]
- Nakazato, R.; Hotta, S.; Yamada, D.; Kou, M.; Nakamura, S.; Takahata, Y.; Tei, H.; Numano, R.; Hida, A.; Shimba, S.; et al. The intrinsic microglial clock system regulates interleukin-6 expression. Glia 2017, 65, 198–208. [Google Scholar] [CrossRef]
- Huang, J.; Peng, X.; Fan, R.; Dong, K.; Shi, X.; Zhang, S.; Yu, X.; Yang, Y. Disruption of Circadian Clocks Promotes Progression of Alzheimer’s Disease in Diabetic Mice. Mol. Neurobiol. 2021, 58, 4404–4412. [Google Scholar] [CrossRef]
- Tagarelli, A.; Piro, A.; Tagarelli, G.; Lagonia, P.; Quattrone, A. Alois Alzheimer: A hundred years after the discovery of the eponymous disorder. Int. J. Biomed. Sci. 2006, 2, 196–204. [Google Scholar] [CrossRef]
- Duyckaerts, C.; Potier, M.C.; Delatour, B. Alzheimer disease models and human neuropathology: Similarities and differences. Acta Neuropathol. 2008, 115, 5–38. [Google Scholar] [CrossRef]
- Olabarria, M.; Noristani, H.N.; Verkhratsky, A.; Rodríguez, J.J. Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia 2010, 58, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Bouvier, D.S.; Jones, E.V.; Quesseveur, G.; Davoli, M.A.; Ferreira, T.A.; Quirion, R.; Mechawar, N.; Murai, K.K. High-Resolution Dissection of Reactive Glial Nets in Alzheimer’s Disease. Sci. Rep. 2016, 6, 24544. [Google Scholar] [CrossRef] [PubMed]
- Koistinaho, M.; Lin, S.; Wu, X.; Esterman, M.; Koger, D.; Hanson, J.; Higgs, R.; Liu, F.; Malkani, S.; Bales, K.R.; et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat. Med. 2004, 10, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Basak, J.M.; Verghese, P.B.; Yoon, H.; Kim, J.; Holtzman, D.M. Low-density lipoprotein receptor represents an apolipoprotein E-independent pathway of Aβ uptake and degradation by astrocytes. J. Biol. Chem. 2012, 287, 13959–13971. [Google Scholar] [CrossRef]
- Thal, D.R. The role of astrocytes in amyloid β-protein toxicity and clearance. Exp. Neurol. 2012, 236, 1–5. [Google Scholar] [CrossRef]
- Xiao, Q.; Yan, P.; Ma, X.; Liu, H.; Perez, R.; Zhu, A.; Gonzalez, E.; Burchett, J.M.; Schuler, D.R.; Cirrito, J.R.; et al. Enhancing astrocytic lysosome biogenesis facilitates Aβ clearance and attenuates amyloid plaque pathogenesis. J. Neurosci. 2014, 34, 9607–9620. [Google Scholar] [CrossRef]
- Söllvander, S.; Nikitidou, E.; Brolin, R.; Söderberg, L.; Sehlin, D.; Lannfelt, L.; Erlandsson, A. Accumulation of amyloid-β by astrocytes result in enlarged endosomes and microvesicle-induced apoptosis of neurons. Mol. Neurodegener. 2016, 11, 38. [Google Scholar] [CrossRef]
- Liu, C.C.; Hu, J.; Zhao, N.; Wang, J.; Wang, N.; Cirrito, J.R.; Kanekiyo, T.; Holtzman, D.M.; Bu, G. Astrocytic LRP1 Mediates Brain Aβ Clearance and Impacts Amyloid Deposition. J. Neurosci. 2017, 37, 4023–4031. [Google Scholar] [CrossRef]
- Lucey, B.P.; Hicks, T.J.; McLeland, J.S.; Toedebusch, C.D.; Boyd, J.; Elbert, D.L.; Patterson, B.W.; Baty, J.; Morris, J.C.; Ovod, V.; et al. Effect of sleep on overnight cerebrospinal fluid amyloid β kinetics. Ann. Neurol. 2018, 83, 197–204. [Google Scholar] [CrossRef]
- Haydon, P.G. Astrocytes and the modulation of sleep. Curr. Opin. Neurobiol. 2017, 44, 28–33. [Google Scholar] [CrossRef]
- Barca-Mayo, O.; Pons-Espinal, M.; Follert, P.; Armirotti, A.; Berdondini, L.; De Pietri Tonelli, D. Astrocyte deletion of Bmal1 alters daily locomotor activity and cognitive functions via GABA signalling. Nat. Commun. 2017, 8, 14336. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.A.H.; Schwarz-Herzke, B.; Rollenhagen, A.; Anstötz, M.; Holub, M.; Lübke, J.; Rose, C.R.; Schnittler, H.J.; von Gall, C. Bmal1-deficiency affects glial synaptic coverage of the hippocampal mossy fiber synapse and the actin cytoskeleton in astrocytes. Glia 2020, 68, 947–962. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Liu, S.; Sothern, R.B.; Xu, S.; Chan, P. Expression of clock genes Per1 and Bmal1 in total leukocytes in health and Parkinson’s disease. Eur. J. Neurol. 2010, 17, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Cheng, C.; Jia, C.; Leng, Y.; Qian, J.; Yu, H.; Liu, Y.; Wang, N.; Yang, Y.; Al-Nusaif, M.; et al. Peripheral Clock System Abnormalities in Patients with Parkinson’s Disease. Front. Aging Neurosci. 2021, 13, 736026. [Google Scholar] [CrossRef] [PubMed]
- Videnovic, A.; Noble, C.; Reid, K.J.; Peng, J.; Turek, F.W.; Marconi, A.; Rademaker, A.W.; Simuni, T.; Zadikoff, C.; Zee, P.C. Circadian melatonin rhythm and excessive daytime sleepiness in Parkinson disease. JAMA Neurol. 2014, 71, 463–469. [Google Scholar] [CrossRef]
- Videnovic, A.; Golombek, D. Circadian and sleep disorders in Parkinson’s disease. Exp. Neurol. 2013, 243, 45–56. [Google Scholar] [CrossRef]
- Lauretti, E.; Di Meco, A.; Merali, S.; Praticò, D. Circadian rhythm dysfunction: A novel environmental risk factor for Parkinson’s disease. Mol. Psychiatry 2017, 22, 280–286. [Google Scholar] [CrossRef]
- Delgado-Lara, D.L.; González-Enríquez, G.V.; Torres-Mendoza, B.M.; González-Usigli, H.; Cárdenas-Bedoya, J.; Macías-Islas, M.A.; de la Rosa, A.C.; Jiménez-Delgado, A.; Pacheco-Moisés, F.; Cruz-Serrano, J.A.; et al. Effect of melatonin administration on the PER1 and BMAL1 clock genes in patients with Parkinson’s disease. Biomed. Pharmacother. 2020, 129, 110485. [Google Scholar] [CrossRef]
- Musiek, E.S.; Lim, M.M.; Yang, G.; Bauer, A.Q.; Qi, L.; Lee, Y.; Roh, J.H.; Ortiz-González, X.; Dearborn, J.T.; Culver, J.P.; et al. Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration. J. Clin. Investig. 2013, 123, 5389–5400. [Google Scholar] [CrossRef]
- Yoo, I.D.; Park, M.W.; Cha, H.W.; Yoon, S.; Boonpraman, N.; Yi, S.S.; Moon, J.S. Elevated CLOCK and BMAL1 Contribute to the Impairment of Aerobic Glycolysis from Astrocytes in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 7862. [Google Scholar] [CrossRef]
- Liu, J.Y.; Xue, J.; Wang, F.; Wang, Y.L.; Dong, W.L. α-Synuclein-Induced Destabilized BMAL1 mRNA Leads to Circadian Rhythm Disruption in Parkinson’s Disease. Neurotox. Res. 2023, 41, 177–186. [Google Scholar] [CrossRef]
- Kanan, M.F.; Sheehan, P.W.; Haines, J.N.; Gomez, P.G.; Dhuler, A.; Nadarajah, C.J.; Wargel, Z.M.; Freeberg, B.M.; Nelvagal, H.R.; Izumo, M.; et al. Neuronal deletion of the circadian clock gene Bmal1 induces cell-autonomous dopaminergic neurodegeneration. JCI Insight 2024, 9, e162771. [Google Scholar] [CrossRef]
- McKee, C.A.; Polino, A.J.; King, M.W.; Musiek, E.S. Circadian clock protein BMAL1 broadly influences autophagy and endolysosomal function in astrocytes. Proc. Natl. Acad. Sci. USA 2023, 120, e2220551120. [Google Scholar] [CrossRef]
- Jiwaji, Z.; Tiwari, S.S.; Avilés-Reyes, R.X.; Hooley, M.; Hampton, D.; Torvell, M.; Johnson, D.A.; McQueen, J.; Baxter, P.; Sabari-Sankar, K.; et al. Reactive astrocytes acquire neuroprotective as well as deleterious signatures in response to Tau and Aß pathology. Nat. Commun. 2022, 13, 135. [Google Scholar] [CrossRef]
- Lee, J.; Moulik, M.; Fang, Z.; Saha, P.; Zou, F.; Xu, Y.; Nelson, D.L.; Ma, K.; Moore, D.D.; Yechoor, V.K. Bmal1 and β-cell clock are required for adaptation to circadian disruption, and their loss of function leads to oxidative stress-induced β-cell failure in mice. Mol. Cell Biol. 2013, 33, 2327–2338. [Google Scholar] [CrossRef]
- Early, J.O.; Menon, D.; Wyse, C.A.; Cervantes-Silva, M.P.; Zaslona, Z.; Carroll, R.G.; Palsson-McDermott, E.M.; Angiari, S.; Ryan, D.G.; Corcoran, S.E.; et al. Circadian clock protein BMAL1 regulates IL-1β in macrophages via NRF2. Proc. Natl. Acad. Sci. USA 2018, 115, E8460–E8468. [Google Scholar] [CrossRef]
- Diaz-Castro, B.; Gangwani, M.R.; Yu, X.; Coppola, G.; Khakh, B.S. Astrocyte molecular signatures in Huntington’s disease. Sci. Transl. Med. 2019, 11, eaaw8546. [Google Scholar] [CrossRef]
- Habib, N.; McCabe, C.; Medina, S.; Varshavsky, M.; Kitsberg, D.; Dvir-Szternfeld, R.; Green, G.; Dionne, D.; Nguyen, L.; Marshall, J.L.; et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci. 2020, 23, 701–706. [Google Scholar] [CrossRef]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.O.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Abjean, L.; Ben Haim, L.; Riquelme-Perez, M.; Gipchtein, P.; Derbois, C.; Palomares, M.A.; Petit, F.; Hérard, A.S.; Gaillard, M.C.; Guillermier, M.; et al. Reactive astrocytes promote proteostasis in Huntington’s disease through the JAK2-STAT3 pathway. Brain 2023, 146, 149–166. [Google Scholar] [CrossRef]
- Arranz, A.M.; De Strooper, B. The role of astroglia in Alzheimer’s disease: Pathophysiology and clinical implications. Lancet Neurol. 2019, 18, 406–414. [Google Scholar] [CrossRef]
- Ceyzériat, K.; Ben Haim, L.; Denizot, A.; Pommier, D.; Matos, M.; Guillemaud, O.; Palomares, M.A.; Abjean, L.; Petit, F.; Gipchtein, P.; et al. Modulation of astrocyte reactivity improves functional deficits in mouse models of Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 104. [Google Scholar] [CrossRef]
- Reichenbach, N.; Delekate, A.; Plescher, M.; Schmitt, F.; Krauss, S.; Blank, N.; Halle, A.; Petzold, G.C. Inhibition of Stat3-mediated astrogliosis ameliorates pathology in an Alzheimer’s disease model. EMBO Mol. Med. 2019, 11, e9665. [Google Scholar] [CrossRef]
- Wojtas, A.M.; Sens, J.P.; Kang, S.S.; Baker, K.E.; Berry, T.J.; Kurti, A.; Daughrity, L.; Jansen-West, K.R.; Dickson, D.W.; Petrucelli, L.; et al. Astrocyte-derived clusterin suppresses amyloid formation in vivo. Mol. Neurodegener. 2020, 15, 71. [Google Scholar] [CrossRef]
- Kraft, A.W.; Hu, X.; Yoon, H.; Yan, P.; Xiao, Q.; Wang, Y.; Gil, S.C.; Brown, J.; Wilhelmsson, U.; Restivo, J.L.; et al. Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J. 2013, 27, 187–198. [Google Scholar] [CrossRef]
- Xu, Z.; Xiao, N.; Chen, Y.; Huang, H.; Marshall, C.; Gao, J.; Cai, Z.; Wu, T.; Hu, G.; Xiao, M. Deletion of aquaporin-4 in APP/PS1 mice exacerbates brain Aβ accumulation and memory deficits. Mol. Neurodegener. 2015, 10, 58. [Google Scholar] [CrossRef]
- Zheng, Y.; Pan, L.; Wang, F.; Yan, J.; Wang, T.; Xia, Y.; Yao, L.; Deng, K.; Zheng, Y.; Xia, X.; et al. Neural function of Bmal1: An overview. Cell Biosci. 2023, 13, 1. [Google Scholar] [CrossRef]
- Izumo, M.; Pejchal, M.; Schook, A.C.; Lange, R.P.; Walisser, J.A.; Sato, T.R.; Wang, X.; Bradfield, C.A.; Takahashi, J.S. Differential effects of light and feeding on circadian organization of peripheral clocks in a forebrain Bmal1 mutant. Elife 2014, 3, e04617. [Google Scholar] [CrossRef]
- Gerstner, J.R.; Smith, G.G.; Lenz, O.; Perron, I.J.; Buono, R.J.; Ferraro, T.N. BMAL1 controls the diurnal rhythm and set point for electrical seizure threshold in mice. Front. Syst. Neurosci. 2014, 8, 121. [Google Scholar] [CrossRef]
- Wu, H.; Liu, Y.; Liu, L.; Meng, Q.; Du, C.; Li, K.; Dong, S.; Zhang, Y.; Li, H.; Zhang, H. Decreased expression of the clock gene Bmal1 is involved in the pathogenesis of temporal lobe epilepsy. Mol. Brain 2021, 14, 113. [Google Scholar] [CrossRef]
- Lembach, A.; Stahr, A.; Ali, A.A.H.; Ingenwerth, M.; von Gall, C. Sex-Dependent Effects of Bmal1-Deficiency on Mouse Cerebral Cortex Infarction in Response to Photothrombotic Stroke. Int. J. Mol. Sci. 2018, 19, 3124. [Google Scholar] [CrossRef] [PubMed]
- Slomnicki, L.P.; Myers, S.A.; Ohri, S.S.; Parsh, M.V.; Andres, K.R.; Chariker, J.H.; Rouchka, E.C.; Whittemore, S.R.; Hetman, M. Improved locomotor recovery after contusive spinal cord injury in Bmal1−/− mice is associated with protection of the blood spinal cord barrier. Sci. Rep. 2020, 10, 14212. [Google Scholar] [CrossRef] [PubMed]
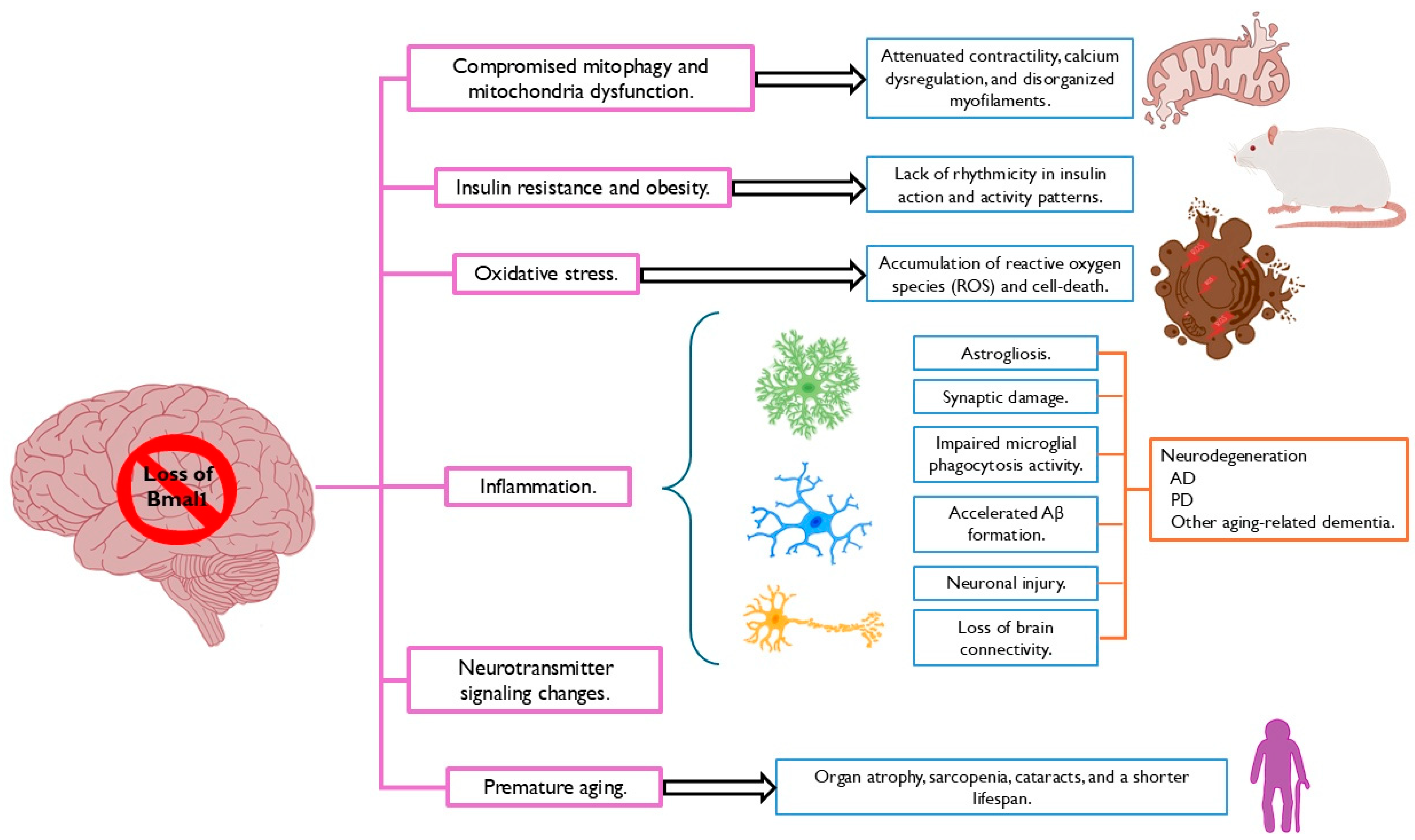
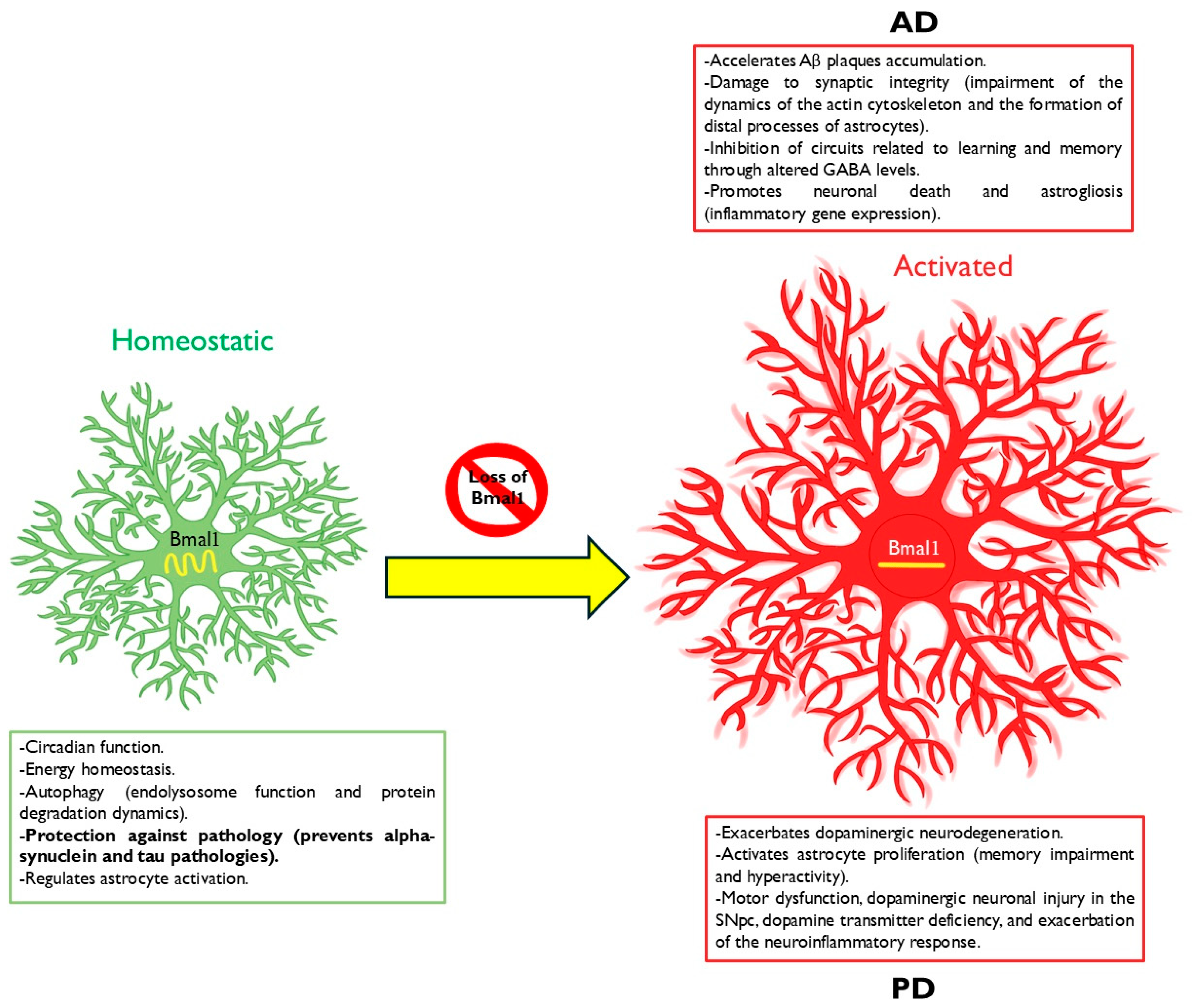
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brash-Arias, D.; García, L.I.; Aranda-Abreu, G.; Toledo-Cárdenas, R.; Pérez-Estudillo, C.; Chi-Castañeda, D. BMAL1 in Astrocytes: A Protective Role in Alzheimer’s and Parkinson’s Disease. Neuroglia 2025, 6, 1. https://doi.org/10.3390/neuroglia6010001
Brash-Arias D, García LI, Aranda-Abreu G, Toledo-Cárdenas R, Pérez-Estudillo C, Chi-Castañeda D. BMAL1 in Astrocytes: A Protective Role in Alzheimer’s and Parkinson’s Disease. Neuroglia. 2025; 6(1):1. https://doi.org/10.3390/neuroglia6010001
Chicago/Turabian StyleBrash-Arias, David, Luis I. García, Gonzalo Aranda-Abreu, Rebeca Toledo-Cárdenas, César Pérez-Estudillo, and Donaji Chi-Castañeda. 2025. "BMAL1 in Astrocytes: A Protective Role in Alzheimer’s and Parkinson’s Disease" Neuroglia 6, no. 1: 1. https://doi.org/10.3390/neuroglia6010001
APA StyleBrash-Arias, D., García, L. I., Aranda-Abreu, G., Toledo-Cárdenas, R., Pérez-Estudillo, C., & Chi-Castañeda, D. (2025). BMAL1 in Astrocytes: A Protective Role in Alzheimer’s and Parkinson’s Disease. Neuroglia, 6(1), 1. https://doi.org/10.3390/neuroglia6010001