
Microglial Dyshomeostasis: A Common Substrate in Neurodevelopmental and Neurodegenerative Diseases


Abstract
1. Introduction
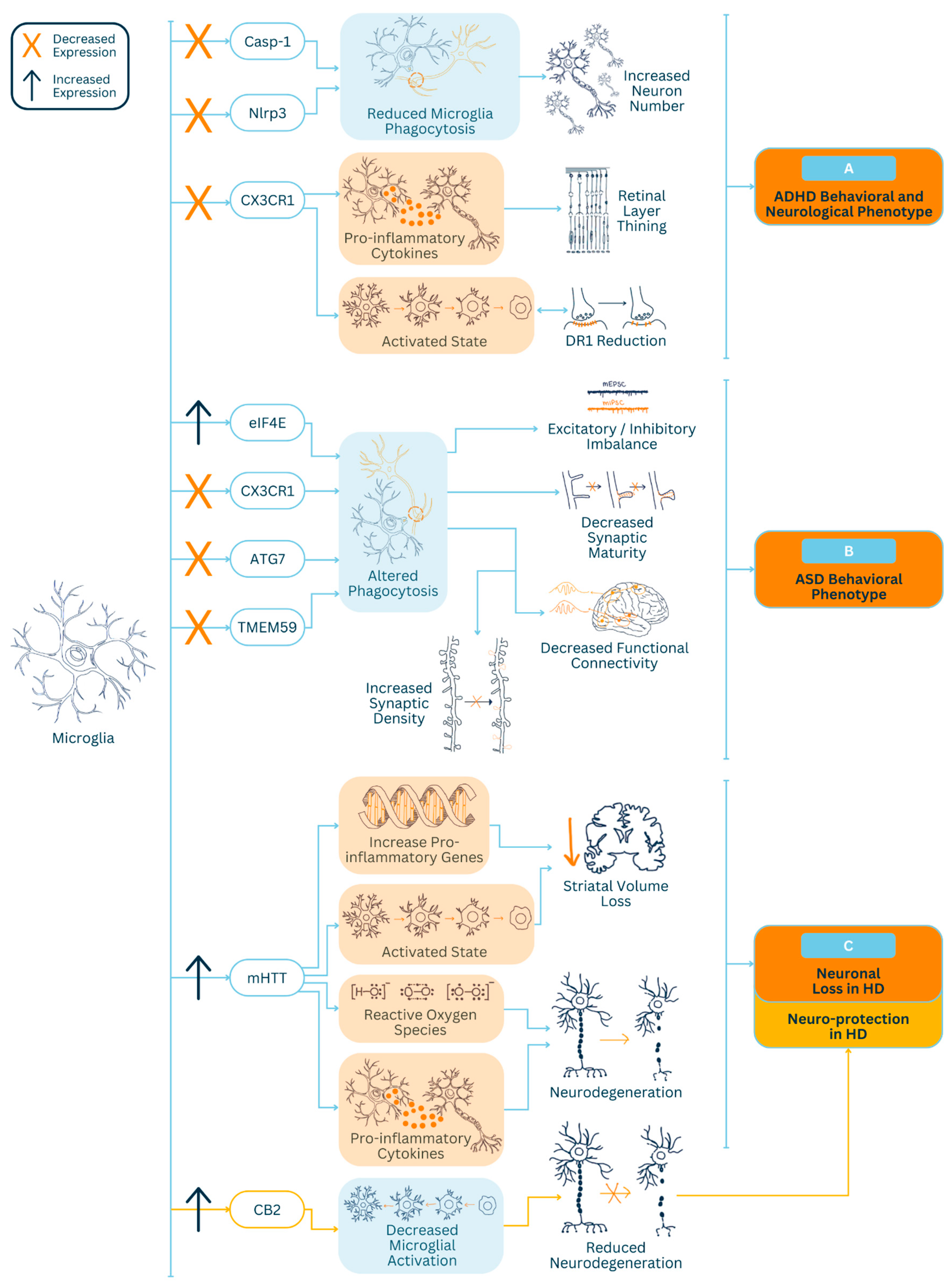
2. Microglial Causative Role in Neurodevelopmental Disorders
2.1. Autism Spectrum Disorder
2.2. Attention-Deficit Hyperactivity Disorder
3. Microglial Causative Role in Neurodegenerative Diseases

{kind=link}
| Study | Animal Model (s) | Microglial Dysfunction | Functional Changes |
|---|---|---|---|
| ASD [11] | Tmem59 KO mice | ↓ synaptic pruning | ↓ synaptic inhibition, ↑ excitatory synaptic density |
| ASD [12] | CX3CR1 KO mice | ↓ synaptic pruning | ↓ functional connectivity, ↓ social interaction |
| ADHD [13] | AHR & WKY rats | ↑ proinflammatory | ↓ retinal layer thinning |
| ASD [14] | Syn-R26 mice | ↓ synaptic pruning | ↓ synaptic inhibition, ↑ excitatory synaptic density |
| ASD [15] | Atg7 KO mice | ↓ synaptic pruning | ↑ immature synaptic density, ↓ functional connectivity |
| ADHD [16] | Human | ↑ activation | ↓ dopamine receptor 1 |
| ADHD [17] | Casp1 KO mice | ↓ microglial phagocytosis | ↑ increased neuronal numbers in the thalamus |
| HD [31] | R6/2 male mice | ↑ neurotoxicity | ↓ striatal volume loss |
| HD [32] | R6/2 male mice w/CB2 KO | ↑ activation | ↑ neurodegeneration |
| HD [33] | BV2 mouse | ↑ proinflammatory | ↑ neurodegeneration |
| HD [34] | Human | ↑ proinflammatory | ↑ neurodegeneration |
| HD [35] | Human | ↑ activation | ↓ dopamine receptor 2 binding |
Huntington’s Disease
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kasparek, T.; Theiner, P.; Filova, A. Neurobiology of ADHD From Childhood to Adulthood: Findings of Imaging Methods. J. Atten. Disord. 2015, 19, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Varghese, M.; Keshav, N.; Jacot-Descombes, S.; Warda, T.; Wicinski, B.; Dickstein, D.L.; Harony-Nicolas, H.; De Rubeis, S.; Drapeau, E.; Buxbaum, J.D.; et al. Autism Spectrum Disorder: Neuropathology and Animal Models. Acta Neuropathol. (Berl.) 2017, 134, 537–566. [Google Scholar] [CrossRef] [PubMed]
- Waldvogel, H.J.; Kim, E.H.; Tippett, L.J.; Vonsattel, J.-P.G.; Faull, R.L.M. The Neuropathology of Huntington’s Disease. Curr. Top. Behav. Neurosci. 2015, 22, 33–80. [Google Scholar] [CrossRef] [PubMed]
- Lukens, J.R.; Eyo, U.B. Microglia and Neurodevelopmental Disorders. Annu. Rev. Neurosci. 2022, 45, 425–445. [Google Scholar] [CrossRef] [PubMed]
- Crotti, A.; Glass, C.K. The Choreography of Neuroinflammation in Huntington’s Disease. Trends Immunol. 2015, 36, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Napoli, I.; Neumann, H. Microglial Clearance Function in Health and Disease. Neuroscience 2009, 158, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- van Furth, R.; Cohn, Z.A.; Hirsch, J.G.; Humphrey, J.H.; Spector, W.G.; Langevoort, H.L. The Mononuclear Phagocyte System: A New Classification of Macrophages, Monocytes, and Their Precursor Cells. Bull. World Health Organ. 1972, 46, 845–852. [Google Scholar] [PubMed]
- Ginhoux, F.; Merad, M. Ontogeny and Homeostasis of Langerhans Cells. Immunol. Cell Biol. 2010, 88, 387–392. [Google Scholar] [CrossRef] [PubMed]
- del Río-Hortega, P. Microglia. Cytol. Cell. Pathol. Nerv. Syst. 1932, 2, 483–534. [Google Scholar]
- Lull, M.E.; Block, M.L. Microglial Activation and Chronic Neurodegeneration. Neurotherapeutics 2010, 7, 354–365. [Google Scholar] [CrossRef]
- Meng, J.; Han, L.; Zheng, N.; Wang, T.; Xu, H.; Jiang, Y.; Wang, Z.; Liu, Z.; Zheng, Q.; Zhang, X.; et al. Microglial Tmem59 Deficiency Impairs Phagocytosis of Synapse and Leads to Autism-Like Behaviors in Mice. J. Neurosci. 2022, 42, 4958–4979. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Paolicelli, R.C.; Sforazzini, F.; Weinhard, L.; Bolasco, G.; Pagani, F.; Vyssotski, A.L.; Bifone, A.; Gozzi, A.; Ragozzino, D.; et al. Deficient Neuron-Microglia Signaling Results in Impaired Functional Brain Connectivity and Social Behavior. Nat. Neurosci. 2014, 17, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Sanches, E.S.; Boia, R.; Leitão, R.A.; Madeira, M.H.; Fontes-Ribeiro, C.A.; Ambrósio, A.F.; Fernandes, R.; Silva, A.P. Attention-Deficit/Hyperactivity Disorder Animal Model Presents Retinal Alterations and Methylphenidate Has a Differential Effect in ADHD versus Control Conditions. Antioxidants 2023, 12, 937. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.-X.; Kim, G.H.; Tan, J.-W.; Riso, A.E.; Sun, Y.; Xu, E.Y.; Liao, G.-Y.; Xu, H.; Lee, S.-H.; Do, N.-Y.; et al. Elevated Protein Synthesis in Microglia Causes Autism-like Synaptic and Behavioral Aberrations. Nat. Commun. 2020, 11, 1797. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-J.; Cho, M.-H.; Shim, W.H.; Kim, J.K.; Jeon, E.-Y.; Kim, D.-H.; Yoon, S.-Y. Deficient Autophagy in Microglia Impairs Synaptic Pruning and Causes Social Behavioral Defects. Mol. Psychiatry 2017, 22, 1576–1584. [Google Scholar] [CrossRef] [PubMed]
- Yokokura, M.; Takebasashi, K.; Takao, A.; Nakaizumi, K.; Yoshikawa, E.; Futatsubashi, M.; Suzuki, K.; Nakamura, K.; Yamasue, H.; Ouchi, Y. In Vivo Imaging of Dopamine D1 Receptor and Activated Microglia in Attention-Deficit/Hyperactivity Disorder: A Positron Emission Tomography Study. Mol. Psychiatry 2021, 26, 4958–4967. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.-C.; Nichols, E.K.; Rauch, I.; Chang, W.-C.; Lin, P.M.; Misra, R.; Kitaoka, M.; Vance, R.E.; Saijo, K. Lytic Cell Death in Specific Microglial Subsets Is Required for Preventing Atypical Behavior in Mice. eNeuro 2021, 8, ENEURO.0342-20.2020. [Google Scholar] [CrossRef] [PubMed]
- Reshef, R.; Kudryavitskaya, E.; Shani-Narkiss, H.; Isaacson, B.; Rimmerman, N.; Mizrahi, A.; Yirmiya, R. The Role of Microglia and Their CX3CR1 Signaling in Adult Neurogenesis in the Olfactory Bulb. eLife 2017, 6, e30809. [Google Scholar] [CrossRef] [PubMed]
- Dixon, M.A.; Greferath, U.; Fletcher, E.L.; Jobling, A.I. The Contribution of Microglia to the Development and Maturation of the Visual System. Front. Cell. Neurosci. 2021, 15, 659843. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s Disease and Its Potential as Therapeutic Target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef]
- Liu, Z.; Ning, J.; Zheng, X.; Meng, J.; Han, L.; Zheng, H.; Zhong, L.; Chen, X.-F.; Zhang, X.; Luo, H.; et al. TMEM59 Interacts with TREM2 and Modulates TREM2-Dependent Microglial Activities. Cell Death Dis. 2020, 11, 678. [Google Scholar] [CrossRef] [PubMed]
- Bruining, H.; Hardstone, R.; Juarez-Martinez, E.L.; Sprengers, J.; Avramiea, A.-E.; Simpraga, S.; Houtman, S.J.; Poil, S.-S.; Dallares, E.; Palva, S.; et al. Measurement of Excitation-Inhibition Ratio in Autism Spectrum Disorder Using Critical Brain Dynamics. Sci. Rep. 2020, 10, 9195. [Google Scholar] [CrossRef]
- Canitano, R.; Palumbi, R. Excitation/Inhibition Modulators in Autism Spectrum Disorder: Current Clinical Research. Front. Neurosci. 2021, 15, 753274. [Google Scholar] [CrossRef]
- Siegel-Ramsay, J.E.; Romaniuk, L.; Whalley, H.C.; Roberts, N.; Branigan, H.; Stanfield, A.C.; Lawrie, S.M.; Dauvermann, M.R. Glutamate and Functional Connectivity—Support for the Excitatory-Inhibitory Imbalance Hypothesis in Autism Spectrum Disorders. Psychiatry Res. Neuroimaging 2021, 313, 111302. [Google Scholar] [CrossRef] [PubMed]
- Cohen, L.D.; Ziv, T.; Ziv, N.E. Synapse Integrity and Function: Dependence on Protein Synthesis and Identification of Potential Failure Points. Front. Mol. Neurosci. 2022, 15, 1038614. [Google Scholar] [CrossRef]
- Guang, S.; Pang, N.; Deng, X.; Yang, L.; He, F.; Wu, L.; Chen, C.; Yin, F.; Peng, J. Synaptopathology Involved in Autism Spectrum Disorder. Front. Cell. Neurosci. 2018, 12, 470. [Google Scholar] [CrossRef]
- Amorim, I.S.; Lach, G.; Gkogkas, C.G. The Role of the Eukaryotic Translation Initiation Factor 4E (eIF4E) in Neuropsychiatric Disorders. Front. Genet. 2018, 9, 424981. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chen, H.; Ding, L.; Huang, J.; Zhang, M.; Liu, Y.; Ma, R.; Zheng, S.; Gong, J.; Piña-Crespo, J.C.; et al. Microglial Targeted Therapy Relieves Cognitive Impairment Caused by Cntnap4 Deficiency. Explor. Beijing China 2023, 3, 20220160. [Google Scholar] [CrossRef]
- Su, Z.; Herholz, K.; Gerhard, A.; Roncaroli, F.; Du Plessis, D.; Jackson, A.; Turkheimer, F.; Hinz, R. [11C]-(R)PK11195 Tracer Kinetics in the Brain of Glioma Patients and a Comparison of Two Referencing Approaches. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 1406–1419. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P.-Y. Inflammasomes: Mechanism of Action, Role in Disease, and Therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef]
- Crapser, J.D.; Ochaba, J.; Soni, N.; Reidling, J.C.; Thompson, L.M.; Green, K.N. Microglial Depletion Prevents Extracellular Matrix Changes and Striatal Volume Reduction in a Model of Huntington’s Disease. Brain 2020, 143, 266–288. [Google Scholar] [CrossRef] [PubMed]
- Palazuelos, J.; Aguado, T.; Pazos, M.R.; Julien, B.; Carrasco, C.; Resel, E.; Sagredo, O.; Benito, C.; Romero, J.; Azcoitia, I.; et al. Microglial CB2 Cannabinoid Receptors Are Neuroprotective in Huntington’s Disease Excitotoxicity. Brain J. Neurol. 2009, 132, 3152–3164. [Google Scholar] [CrossRef] [PubMed]
- Crotti, A.; Benner, C.; Kerman, B.; Gosselin, D.; Lagier-Tourenne, C.; Zuccato, C.; Cattaneo, E.; Gage, F.H.; Cleveland, D.W.; Glass, C.K. Mutant Huntingtin Promotes Autonomous Microglia Activation via Myeloid Lineage-Determining Factors. Nat. Neurosci. 2014, 17, 513–521. [Google Scholar] [CrossRef] [PubMed]
- O’Regan, G.C.; Farag, S.H.; Casey, C.S.; Wood-Kaczmar, A.; Pocock, J.M.; Tabrizi, S.J.; Andre, R. Human Huntington’s Disease Pluripotent Stem Cell-Derived Microglia Develop Normally but Are Abnormally Hyper-Reactive and Release Elevated Levels of Reactive Oxygen Species. J. Neuroinflamm. 2021, 18, 94. [Google Scholar] [CrossRef] [PubMed]
- Pavese, N.; Gerhard, A.; Tai, Y.F.; Ho, A.K.; Turkheimer, F.; Barker, R.A.; Brooks, D.J.; Piccini, P. Microglial Activation Correlates with Severity in Huntington Disease: A Clinical and PET Study. Neurology 2006, 66, 1638–1643. [Google Scholar] [CrossRef] [PubMed]
- Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The Role of Microglia and Astrocytes in Huntington’s Disease. Front. Mol. Neurosci. 2019, 12, 258. [Google Scholar] [CrossRef]
- Luo, Y.; Lv, K.; Du, Z.; Zhang, D.; Chen, M.; Luo, J.; Wang, L.; Liu, T.; Gong, H.; Fan, X. Minocycline Improves Autism-Related Behaviors by Modulating Microglia Polarization in a Mouse Model of Autism. Int. Immunopharmacol. 2023, 122, 110594. [Google Scholar] [CrossRef]
- Saft, C.; von Hein, S.M.; Lücke, T.; Thiels, C.; Peball, M.; Djamshidian, A.; Heim, B.; Seppi, K. Cannabinoids for Treatment of Dystonia in Huntington’s Disease. J. Huntingt. Dis. 2018, 7, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Girgis, R.R.; Van Snellenberg, J.X.; Glass, A.; Kegeles, L.S.; Thompson, J.L.; Wall, M.; Cho, R.Y.; Carter, C.S.; Slifstein, M.; Abi-Dargham, A.; et al. A Proof-of-Concept, Randomized Controlled Trial of DAR-0100A, a Dopamine-1 Receptor Agonist, for Cognitive Enhancement in Schizophrenia. J. Psychopharmacol. Oxf. Engl. 2016, 30, 428–435. [Google Scholar] [CrossRef]
- Singh, N.; Das, B.; Zhou, J.; Hu, X.; Yan, R. Targeted BACE-1 Inhibition in Microglia Enhances Amyloid Clearance and Improved Cognitive Performance. Sci. Adv. 2022, 8, eabo3610. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furlan, V.A.; MacAuslan, D.; Ha, K.; Patel, N.; Adam, S.; Zanagar, B.; Venugopal, S. Microglial Dyshomeostasis: A Common Substrate in Neurodevelopmental and Neurodegenerative Diseases. Neuroglia 2024, 5, 119-128. https://doi.org/10.3390/neuroglia5020009
Furlan VA, MacAuslan D, Ha K, Patel N, Adam S, Zanagar B, Venugopal S. Microglial Dyshomeostasis: A Common Substrate in Neurodevelopmental and Neurodegenerative Diseases. Neuroglia. 2024; 5(2):119-128. https://doi.org/10.3390/neuroglia5020009
Chicago/Turabian StyleFurlan, Vada Andree, Daria MacAuslan, Khiem Ha, Nitish Patel, Shawn Adam, Beylem Zanagar, and Sharmila Venugopal. 2024. "Microglial Dyshomeostasis: A Common Substrate in Neurodevelopmental and Neurodegenerative Diseases" Neuroglia 5, no. 2: 119-128. https://doi.org/10.3390/neuroglia5020009
APA StyleFurlan, V. A., MacAuslan, D., Ha, K., Patel, N., Adam, S., Zanagar, B., & Venugopal, S. (2024). Microglial Dyshomeostasis: A Common Substrate in Neurodevelopmental and Neurodegenerative Diseases. Neuroglia, 5(2), 119-128. https://doi.org/10.3390/neuroglia5020009