Abstract
The blood–brain barrier (BBB) is a highly intricate neurovascular structure that plays a crucial role in maintaining neural homeostasis by selectively allowing certain molecules to enter the central nervous system (CNS). However, in the context of Alzheimer’s Disease (AD), a progressive neurodegenerative disorder characterized by a gradual decline in cognitive function, the BBB’s functionality becomes impaired. This impairment leads to the breakdown of the barrier and disrupts its ability to regulate molecular transport effectively. Consequently, cellular infiltration into the CNS occurs, along with aberrant signaling and clearance of molecules, ultimately contributing to neurological deficits. One of the key factors implicated in the failure of amyloid-beta (Aβ) transport, a hallmark of AD, is the decreased expression of low-density lipoprotein receptor-related protein 1 (LRP1). LRP1 plays a crucial role in facilitating the transport of Aβ across the BBB. Additionally, the increased levels of the receptor for advanced glycation end products (RAGE) further contribute to the deregulation of the BBB in AD. These molecular imbalances significantly impact Aβ clearance and contribute to the development and progression of AD. In this review, we aimed to summarize the critical aspects of Aβ transporters in the BBB that become dysfunctional during the pathogenesis of AD.
1. Introduction
The blood–brain barrier (BBB) is a complex neurovascular structure responsible for the control of neural homeostasis through the selection of molecules that can reach the central nervous system (CNS). It is composed of brain endothelial cells (ECs), pericytes, astrocytes, microglial cells, and smooth muscle cells (SMCs), forming a continuous endothelial membrane within brain microvessels that has sealed cell-to-cell contacts, and forming the neurovascular unit (NVU) that contributes to the regulation of BBB permeability [1]. The BBB is a unique structure where tight junctions and absence of fenestrations result in an almost impenetrable barrier, essential to protect the brain from toxins and other harmful substances. However, it also works as a hermetic barrier, preventing the success of therapies directed to brain disorders through the selection of drugs [2].
Cerebral blood vessels line up with the major brain circuits related to sensation, memory, and motion, suggesting that the cerebrovascular system plays an important role in normal CNS functioning [3]. At the capillary level, BBB integrity is maintained by pericytes [4] that, together with the SMCs and ECs, express many different transcripts encoding a large number of transporters, receptors, active efflux pumps, ion channels, and regulatory molecules [5], whose expression patterns vary along the capillaries [6]. Under disease conditions, BBB breakdown and dysfunction leads to NVU dysregulation and defective signaling, resulting in cellular infiltration into the CNS, along with the aberrant transport and clearance of molecules, contributing to neurological deficits [7].
Neuroinflammation is one of the main consequences of BBB breakdown, resulting in drastic changes in the neural environment, due to microglia activation and the release of several cytokines and oxidative compounds [8]. These environmental changes increase the expression of cell adhesion molecules in ECs, alters the pattern and function of BBB transporters, receptors, and channels, and disrupt tight and adherent junctions, resulting in higher rates of immune cell recruitment and infiltration into the CNS [9]. Infiltrated leukocytes are activated, contributing to the maintenance of a pro-inflammatory environment and leading to NVU disarrangement, along with the detachment of pericytes and astrocytic endfeet from the EC layer. With loss of EC cell-to-cell contacts and NVU disassembly, the BBB loses its barrier capacity, and a large group of molecules can pass freely through the BBB into the CNS [10].
The activation of kinin receptors has been associated with BBB breakdown and neuroinflammation, but the direct role of this activity in the inflammatory profile and permeability of brain microvascular cells has not been fully studied [11]. It has already been observed that activation of the B1 kinin receptor (B1R) in the endothelial cells of brain microcapillaries can lead to an increase in the secretion of pro-inflammatory cytokines (e.g., IL-6 and IL-8) associated with inflammation of endothelial cells and leukocyte adhesion and recruitment, in addition to increased paracellular permeability due to the loss of the tight junction protein [12].
In a pro-inflammatory environment, several cellular changes are observed. Astrocytes and microglia are activated, turning into an inflammatory phenotype, releasing cytokines, such as IL-1, IL-1β, IL-6, IL-18, and tumor necrosis factor (TNF), chemokines, such as C-C motif chemokine ligand 1 (CCL1), CCL5, and C-X-C motif chemokine ligand 1 (CXCL1), small-molecule messengers, including prostaglandins and nitric oxide (NO), and reactive oxygen species by innate immune cells in the CNS, resulting in neuron mitochondrial dysfunction, generating oxidative stress and neuron dysfunction, and death [13], that is the main consequence of neurodegenerative diseases, such as Parkinson’s disease (PD) and Alzheimer’s disease (AD).
2. Pathogenesis of AD
AD is a progressive neurodegenerative disorder and a disease of aging, being characteristic in people over 60 years old, with a higher incidence in elderly people over 75 years old, which is clinically characterized by progression from incipient cognitive impairment to severe cognitive damage, including memory impairment, language deterioration, visuospatial deficits, emotional and thinking problems, and a decrease in motivation [14]. Epidemiological data show that the incidence of AD in the world population has been growing significantly in the last two decades, and projections show that the trend is for an even more accentuated increase in the coming years [15].
It is a disease that is difficult to diagnose early, being mostly identified when the patient begins to develop more acute symptoms of dementia. However, at this stage, the pathology is already well developed, and the patient already has an accumulation of amyloid-β (Aβ) plaques, tau-mediated neuronal dysfunction, and structural changes in the brain [16]. Cerebral plaques containing aggregates of various Aβ peptides derived from the amyloid precursor protein (APP), as well as from the neurofibrillary tangles (NFTs) containing hyperphosphorylated and aggregated tau, are the main hallmarks of AD pathology [17]. Recent studies have shown that amyloid pathology begins earlier than tau pathology in the course of AD development. The amyloid cascade hypothesis states that Aβ becomes abnormal, and that this change triggers cortical tau pathology and tau-mediated neurodegeneration [18,19,20]. Physiologically, in healthy neurons, APP will be digested by α and γ-secretase enzymes that produce soluble polypeptides that can be broken and recycled in the cell. However, when β-secretase binds to γ-secretase, instead of α-secretase, the resulting product is Aβ, an insoluble peptide, which clumps together and forms Aβ plaques, which is neurotoxic [21]. Aβ plaques may cause some problems in the cell, such as the disruption of synapses if they are located between two healthy neurons, impairing memory and generating immune responses, leading to inflammation and damaging the surrounding neurons [22]. Aβ plaques may also cause angiopathy when deposited on the outer side of blood vessels, causing either hemorrhage or the rupture of the vessel [23]. Under normal circumstances, Aβ clearance occurs rapidly. It is estimated that in 13 min Aβ is removed from the brain via transport across the BBB into the bloodstream by the low-density lipoprotein receptor-related protein 1 (LRP1), preventing the accumulation of Aβ in the brain and, consequently, the formation of Aβ plaques [24]. All these phenomena take place in the extracellular compartment. Inside the cell, problems are generated by the NFTs.
NFTs are neurofibrillary tangles of phosphorylated tau proteins that break off the surface of axonal microtubules where they stabilize their packaged structure. Tau binds to the carboxy-terminal half of the microtubules, which has three or four semi-homologous repeats of thirty-one or thirty-two amino acids that are encoded by exon 10 [25]. The lack of tau proteins in the axonal microtubules weakens their structure, which may lead to the loss of their signaling function, since the axon is the main route for transporting molecules to different regions of the cell. It has been already described in the literature that Aβ plaques formed in the extracellular space trigger intracellular signaling that leads to the activation of kinases that, in turn, phosphorylate tau, and lead to the formation of the NFTs [26].
3. Aβ Clearance through the BBB
LRP1 is mainly localized to the abluminal side of the BBB and is the key receptor mediating Aβ transcytosis across the BBB into the circulation [24]. Each species of Aβ has a different transport rate via LRP1-mediated clearance; Aβ40 is cleared at the fastest rate, and Aβ42 is cleared at a faster rate than the vasculotropic Dutch Aβ mutants [27]. Brain-derived Aβ binds to LRP1 at the abluminal side of the BBB and is rapidly internalized into ECs and cleared into the bloodstream. Some EC molecules participate in this process: endothelial cell-dependent clathrin/phosphatidylinositol-binding clathrin assembly protein (PICALM)-mediated internalization of LRP1–Aβ complexes [28]. Clathrin rapidly dissociates from internalized vesicles, and PICALM continues to drive intracellular traffic of Aβ-containing endocytic vesicles through the endothelium, fusing them first with the Rab5-positive and later with the Rab11-positive endosomes, mediating Aβ exocytosis on the luminal side of the BBB [28].
There are many genetic risk factors and brain endothelial genes that can influence LRP1-mediated Aβ clearance, leading to its accumulation in the brain. Apolipoprotein E (APOE) is one of the key factors in controlling Aβ accumulation since its three isoforms, APOE2, APOE3, and APOE4, interact with Aβ in different ways, regulating its metabolism and transport through the BBB [29]. APOE4 is the major risk factor for AD, contributing to vascular and neuronal dysfunction through the Aβ-dependent and independent pathways. Rapid BBB clearance of Aβ complexes with APOE2 and APOE3 is mediated by LRP1; on the other hand, APOE4-Aβ complexes do not interact with LRP1 and are removed via the much slower, very low-density lipoprotein receptor (VLDLR)-mediated internalization and transcytosis, contributing to Aβ accumulation in the brain (Figure 1) [30,31].
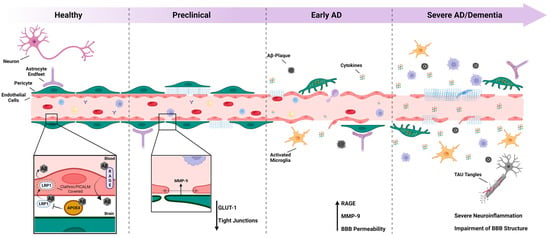
Figure 1.
The blood–brain barrier and Alzheimer’s disease: The blood–brain barrier (BBB) is a dynamic and vital structure that plays a crucial role in maintaining the homeostasis of the central nervous system (CNS). However, in the context of Alzheimer’s disease (AD), the BBB’s integrity becomes compromised, leading to chronic neuroinflammation and the accumulation of Aβ and tau proteins in the brain. During the preclinical phase of AD, the tight junctions between endothelial cells of the BBB start to deteriorate, resulting in the increased permeability of blood vessels. This breakdown is followed by the uncoupling of pericytes, further exacerbating the passage of cytokines and Aβ plaques between the brain tissue and the vessel lumen. As the disease progresses to the advanced stage, neurons are adversely affected, resulting in the formation of tau tangles, and a severe neuroinflammatory environment is established. At this point, the BBB experiences an almost complete rupture, severely compromising its function. Overall, the progressive loss of BBB integrity in AD leads to chronic neuroinflammation and the accumulation of the Aβ and tau proteins, and ultimately contributes to the pathogenesis of the disease.
In AD brain ECs, PICALM levels are reduced, which can lead to Aβ accumulation in the brain by hindering LRP1-mediated Aβ transport, since the Rab5-positive early-endosome ends up fusing with the Rab7-positive late endosome, which guide endocytic vesicles to lysosomes, resulting in minimal degradation of Aβ during its transcytosis across the BBB. Several AD-associated single-nucleotide polymorphisms (SNPs) have been described as blockers of PICALM expression, impairing Aβ clearance [28].
Clusterin (CLU), a gene which encodes a heterodimeric-secreted chaperone glycoprotein, is the most abundantly expressed apolipoprotein in the brain, being involved with the clearance of extracellular misfolded proteins, and with the regulation of apoptosis, inflammation, and cancer [32]. Clusterin can bind to Aβ preventing its aggregation and export it via LRP2 through the BBB, promoting their clearance from the brain significantly faster [33]. Animal models of transgenic AD mice have shown that the lack or decrease of clusterin results in increased Aβ aggregates in the brain [34].
Mesenchyme homeobox gene 2 (MEOX2) is substantially reduced in AD human brain ECs, which leads to aberrant angiogenesis, vascular regression, endothelial hypoplasia, and accelerated proteasomal degradation of LRP1, contributing to brain hypoperfusion and less LRP1-mediated Aβ clearance from the brain [35]. The glucose transporter GLUT1 is also downregulated in AD brain ECs, causing decreased glucose uptake and LRP1 downregulation via sterol-binding protein-2 (SREBP2)-dependent transcription, resulting in Aβ accumulation [36].
In vascular smooth muscle cells (VSMCs), serum response factor (SRF) and myocardin (MYOD) are upregulated in AD-derived VSMCs. Overexpression of SRF and MYOD initiates a hypercontractile phenotype in cerebral arteries via contractile proteins and Ca2+ homeostasis genes that are regulated by SRF/MYOD, resulting in cerebral hypoperfusion, diminished neurovascular coupling, and transactivation of SREBP2, a transcriptional suppressor of LRP1, reducing LRP1 expression and diminishing Aβ clearance via VSMCs in small penetrating vessels [37].
P-glycoprotein (P-gp), encoded by the ABCB1 gene, also known as multidrug resistance protein 1 (MDR1), is an ATP-binding cassette transporter responsible for decreased drug accumulation in multidrug-resistant cells and often mediates the development of resistance to anticancer drugs [38]. It is highly expressed on the luminal side of the BBB [39], facilitating the clearance of Aβ from the brain [40], and its expression and activity in the brain are inversely correlated with aging, Aβ deposition and AD [41]. Aβ itself has been found to compromise the expression of P-gp, thereby exacerbating Aβ deposition and disease [42].
4. Aβ Uptake through the BBB
Receptor for advanced glycation end products (RAGE) is a cell-surface receptor of the immunoglobulin (IgG) superfamily. It transports Aβ into the brain and accelerates Aβ pathology in AD mouse models. Most cell populations involved with the NVU, such as ECs, VSMCs, pericytes, glial cells, and neurons, express low levels of RAGE that can bind to monomeric Aβ via its V domain, and to aggregated Aβ via its C1 domain [43]. RAGE also mediates Aβ-induced neurotoxicity directly by causing oxidative stress and indirectly by activating microglia, leading to neuroinflammation, and its expression is increased in the ECs and neurons in an Aβ-rich environment, where it mediates the transport of circulating Aβ from the luminal side of the ECs across the BBB into the brain [44]. Blockage or reduced RAGE expression in the ECs may be a potential target to control Aβ influx and toxicity in AD. Contrary to RAGE, the soluble extracellular domain of RAGE (sRAGE), found circulating in the plasma, sequesters circulating RAGE ligands, including Aβ, preventing their interaction with cell-surface RAGE [45]. In AD patients, levels of plasma sRAGE is reduced, indicating that the lack of sRAGE may increase Aβ accumulation (Figure 1), and experimental models using AD mice with sRAGE inhibited showed Aβ accumulation in the brain parenchyma, reinforcing that sRAGE may be a potential therapeutic target for AD [46].
5. The Role of Astrocytes in AD: Implications of Aβ and Tau Pathologies
Astrocytes are a key component of the central nervous system, playing an essential role in maintaining normal brain function. In conjunction with endothelial cells, they comprise the BBB, where, by secreting a range of factors, assumes a pivotal responsibility in upholding the functionality and integrity of the BBB. In addition to their role in BBB homeostasis, these cells are involved in several other functions, such as providing metabolic support, modulating the immune response, and regulating the extracellular environment. Astrocytes are also critical for synaptic plasticity and neuronal communication, and they can modulate the activity of neurons by releasing neurotransmitters and gliotransmitters [47].
One of the key functions of astrocytes is their role in maintaining the extracellular environment. These cells are involved in the regulation of the pH, ionic composition, and osmolarity of the extracellular fluid, which are critical for proper neuronal function. Astrocytes are also involved in the clearance of neurotransmitters, such as glutamate, from the synaptic cleft, preventing the overstimulation of neurons that could lead to excitotoxicity [48]. In addition, astrocytes are involved in the uptake and metabolism of glucose and other metabolites, providing neurons with the energy they need to function properly [49].
Alterations in astrocytic function have been implicated in several neurological disorders, such as Alzheimer’s disease (AD), Parkinson’s disease, and epilepsy. For example, in AD, astrocytes become reactive and release pro-inflammatory cytokines, contributing to the neuroinflammation and neuronal damage seen in the disease [50].
In AD, the accumulation of Aβ oligomers (AβOs) in the brain can alter astrocytic function and contribute to neuroinflammation and neurodegeneration. Aβ oligomers can activate astrocytes, inducing an inflammatory response characterized by the release of pro-inflammatory cytokines, chemokines, and reactive oxygen species. This inflammatory response can lead to the activation of microglia and the further release of pro-inflammatory cytokines, further exacerbating neuroinflammation and neuronal dysfunction. In addition to the pro-inflammatory response, astrocytes can also release glutamate in response to Aβ oligomers, which can lead to neuronal hyperexcitability and further neurotoxicity [51,52].
Furthermore, AβOs can inhibit the production of neurotrophic factors, such as brain-derived neurotrophic factor (BDNF), by astrocytes, leading to impaired neuronal survival and plasticity [53,54].
The interaction between astrocytes and Aβ oligomers is complex and not yet fully understood. However, recent studies have shed light on some of the mechanisms through which AβOs alter astrocytic function. Our group was among the first to demonstrate that AβOs bind to the surface of astrocytes and are internalized, resulting in elevated levels of reactive oxygen species (ROS) and nitrite [55]. For example, Aβ oligomers can bind to several receptors on astrocytes, including Toll-like receptor 4 (TLR4), scavenger receptor class B type I (SR-BI), and receptor for advanced glycation end products (RAGE), leading to the activation of intracellular signaling pathways and the induction of an inflammatory response. Aβ oligomers can also inhibit the activity of neprilysin, a key enzyme involved in Aβ clearance, in astrocytes, leading to further Aβ accumulation and toxicity [56].
Furthermore, Alzheimer’s disease is characterized by the presence of tau protein aggregates in addition to beta-amyloid protein aggregation. While Aβ forms plaques outside of neurons, tau forms neurofibrillary tangles inside them. Both proteins have been implicated in the death of brain cells and the subsequent cognitive decline observed in AD patients. In Alzheimer’s disease, the accumulation of tau in the astrocytes of the dentate gyrus leads to neuronal dysfunction and memory deficits [57].
A recent study using TRAP-seq translatome analysis of astrocytes in mice with Aβ and tau pathology found that while only Aβ influenced the expression of AD-risk genes, both pathologies induced age-dependent changes with overlapping signatures similar to those found in human post-mortem AD astrocytes. This signature included the repression of bioenergetic and translation machinery, inflammation pathways, and protein degradation/proteostasis genes, which were enriched in targets of the inflammatory mediator Spi1 and the stress-activated cytoprotective Nrf2. Thus, both Aβ and tau induce overlapping astrocytic profiles that can have both harmful and adaptive-protective effects. Understanding the effects of Aβ and tau on astrocytes is crucial for developing effective therapies for not only AD, but also for other neurodegenerative diseases [58].
6. The Role of Astrocytes in Synaptic Dysfunction in AD
One of the hallmark features of AD is the loss of synapses in the brain, which has been thought to contribute to the cognitive deficits observed in affected individuals. While neurons have traditionally been considered the primary drivers of synapse loss in AD, recent evidence suggests that astrocytes, a type of glial cell in the brain, may also play an important role [59].
Several studies have demonstrated that astrocytes are closely associated with synapses and are critical for their maintenance and plasticity. In the context of AD, astrocytes have been shown to become reactive and secrete pro-inflammatory cytokines, which can contribute to synapse loss and neurodegeneration [60]. Moreover, astrocytes have been found to play a key role in regulating the clearance of the Aβ protein, a hallmark feature of AD pathology. Dysregulation of Aβ clearance via astrocytes can lead to the accumulation of toxic Aβ aggregates, which can trigger inflammation and synapse loss [61].
While the precise mechanisms by which astrocytes contribute to synapse loss in AD are still being elucidated, recent studies have provided insights into potential therapeutic strategies. For example, one study demonstrated that enhancing the astrocytic clearance of Aβ using a small-molecule drug improved synaptic plasticity and cognitive function in AD mouse models [62]. Another study showed that inhibiting astrocytic pro-inflammatory cytokine production reduced synapse loss and improved cognitive function in a mouse model of AD [63].
Yue and colleagues demonstrated, utilizing an in vitro model, that AβO42 itself does not cause damage to endothelial cells. The conditioned medium of astrocytes, which were previously exposed to AβO42, exhibited the ability to reduce the expression of ZO-1 and Claudin-5, as well as inhibit the phosphorylation of VEGFR2, eNOS, and the signaling pathways of ERK/MAPK and PI3K/AKT [64]. These results suggest that astrocytes could be one of the main sources of factors that could promote BBB disruption. Indeed, a recent report has shown that APOE4, one of the main factors that lead to Aβ accumulation, secreted by astrocytes can impair BBB integrity. APOE4 knock-in mice exhibited an increased BBB permeability with a reduction in the number of astrocytic endfeet, disrupted tight junctions, and an increase in the level of MMP-9 expression [65]. This disruption of the BBB was rescued after one month through specifically knocking out APOE4 from the astrocytes [65]. In the same direction, Kim and coworkers showed that canonical inflammatory factors alone, such as IL1α, TNF, and C1q, were not able to induce the disruption of BBB endothelial cells in vitro [66]. Indeed, contact of endothelial cells with reactive astrocytes in vitro was necessary to induce BBB disruption-like responses and inhibition of astrocytic STAT3 abolishes inflammatory effects on BBB disruption [66].
7. Targeting the BBB and Reactive Astrocytes in AD: Promising Therapeutic Strategies
Clinical strategies for using the BBB in AD treatment are based on trying to control its permeability, opening the BBB in a transient and reversible manner. Studies in rodents and non-human primates have shown that opening the BBB facilitates the efflux of Aβ and tau into the bloodstream, improving the prognosis of animals with AD [67]; this is the reason why numerous clinical trials use focused ultrasound (FUS) technology, which allows the reversible and localized opening of the BBB with little or no adverse effects. However, so far, despite fine control of BBB permeability, clinical outcomes in patients with AD have not been promising, as opening the BBB has not affected the levels of AD markers [68]. Another possibility is the increased influx of drugs or monoclonal antibodies into the brain tissue when the BBB is open. Studies have shown that the influx of these molecules increases considerably when the BBB is opened using the FUS technique, favoring the pharmacological treatment of patients [69]. Table 1 summarizes some of the ongoing or completed clinical trials that use the aforementioned technologies to alter BBB physiology.

Table 1.
List summarizing several ongoing or completed clinical trials that have specifically targeted the blood–brain barrier (BBB) or reactive astrocytes.
Reactive astrogliosis is a hallmark of many neurological diseases, including Alzheimer’s disease. In response to injury or disease, astrocytes undergo morphological and functional changes that can be classified into two main types: A1 and A2. A1 astrocytes are characterized by the upregulation of genes involved in neuroinflammation and phagocytosis, while A2 astrocytes are associated with the production of neurotrophic factors and a decrease in inflammation. In AD, astrocytes display a mixed A1/A2 phenotype, with a predominant A1-like response observed in areas of high amyloid-beta pathology [72,73].
Pharmacological strategies to modulate astrocyte reactivity in AD have been a recent focus of study. One promising approach involves drugs that target TLR4, a receptor involved in the induction of an A1 astrocyte phenotype mediated by microglia. Such drugs have shown potential in reducing neuroinflammation and improving cognitive function in animal models of AD [74].
In a recent study, the authors aimed to investigate the role of the JAK2-STAT3 pathway in controlling astrocyte reactivity in Alzheimer’s disease and to evaluate its potential as a therapeutic target. They found that the JAK2-STAT3 pathway plays a crucial role in the induction and maintenance of reactive astrocytes in vivo, and its inhibition can significantly reduce astrocyte reactivity and amyloid deposition, and improve cognitive deficits in AD mouse models. These findings suggest that targeting the JAK2-STAT3 pathway may be a promising strategy for treating AD [75].
In AD, microglia convert resting astrocytes into reactive astrocytes, making them a major therapeutic target. A recent study has demonstrated that NLY01, a glucagon-like peptide-1 receptor (GLP-1R) agonist, can block microglia-induced astrocyte activation and preserve neurons in AD models, improving spatial learning and memory. The GLP-1 pathway plays a critical role in microglia-reactive astrocyte-associated neuroinflammation in AD, and the effects of NLY01 are mediated through direct actions on Aβ-induced GLP-1R+ microglia, inhibiting astrocyte reactivity. Therefore, drugs that activate GLP-1R, such as NLY01, could represent a promising therapeutic strategy for the treatment of AD and other neurodegenerative diseases [76].
While the presence of reactive astrocytes in Alzheimer’s pathology has been extensively investigated, the impact of the absence of astrocytes in organotypic culture models has been used to understand the role of neuroglial interactions in Alzheimer’s pathology. To investigate the role of astrocytes in AD, researchers have developed an ex vivo model using brain slices from transgenic mice that overexpress Aβ. By selectively ablating astrocytes using a pharmacological agent, these researchers were able to demonstrate that astrocytes are critical for the degradation of Aβ and for the maintenance of synaptic connectivity. Specifically, the pharmacological ablation of astrocytes led to a significant increase in Aβ levels in the brain slices, indicating that astrocytes play a key role in clearing Aβ from the brain. In addition, the loss of astrocytes resulted in a significant decrease in the density of the dendritic spines, which are critical for synaptic connectivity and plasticity [77].
In addition to pharmacological strategies, non-pharmacological interventions, such as exercise, have also been shown to modulate astrocyte reactivity in AD. Exercise has been shown to reduce neuroinflammation and improve cognitive function in mouse models of AD, possibly by promoting an A2-like response in astrocytes [78,79]. Similarly, cognitive stimulation has also been shown to reduce neuroinflammation and improve cognitive function in mouse models of AD [80,81]. Although these results are promising, future studies are needed to confirm the involvement of astrocyte reactivity as the central mechanism underlying the observed benefits in these approaches.
Another strategy that has recently gained a lot of space in clinical trials is the imaging of astrocytes and microglia cells using the positron emission tomography (PET) technique in an attempt to identify the onset of neuroinflammation early [82,83]. The results are promising, as increasingly specific markers are being developed, allowing for the imaging and quantification of activated cells [84]. The objective is to establish a quantitative cut-off point that allows, with imaging, to identify at which stage of the disease the patient is in, and which cells specifically are involved in this process, in addition to better understanding the pathogenesis of AD, identifying new, early therapeutic targets (Table 1).
Advanced imaging techniques and fluid biomarkers have provided valuable tools to measure astrocyte reactivity in vivo, allowing researchers to gain insights into the dynamics of astrocytic responses throughout the course of AD. Emerging evidence suggests that astrocyte dysfunction in AD could be a contributing factor to the observed hypometabolism detected via [18F]FDG-PET imaging. This hypometabolism is a characteristic metabolic impairment seen in the brains of AD patients and has been linked to the functional alterations in astrocytes [85].
In a recent study, researchers found that elevated levels of glial fibrillary acidic protein (GFAP) and chitinase-3-like protein 1 (YKL-40) in the CSF were associated with increased amyloid-β accumulation in the brain; in contrast, higher levels of YKL-40 in the CSF were linked to elevated tau pathology. Structural equation modeling revealed that CSF GFAP and YKL-40 play a role in mediating the effects of Aβ and tau on hippocampal atrophy, ultimately contributing to cognitive impairment. These findings suggest the existence of distinct astrocyte biomarker signatures in response to Aβ and tau, providing valuable new insights into the intricate mechanisms underlying reactive astrogliosis and the development of Alzheimer’s disease [86,87].
Currently, there are no clinical studies investigating the impact of blocking or reversing astrocytic reactivity in Alzheimer’s disease. However, significant progress has been made in understanding this area through preclinical models, which serve as crucial tools for studying cellular and molecular mechanisms in the context of Alzheimer’s disease. Further exploration of the literature is needed to gain deeper insights, such as understanding the molecular mechanisms governing the conversion of A1 into A2 astrocytes, which may aid in the recovery or protection of synaptic deficits. Additionally, investigating the inflammatory signaling of astrocytes and microglia crosstalk will be crucial in triggering the inflammatory response and identifying multi-target drugs to control inflammation, astrocytic reactivity, and amyloid and tau pathology.
Astrocytes offer great promise as potential targets for the development of novel and specific fluid or imaging biomarkers to detect preclinical AD. Their involvement in the pathological processes of AD makes them valuable candidates for identifying the early signs of the disease, aiding in early diagnosis and intervention. Moreover, pharmacologically targeting astrocytes could open up promising avenues for developing effective treatments to combat AD. By understanding and modulating the astrocytic responses associated with AD, researchers may unlock new therapeutic possibilities to halt or slow down the progression of this devastating neurodegenerative disorder.
8. Conclusions
The BBB is vital for the proper functioning of the CNS as it is the main regulator of the nervous microenvironment, regulating the traffic of several molecules. BBB dysregulations are involved with the pathophysiology of a series of diseases, including AD. So far, it is not clear whether dysfunctions in the BBB are causes or consequences of AD, but we already know that it plays a vital role in the development and progression of this neurodegenerative disease, and that some possible therapeutic targets are involved in the processes of the transport of molecules across the BBB. Therefore, unraveling the processes of trafficking molecules across the BBB that are modified in the context of AD, and identifying possible therapeutic targets, can be a good strategy to combat this disease.
Author Contributions
Writing—original draft preparation, J.V.R.C., C.B., L.P.D. and F.A.M.; writing—review and editing, F.A.M.; supervision, F.A.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kunigelis, K.E.; Vogelbaum, M.A. Therapeutic Delivery to Central Nervous System. Neurosurg. Clin. N. Am. 2021, 32, 291–303. [Google Scholar] [CrossRef]
- Terstappen, G.C.; Meyer, A.H.; Bell, R.D.; Zhang, W. Strategies for delivering therapeutics across the blood–brain barrier. Nat. Rev. Drug Discov. 2021, 20, 362–383. [Google Scholar] [CrossRef]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood–brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef]
- Zeisel, A.; Muñoz-Manchado, A.B.; Codeluppi, S.; Lönnerberg, P.; La Manno, G.; Juréus, A.; Marques, S.; Munguba, H.; He, L.; Betsholtz, C.; et al. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 2015, 347, 1138–1142. [Google Scholar] [CrossRef] [PubMed]
- Vanlandewijck, M.; He, L.; Mäe, M.A.; Andrae, J.; Ando, K.; Del Gaudio, F.; Nahar, K.; Lebouvier, T.; Laviña, B.; Gouveia, L.; et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature 2018, 554, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Zhao, Z.; Zlokovic, B.V. Alzheimer’s disease: A matter of blood–brain barrier dysfunction? J. Exp. Med. 2017, 214, 3151–3169. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.M. The Role of the Blood Brain Barrier in Neurodegenerative Disorders and their Treatment. J. Alzheimer’s Dis. 2011, 24, 643–656. [Google Scholar] [CrossRef]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Takata, F.; Nakagawa, S.; Matsumoto, J.; Dohgu, S. Blood-Brain Barrier Dysfunction Amplifies the Development of Neuroinflammation: Understanding of Cellular Events in Brain Microvascular Endothelial Cells for Prevention and Treatment of BBB Dysfunction. Front. Cell. Neurosci. 2021, 15, 661838. [Google Scholar] [CrossRef]
- Sikpa, D.; Whittingstall, L.; Savard, M.; Lebel, R.; Côté, J.; McManus, S.; Chemtob, S.; Fortin, D.; Lepage, M.; Gobeil, F. Pharmacological Modulation of Blood–Brain Barrier Permeability by Kinin Analogs in Normal and Pathologic Conditions. Pharmaceuticals 2020, 13, 279. [Google Scholar] [CrossRef] [PubMed]
- Mugisho, O.O.; Robilliard, L.D.; Nicholson, L.F.B.; Graham, E.S.; O’Carroll, S.J. Bradykinin receptor-1 activation induces inflammation and increases the permeability of human brain microvascular endothelial cells. Cell Biol. Int. 2019, 44, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Eratne, D.; Loi, S.M.; Farrand, S.; Kelso, W.; Velakoulis, D.; Looi, J.C. Alzheimer’s disease: Clinical update on epidemiology, pathophysiology and diagnosis. Australas. Psychiatry 2018, 26, 347–357. [Google Scholar] [CrossRef] [PubMed]
- 2023 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2023, 19, 1598–1695. [CrossRef]
- Drew, L. An age-old story of dementia. Nature 2018, 559, S2–S3. [Google Scholar] [CrossRef]
- Hung, A.; Liang, Y.; Chow, T.C.; Tang, H.; Wu, S.L.; Wai, M.; Yew, D. Mutated tau, amyloid and neuroinflammation in Alzheimer disease—A brief review. Prog. Histochem. Cytochem. 2016, 51, 1–8. [Google Scholar] [CrossRef]
- van der Kant, R.; Goldstein, L.S.B.; Ossenkoppele, R. Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat. Rev. Neurosci. 2019, 21, 21–35. [Google Scholar] [CrossRef]
- Karran, E.; De Strooper, B. The amyloid cascade hypothesis: Are we poised for success or failure? J. Neurochem. 2016, 139, 237–252. [Google Scholar] [CrossRef]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef]
- Ashrafian, H.; Zadeh, E.H.; Khan, R.H. Review on Alzheimer’s disease: Inhibition of amyloid beta and tau tangle formation. Int. J. Biol. Macromol. 2020, 167, 382–394. [Google Scholar] [CrossRef]
- Sigurdsson, E.M.; Knudsen, E.; Asuni, A.; Fitzer-Attas, C.; Sage, D.; Quartermain, D.; Goni, F.; Frangione, B.; Wisniewski, T. An Attenuated Immune Response Is Sufficient to Enhance Cognition in an Alzheimer’s Disease Mouse Model Immunized with Amyloid-β Derivatives. J. Neurosci. 2004, 24, 6277–6282. [Google Scholar] [CrossRef] [PubMed]
- Kurz, C.; Walker, L.; Rauchmann, B.; Perneczky, R. Dysfunction of the blood–brain barrier in Alzheimer’s disease: Evidence from human studies. Neuropathol. Appl. Neurobiol. 2021, 48, e12782. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Zlokovic, B.V. Role of the Blood-Brain Barrier in the Pathogenesis of Alzheimers Disease. Curr. Alzheimer Res. 2007, 4, 191–197. [Google Scholar] [CrossRef]
- Boutajangout, A.; Wisniewski, T. Tau-Based Therapeutic Approaches for Alzheimer’s Disease—A Mini-Review. Gerontology 2014, 60, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S. Molecular and Cellular Basis of Neurodegeneration in Alzheimer’s Disease. Mol. Cells 2017, 40, 613–620. [Google Scholar] [CrossRef]
- Bell, R.D.; Sagare, A.P.; E Friedman, A.; Bedi, G.S.; Holtzman, D.M.; Deane, R.; Zlokovic, B.V. Transport Pathways for Clearance of Human Alzheimer’s Amyloid β-Peptide and Apolipoproteins E and J in the Mouse Central Nervous System. J. Cereb. Blood Flow Metab. 2006, 27, 909–918. [Google Scholar] [CrossRef]
- Zhao, Z.; Sagare, A.P.; Ma, Q.; Halliday, M.R.; Kong, P.; Kisler, K.; A Winkler, E.; Ramanathan, A.; Kanekiyo, T.; Bu, G.; et al. Central role for PICALM in amyloid-β blood-brain barrier transcytosis and clearance. Nat. Neurosci. 2015, 18, 978–987. [Google Scholar] [CrossRef]
- Huang, Y.; Mahley, R.W. Apolipoprotein E: Structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol. Dis. 2014, 72 Pt A, 3–12. [Google Scholar] [CrossRef]
- Tai, L.M.; Mehra, S.; Shete, V.; Estus, S.; Rebeck, G.W.; Bu, G.; LaDu, M.J. Soluble apoE/Aβ complex: Mechanism and therapeutic target for APOE4-induced AD risk. Mol. Neurodegener. 2014, 9, 2. [Google Scholar] [CrossRef]
- Nelson, A.; Sagare, A.; Zlokovic, B. Blood–Brain Barrier Transport of Alzheimer’s Amyloid β-Peptide. In Developing Therapeutics for Alzheimer’s Disease; Elsevier: Amsterdam, The Netherlands, 2016; pp. 251–270. [Google Scholar] [CrossRef]
- Narayan, P.; Orte, A.; Clarke, R.W.; Bolognesi, B.; Hook, S.; Ganzinger, K.A.; Meehan, S.; Wilson, M.R.; Dobson, C.M.; Klenerman, D. The extracellular chaperone clusterin sequesters oligomeric forms of the amyloid-β1−40 peptide. Nat. Struct. Mol. Biol. 2011, 19, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Narayan, P.; Meehan, S.; Carver, J.A.; Wilson, M.R.; Dobson, C.M.; Klenerman, D. Amyloid-β Oligomers are Sequestered by both Intracellular and Extracellular Chaperones. Biochemistry 2012, 51, 9270–9276. [Google Scholar] [CrossRef] [PubMed]
- DeMattos, R.B.; Cirrito, J.R.; Parsadanian, M.; May, P.C.; A O’Dell, M.; Taylor, J.W.; Harmony, J.A.; Aronow, B.J.; Bales, K.R.; Paul, S.M.; et al. ApoE and Clusterin Cooperatively Suppress Aβ Levels and Deposition: Evidence that ApoE Regulates Extracellular Aβ Metabolism In Vivo. Neuron 2004, 41, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Guo, H.; Chow, N.; Sallstrom, J.; Bell, R.D.; Deane, R.; I Brooks, A.; Kanagala, S.; Rubio, A.; Sagare, A.; et al. Role of the MEOX2 homeobox gene in neurovascular dysfunction in Alzheimer disease. Nat. Med. 2005, 11, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Hunt, A.; Schönknecht, P.; Henze, M.; Seidl, U.; Haberkorn, U.; Schröder, J. Reduced cerebral glucose metabolism in patients at risk for Alzheimer’s disease. Psychiatry Res. Neuroimaging 2007, 155, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Deane, R.; Chow, N.; Long, X.; Sagare, A.; Singh, I.; Streb, J.W.; Guo, H.; Rubio, A.; Van Nostrand, W.; et al. SRF and myocardin regulate LRP-mediated amyloid-β clearance in brain vascular cells. Nature 2008, 11, 143–153. [Google Scholar] [CrossRef]
- Li, M.; Shang, D.-S.; Zhao, W.-D.; Tian, L.; Li, B.; Fang, W.-G.; Zhu, L.; Man, S.-M.; Chen, Y.-H. Amyloid β Interaction with Receptor for Advanced Glycation End Products Up-Regulates Brain Endothelial CCR5 Expression and Promotes T Cells Crossing the Blood-Brain Barrier. J. Immunol. 2009, 182, 5778–5788. [Google Scholar] [CrossRef]
- Jedlitschky, G.; Vogelgesang, S.; Kroemer, H.K. MDR1–P-glycoprotein (ABCB1)-Mediated Disposition of Amyloid-? Peptides: Implications for the Pathogenesis and Therapy of Alzheimer’s Disease. Clin. Pharmacol. Ther. 2010, 88, 441–443. [Google Scholar] [CrossRef]
- Bendayan, R.; Ronaldson, P.T.; Gingras, D.; Bendayan, M. In situ localization of P-glycoprotein (ABCB1) in human and rat brain. J. Histochem. Cytochem. 2006, 54, 1159–1167. [Google Scholar] [CrossRef]
- Ding, Y.; Zhong, Y.; Baldeshwiler, A.; Abner, E.L.; Bauer, B.; Hartz, A.M.S. Protecting P-glycoprotein at the blood–brain barrier from degradation in an Alzheimer’s disease mouse model. Fluids Barriers CNS 2021, 18, 10. [Google Scholar] [CrossRef]
- Chai, A.B.; Hartz, A.M.S.; Gao, X.; Yang, A.; Callaghan, R.; Gelissen, I.C. New Evidence for P-gp-Mediated Export of Amyloid-β Peptides in Molecular, Blood-Brain Barrier and Neuronal Models. Int. J. Mol. Sci. 2020, 22, 246. [Google Scholar] [CrossRef]
- van Assema, D.M.E.; Lubberink, M.; Bauer, M.; van der Flier, W.M.; Schuit, R.C.; Windhorst, A.D.; Comans, E.F.I.; Hoetjes, N.J.; Tolboom, N.; Langer, O.; et al. Blood–brain barrier P-glycoprotein function in Alzheimer’s disease. Brain 2011, 135, 181–189. [Google Scholar] [CrossRef]
- Deane, R.; Singh, I.; Sagare, A.P.; Bell, R.D.; Ross, N.T.; LaRue, B.; Love, R.; Perry, S.; Paquette, N.; Deane, R.J.; et al. A multimodal RAGE-specific inhibitor reduces amyloid β–mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Investig. 2012, 122, 1377–1392. [Google Scholar] [CrossRef]
- Wei, S.; Shang, S.; Dang, L.; Gao, F.; Gao, Y.; Gao, L.; Chen, C.; Huo, K.; Wang, J.; Wang, J.; et al. Blood Triglyceride and High-Density Lipoprotein Levels Are Associated with Plasma Amyloid-β Transport: A Population-Based Cross-Sectional Study. J. Alzheimer’s Dis. 2021, 84, 303–314. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Othman, I.; Aamir, K.; Shaikh, M.F. Impact of HMGB1, RAGE, and TLR4 in Alzheimer’s Disease (AD): From Risk Factors to Therapeutic Targeting. Cells 2020, 9, 383. [Google Scholar] [CrossRef]
- Fang, F.; Yu, Q.; Arancio, O.; Chen, D.; Gore, S.S.; Yan, S.S.; Yan, S.F. RAGE mediates Aβ accumulation in a mouse model of Alzheimer’s disease via modulation of β- and γ-secretase activity. Hum. Mol. Genet. 2018, 27, 1002–1014. [Google Scholar] [CrossRef]
- Kim, Y.; Park, J.; Choi, Y.K. The Role of Astrocytes in the Central Nervous System Focused on BK Channel and Heme Oxygenase Metabolites: A Review. Antioxidants 2019, 8, 121. [Google Scholar] [CrossRef]
- Parpura, V.; Verkhratsky, A. Homeostatic function of astrocytes: Ca2+ and Na+ signalling. Transl. Neurosci. 2012, 3, 334–344. [Google Scholar] [CrossRef]
- Bak, L.K.; Schousboe, A.; Waagepetersen, H.S. The glutamate/GABA-glutamine cycle: Aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 2006, 98, 641–653. [Google Scholar] [CrossRef]
- Heneka, M.T.; O’banion, M.K.; Terwel, D.; Kummer, M.P. Neuroinflammatory processes in Alzheimer’s disease. J. Neural Transm. 2010, 117, 919–947. [Google Scholar] [CrossRef]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef]
- Pajarillo, E.; Rizor, A.; Lee, J.; Aschner, M.; Lee, E. The role of astrocytic glutamate transporters GLT-1 and GLAST in neurological disorders: Potential targets for neurotherapeutics. Neuropharmacology 2019, 161, 107559. [Google Scholar] [CrossRef]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer’s disease. J. Neurochem. 2005, 93, 1412–1421. [Google Scholar] [CrossRef]
- Gao, L.; Zhang, Y.; Sterling, K.; Song, W. Brain-derived neurotrophic factor in Alzheimer’s disease and its pharmaceutical potential. Transl. Neurodegener. 2022, 11, 4. [Google Scholar] [CrossRef]
- Diniz, L.P.; Tortelli, V.; Matias, I.; Morgado, J.; Araujo, A.P.B.; Melo, H.M.; da Silva, G.S.S.; Alves-Leon, S.V.; de Souza, J.M.; Ferreira, S.T.; et al. Astrocyte Transforming Growth Factor Beta 1 Protects Synapses against Aβ Oligomers in Alzheimer’s Disease Model. J. Neurosci. 2017, 37, 6797–6809. [Google Scholar] [CrossRef]
- Ries, M.; Sastre, M. Mechanisms of Aβ Clearance and Degradation by Glial Cells. Front. Aging Neurosci. 2016, 8, 160. [Google Scholar] [CrossRef]
- Richetin, K.; Steullet, P.; Pachoud, M.; Perbet, R.; Parietti, E.; Maheswaran, M.; Eddarkaoui, S.; Bégard, S.; Pythoud, C.; Rey, M.; et al. Tau accumulation in astrocytes of the dentate gyrus induces neuronal dysfunction and memory deficits in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1567–1579. [Google Scholar] [CrossRef]
- Jiwaji, Z.; Tiwari, S.S.; Avilés-Reyes, R.X.; Hooley, M.; Hampton, D.; Torvell, M.; Johnson, D.A.; McQueen, J.; Baxter, P.; Sabari-Sankar, K.; et al. Reactive astrocytes acquire neuroprotective as well as deleterious signatures in response to Tau and Aß pathology. Nat. Commun. 2022, 13, 135. [Google Scholar] [CrossRef]
- Subramanian, J.; Savage, J.C.; Tremblay, M. Synaptic Loss in Alzheimer’s Disease: Mechanistic Insights Provided by Two-Photon in vivo Imaging of Transgenic Mouse Models. Front. Cell. Neurosci. 2020, 14, 592607. [Google Scholar] [CrossRef]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimer’s Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Jackson, J.; Jambrina, E.; Li, J.; Marston, H.; Menzies, F.; Phillips, K.; Gilmour, G. Targeting the Synapse in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 735. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.; Yang, F.; Zhu, W.; Cai, M.; Li, X.-T.; Zhang, J.-S.; Yu, Z.-H.; Zhang, W.; Cai, D.-F. Bilobalide inhibits inflammation and promotes the expression of Aβ degrading enzymes in astrocytes to rescue neuronal deficiency in AD models. Transl. Psychiatry 2021, 11, 542. [Google Scholar] [CrossRef]
- Kuwar, R.; Rolfe, A.; Di, L.; Blevins, H.; Xu, Y.; Sun, X.; Bloom, G.S.; Zhang, S.; Sun, D. A Novel Inhibitor Targeting NLRP3 Inflammasome Reduces Neuropathology and Improves Cognitive Function in Alzheimer’s Disease Transgenic Mice. J. Alzheimer’s Dis. 2021, 82, 1769–1783. [Google Scholar] [CrossRef]
- Yue, Q.; Zhou, X.; Zhang, Z.; Hoi, M.P.M. Murine Beta-Amyloid (1–42) Oligomers Disrupt Endothelial Barrier Integrity and VEGFR Signaling via Activating Astrocytes to Release Deleterious Soluble Factors. Int. J. Mol. Sci. 2022, 23, 1878. [Google Scholar] [CrossRef]
- Jackson, R.J.; Meltzer, J.C.; Nguyen, H.; Commins, C.; E Bennett, R.; Hudry, E.; Hyman, B.T. APOE4 derived from astrocytes leads to blood–brain barrier impairment. Brain 2021, 145, 3582–3593. [Google Scholar] [CrossRef]
- Kim, H.; Leng, K.; Park, J.; Sorets, A.G.; Kim, S.; Shostak, A.; Embalabala, R.J.; Mlouk, K.; Katdare, K.A.; Rose, I.V.L.; et al. Reactive astrocytes transduce inflammation in a blood-brain barrier model through a TNF-STAT3 signaling axis and secretion of alpha 1-antichymotrypsin. Nat. Commun. 2022, 13, 6581. [Google Scholar] [CrossRef]
- Pandit, R.; Leinenga, G.; Götz, J. Repeated ultrasound treatment of tau transgenic mice clears neuronal tau by autophagy and improves behavioral functions. Theranostics 2019, 9, 3754–3767. [Google Scholar] [CrossRef] [PubMed]
- Lipsman, N.; Meng, Y.; Bethune, A.J.; Huang, Y.; Lam, B.; Masellis, M.; Herrmann, N.; Heyn, C.; Aubert, I.; Boutet, A.; et al. Blood–brain barrier opening in Alzheimer’s disease using MR-guided focused ultrasound. Nat. Commun. 2018, 9, 2336. [Google Scholar] [CrossRef]
- Epelbaum, S.; Burgos, N.; Canney, M.; Matthews, D.; Houot, M.; Santin, M.D.; Desseaux, C.; Bouchoux, G.; Stroer, S.; Martin, C.; et al. Pilot study of repeated blood-brain barrier disruption in patients with mild Alzheimer’s disease with an implantable ultrasound device. Alzheimers Res. Ther. 2022, 14, 40. [Google Scholar] [CrossRef]
- Rezai, A.R.; Ranjan, M.; D’Haese, P.F.; Haut, M.W.; Carpenter, J.; Najib, U.; Mehta, R.I.; Chazen, J.L.; Zibly, Z.; Yates, J.R.; et al. Noninvasive hippocampal blood-brain barrier opening in Alzheimer’s disease with focused ultrasound. Proc. Natl. Acad. Sci. USA 2020, 117, 9180–9182. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.I.; Carpenter, J.S.; Mehta, R.I.; Haut, M.W.; Wang, P.; Ranjan, M.; Najib, U.; D’haese, P.-F.; Rezai, A.R. Ultrasound-mediated blood–brain barrier opening uncovers an intracerebral perivenous fluid network in persons with Alzheimer’s disease. Fluids Barriers CNS 2023, 20, 46. [Google Scholar] [CrossRef] [PubMed]
- Lazic, A.; Balint, V.; Ninkovic, D.S.; Peric, M.; Stevanovic, M. Reactive and Senescent Astroglial Phenotypes as Hallmarks of Brain Pathologies. Int. J. Mol. Sci. 2022, 23, 4995. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, Y.; Xu, C.; Zhang, H.; Lin, C. TLR4 Targeting as a Promising Therapeutic Strategy for Alzheimer Disease Treatment. Front. Neurosci. 2020, 14, 602508. [Google Scholar] [CrossRef]
- Ceyzériat, K.; Ben Haim, L.; Denizot, A.; Pommier, D.; Matos, M.; Guillemaud, O.; Palomares, M.-A.; Abjean, L.; Petit, F.; Gipchtein, P.; et al. Modulation of astrocyte reactivity improves functional deficits in mouse models of Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 104. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-S.; Kam, T.-I.; Lee, S.; Park, H.; Oh, Y.; Kwon, S.-H.; Song, J.-J.; Kim, D.; Kim, H.; Jhaldiyal, A.; et al. Blocking microglial activation of reactive astrocytes is neuroprotective in models of Alzheimer’s disease. Acta Neuropathol. Commun. 2021, 9, 78. [Google Scholar] [CrossRef]
- Davis, N.; Mota, B.C.; Stead, L.; Palmer, E.O.C.; Lombardero, L.; Rodríguez-Puertas, R.; de Paola, V.; Barnes, S.J.; Sastre, M. Pharmacological ablation of astrocytes reduces Aβ degradation and synaptic connectivity in an ex vivo model of Alzheimer’s disease. J. Neuroinflamm. 2021, 18, 73. [Google Scholar] [CrossRef]
- Kelly, M. Exercise-Induced Modulation of Neuroinflammation in Models of Alzheimer’s Disease. Brain Plast. 2018, 4, 81–94. [Google Scholar] [CrossRef]
- Ribarič, S. Physical Exercise, a Potential Non-Pharmacological Intervention for Attenuating Neuroinflammation and Cognitive Decline in Alzheimer’s Disease Patients. Int. J. Mol. Sci. 2022, 23, 3245. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Moreno, J.; Satorres, E.; Soria-Urios, G.; Meléndez, J.C. Cognitive Stimulation in Moderate Alzheimer’s Disease. J. Appl. Gerontol. 2022, 41, 1934–1941. [Google Scholar] [CrossRef] [PubMed]
- Stekic, A.; Zeljkovic, M.; Kontic, M.Z.; Mihajlovic, K.; Adzic, M.; Stevanovic, I.; Ninkovic, M.; Grkovic, I.; Ilic, T.V.; Nedeljkovic, N.; et al. Intermittent Theta Burst Stimulation Ameliorates Cognitive Deficit and Attenuates Neuroinflammation via PI3K/Akt/mTOR Signaling Pathway in Alzheimer’s-Like Disease Model. Front. Aging Neurosci. 2022, 14, 889983. [Google Scholar] [CrossRef] [PubMed]
- Luzi, F.; Savickas, V.; Taddei, C.; Hader, S.; Singh, N.; Gee, A.D.; Bongarzone, S. Radiolabeling of [11C]FPS-ZM1, a receptor for advanced glycation end products-targeting positron emission tomography radiotracer, using a [11C]CO2-to-[11C]CO chemical conversion. Futur. Med. Chem. 2020, 12, 511–521. [Google Scholar] [CrossRef]
- Barca, C.; Foray, C.; Hermann, S.; Döring, C.; Schäfers, M.; Jacobs, A.H.; Zinnhardt, B. Characterization of the inflammatory post-ischemic tissue by full volumetric analysis of a multimodal imaging dataset. Neuroimage 2020, 222, 117217. [Google Scholar] [CrossRef]
- Zhou, R.; Ji, B.; Kong, Y.; Qin, L.; Ren, W.; Guan, Y.; Ni, R. PET Imaging of Neuroinflammation in Alzheimer’s Disease. Front. Immunol. 2021, 12, 739130. [Google Scholar] [CrossRef]
- Livingston, N.R.; Calsolaro, V.; Hinz, R.; Nowell, J.; Raza, S.; Gentleman, S.; Tyacke, R.J.; Myers, J.; Venkataraman, A.V.; Perneczky, R.; et al. Relationship between astrocyte reactivity, using novel 11C-BU99008 PET, and glucose metabolism, grey matter volume and amyloid load in cognitively impaired individuals. Mol. Psychiatry 2022, 27, 2019–2029. [Google Scholar] [CrossRef]
- Ferrari-Souza, J.P.; Ferreira, P.C.L.; Bellaver, B.; Tissot, C.; Wang, Y.-T.; Leffa, D.T.; Brum, W.S.; Benedet, A.L.; Ashton, N.J.; De Bastiani, M.A.; et al. Astrocyte biomarker signatures of amyloid-β and tau pathologies in Alzheimer’s disease. Mol. Psychiatry 2022, 27, 4781–4789. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).